
Kinesin Eg5 Selective Inhibition by Newly Synthesized Molecules as an Alternative Approach to Counteract Breast Cancer Progression: An In Vitro Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
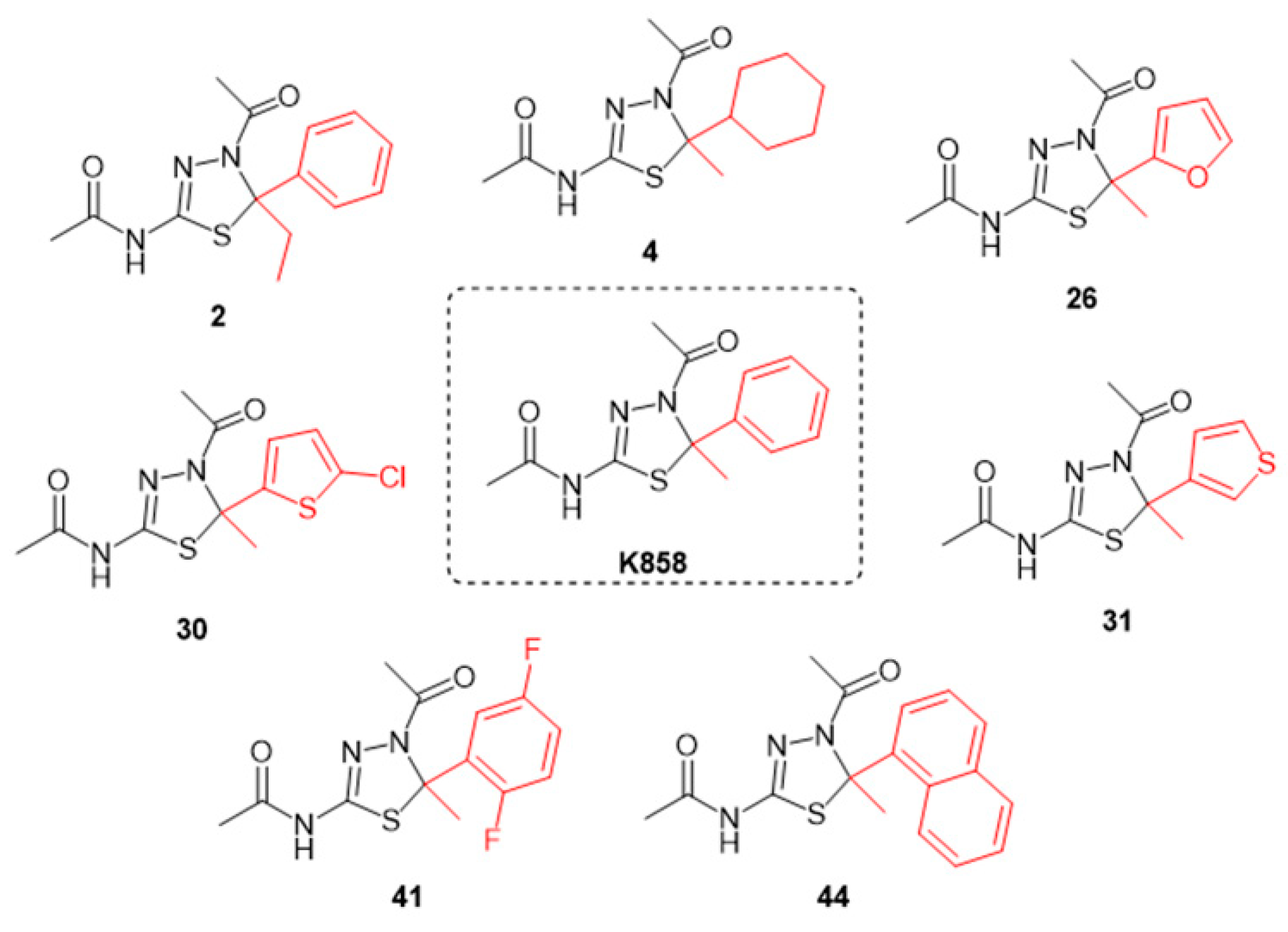
2.1. Chemistry
2.2. Cell Cultures
2.3. MTT Assay
2.4. Cytotoxicity Assay (LDH Assay)
2.5. Transwell Invasion and Migration Assay
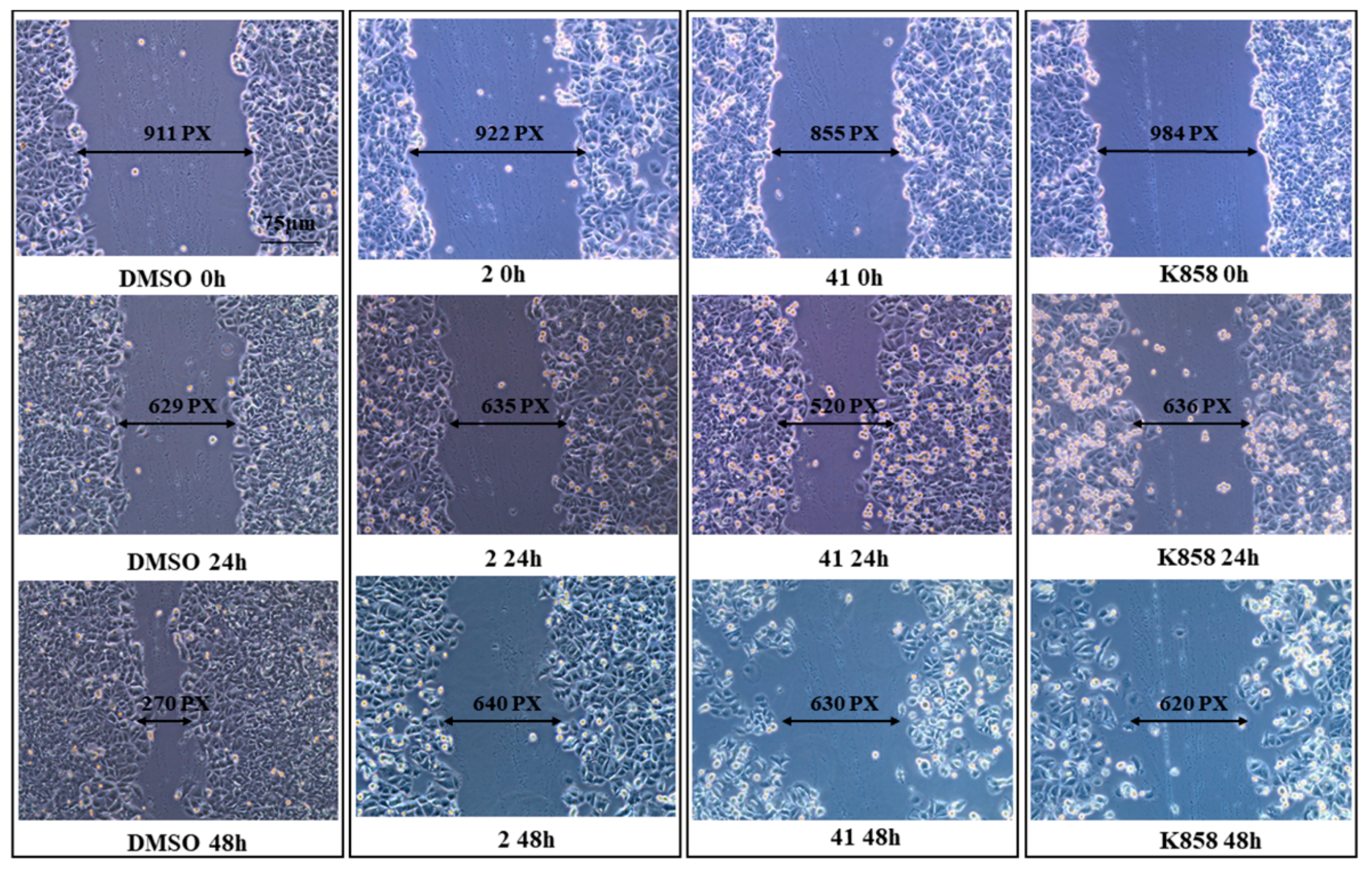
2.6. Wound Healing
2.7. Protein Extraction and Western Blot analysis
2.8. Detection of Apoptosis and Necrosis by Flow Cytometry
2.9. Statistical Analysis
3. Results
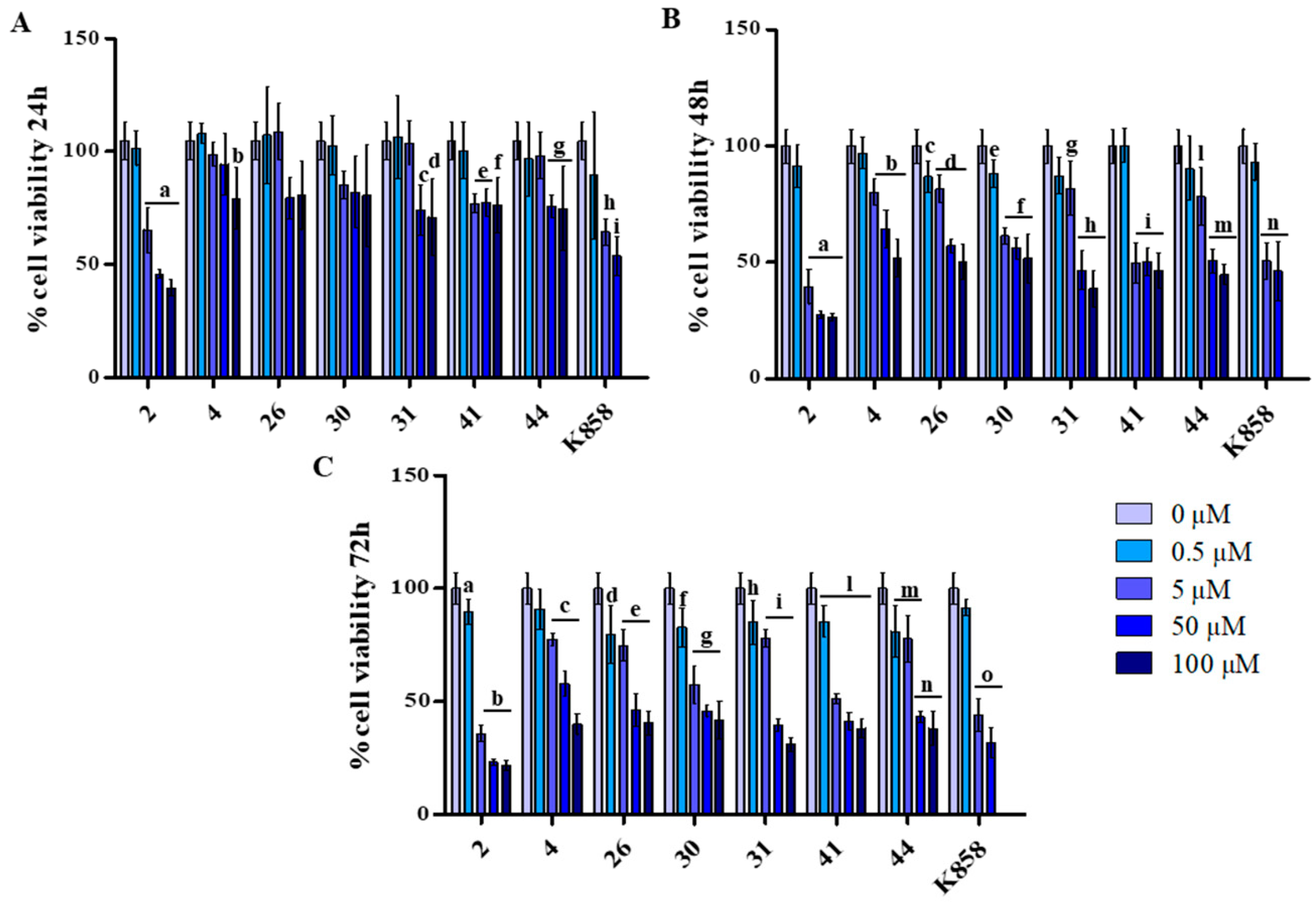
3.1. Effect of K858 and Its Analogues on MCF7 Cell Viability
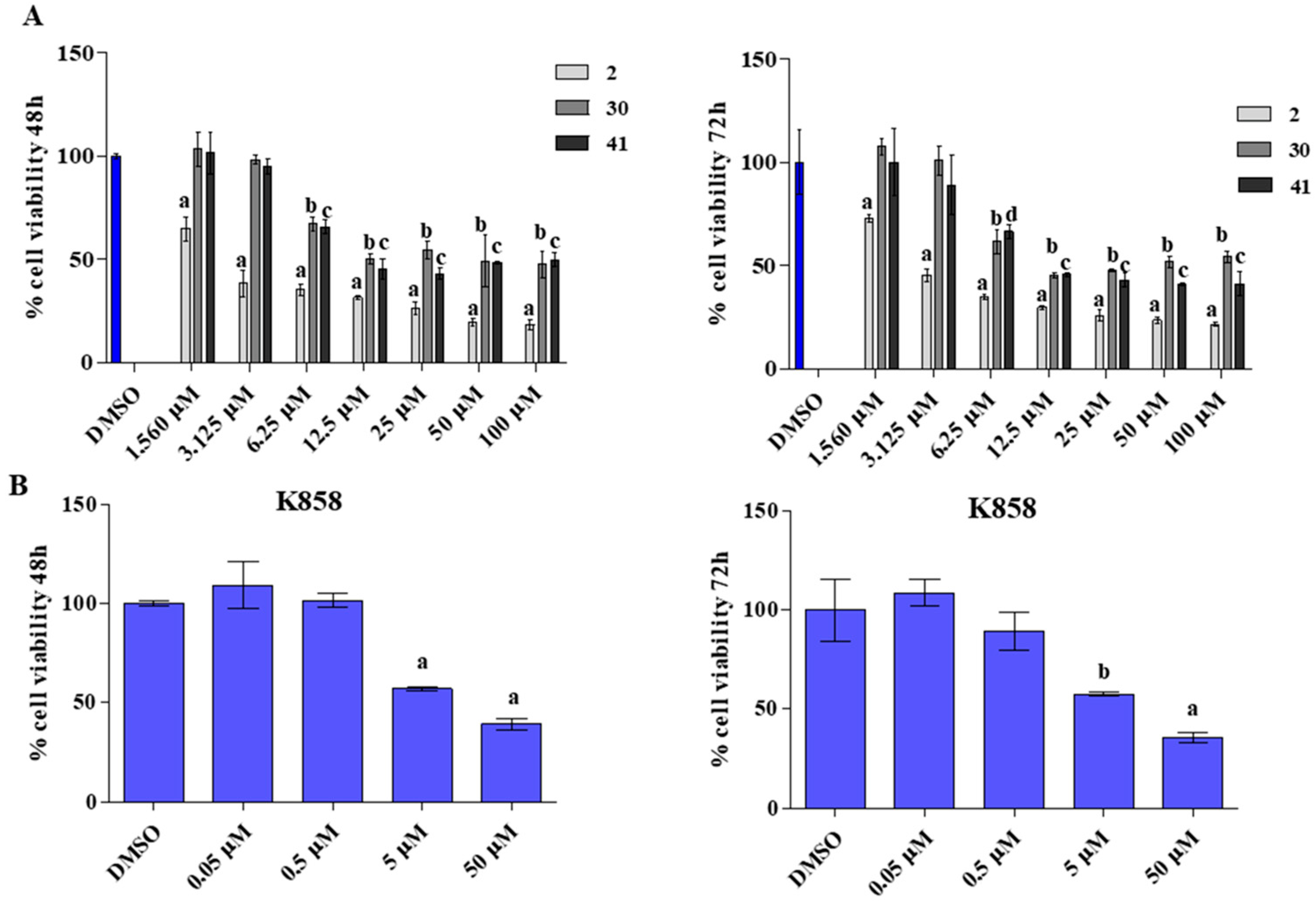
3.2. Effect of 2, 41, and K858 on non-Tumoral Cells Viability
3.3. Effect of 2, 41, and K858 on MCF7 Cytotoxicity
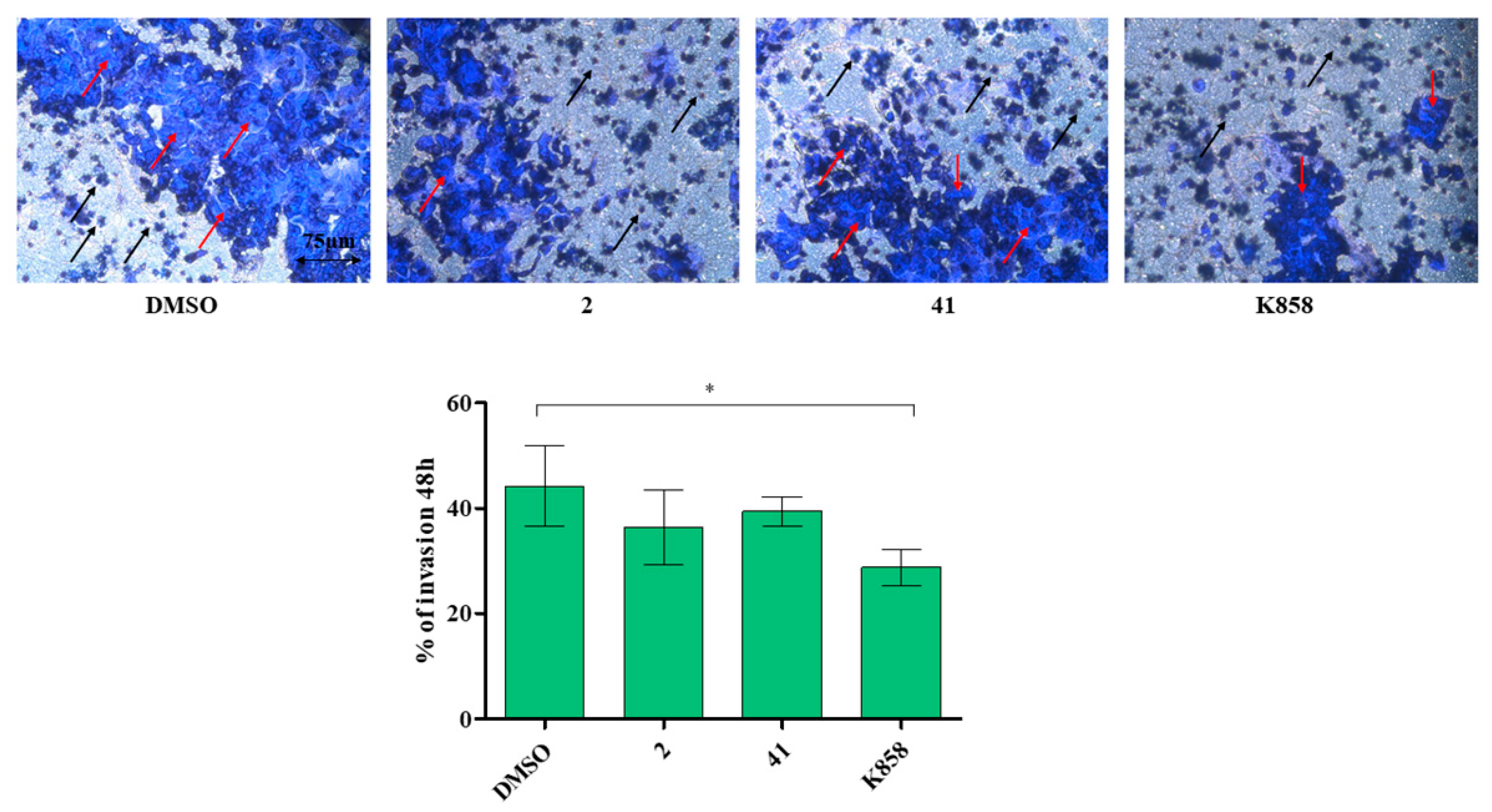
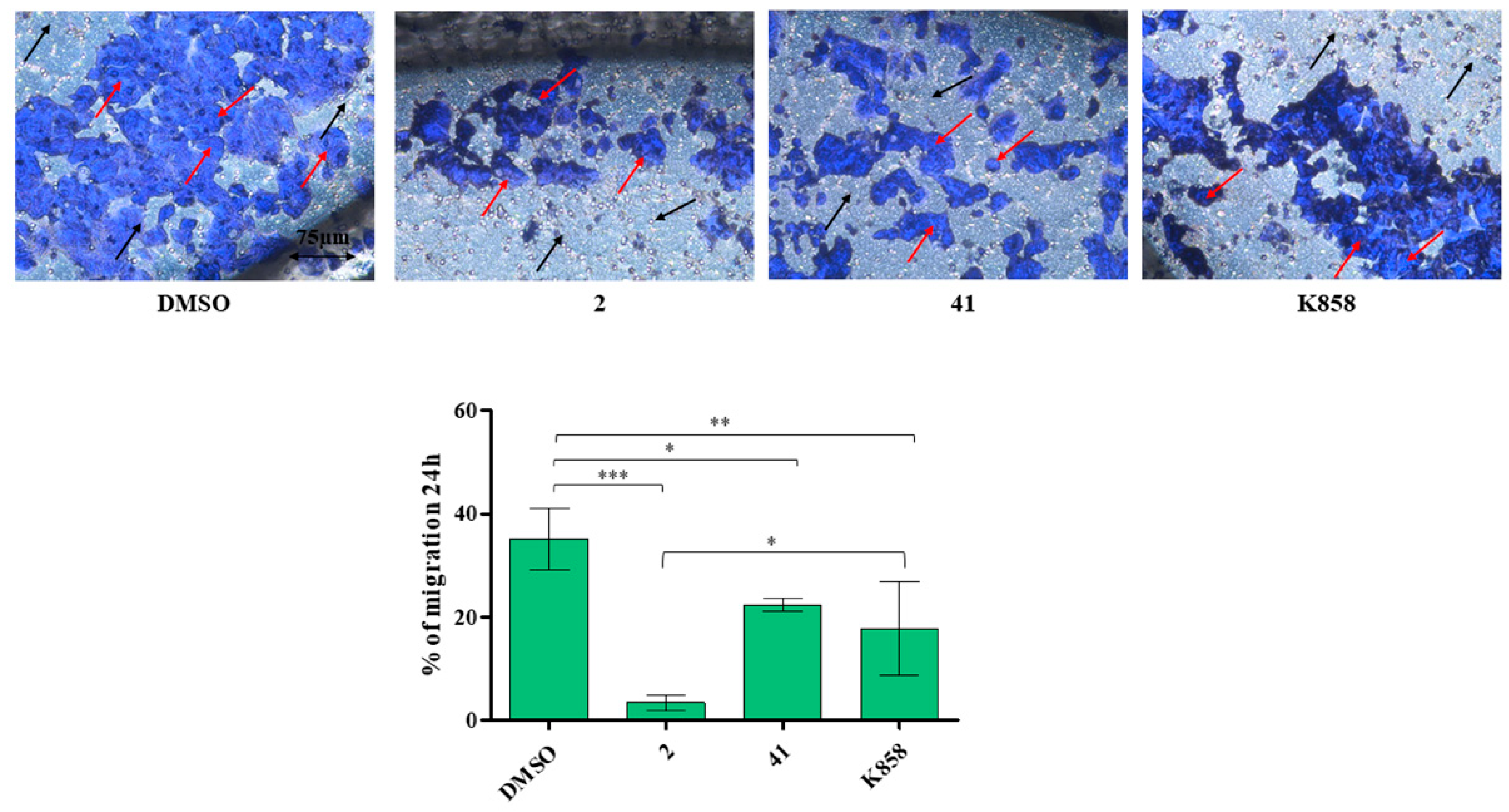
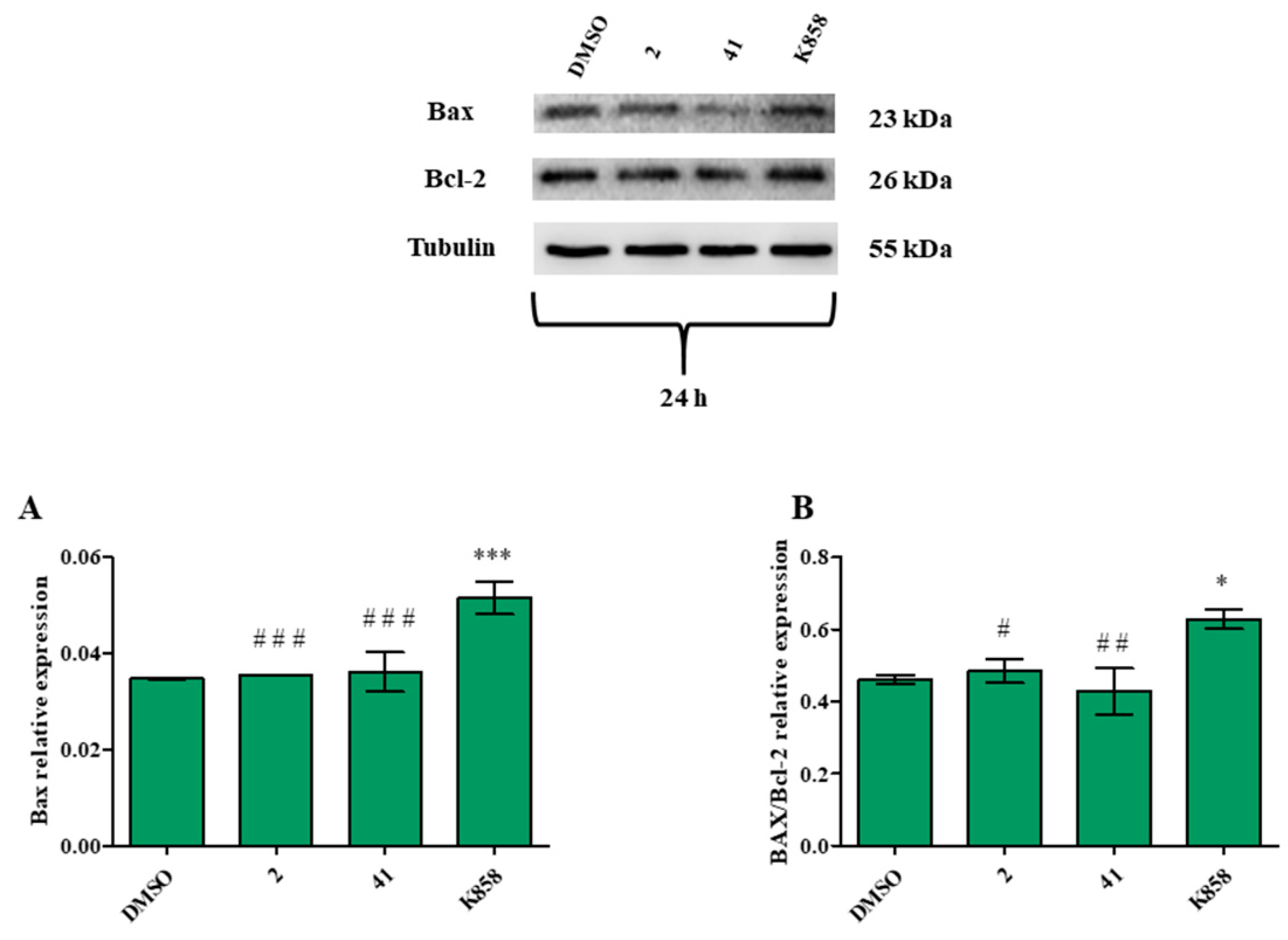
3.4. Effect of Eg5 Inhibitors on MCF7 Cell Invasion, Migration and on NF-kB, MMP-9, HIF-1α, and VEGF Proteins Expression
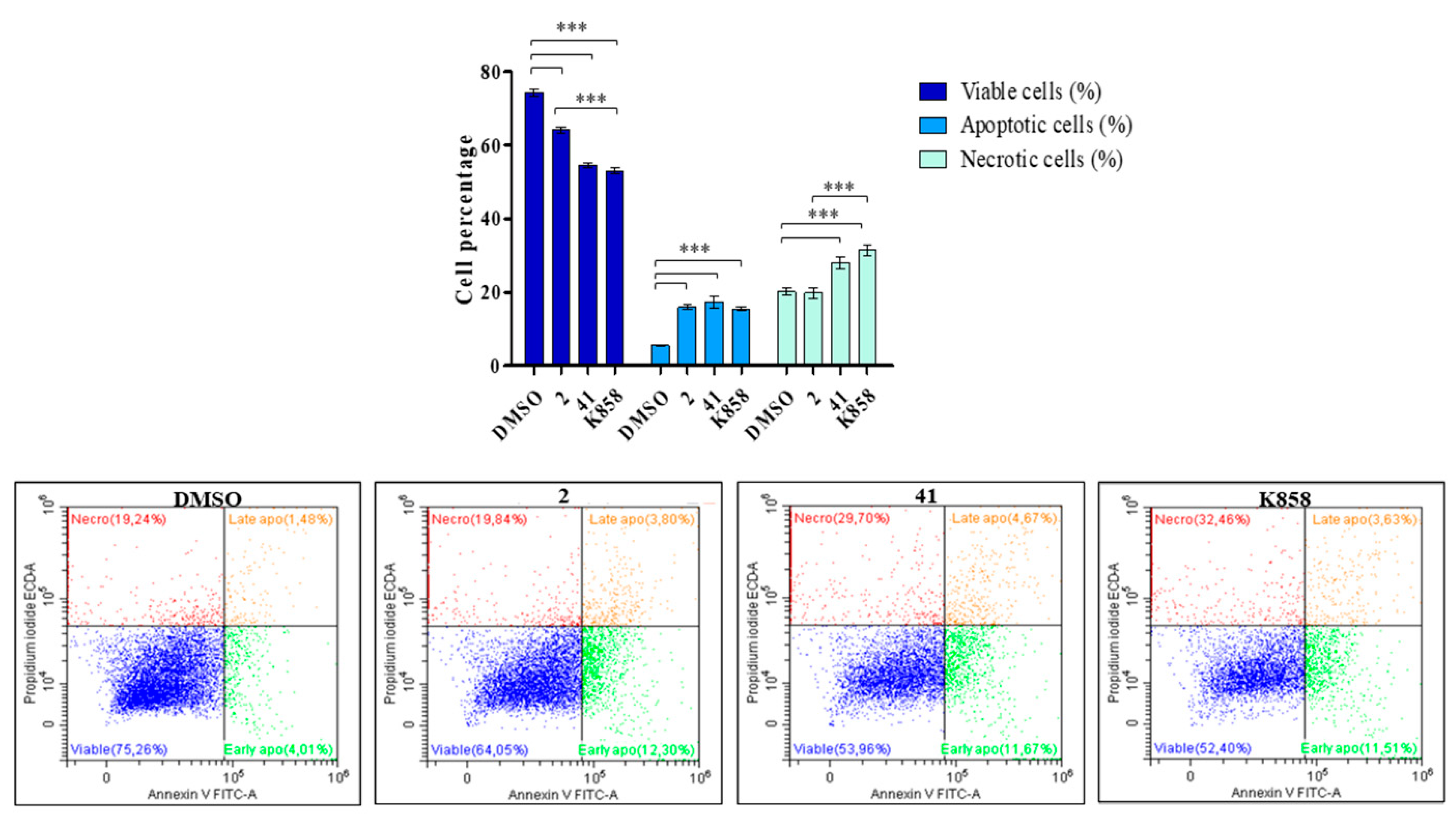
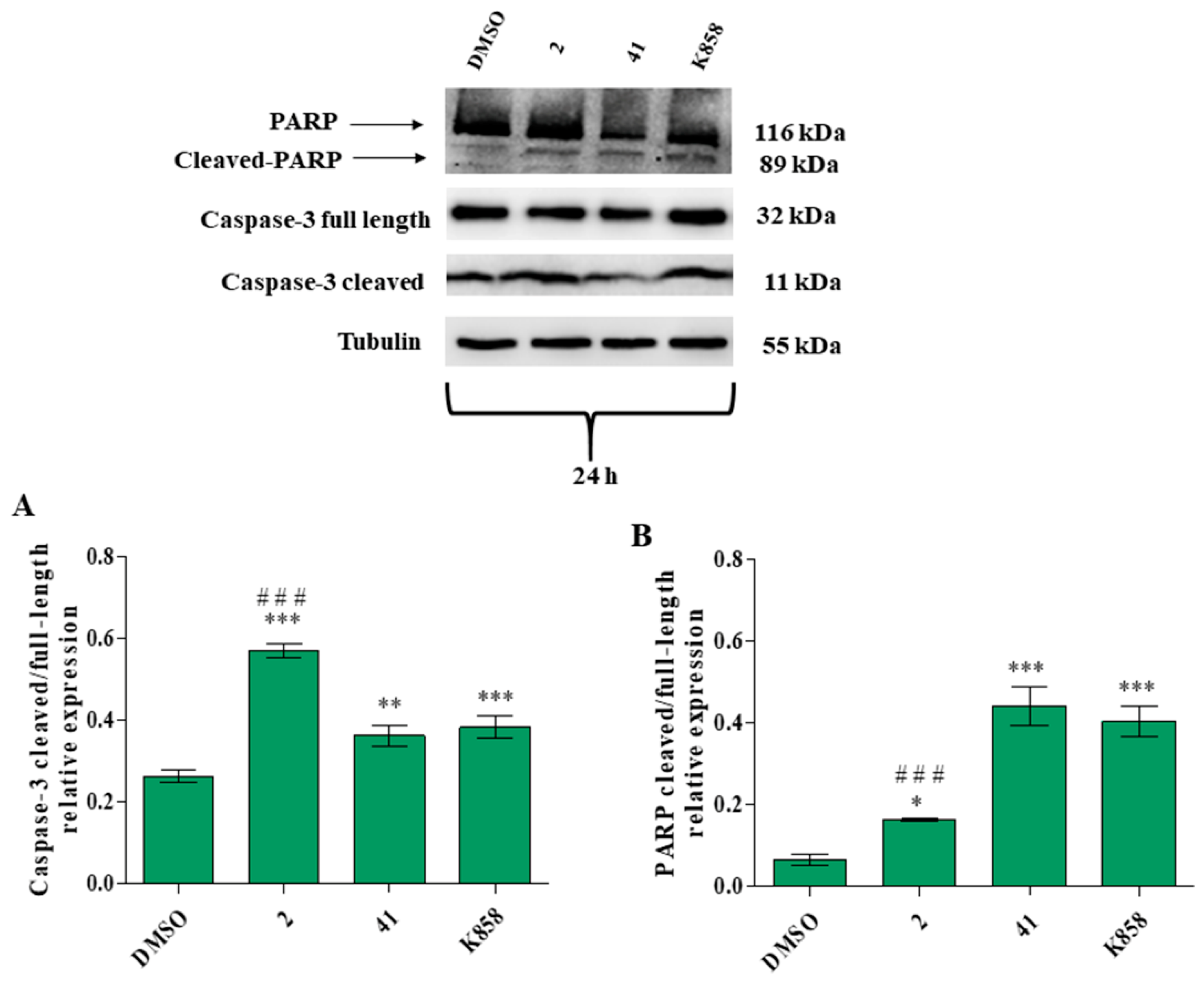
3.5. Evaluation of MCF7 Apoptosis and Necrosis Induction by 2, 41, and K858 Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Fisusi, F.A.; Akala, E.O. Drug Combinations in Breast Cancer Therapy. Pharm. Nanotechnol. 2019, 7, 3–23. [Google Scholar] [CrossRef]
- Hunter, B.; Allingham, J.S. These motors were made for walking. Protein Sci. 2020, 29, 1707–1723. [Google Scholar] [CrossRef]
- Miki, H.; Okada, Y.; Hirokawa, N. Analysis of the kinesin superfamily: Insights into structure and function. Trends Cell Biol. 2005, 15, 467–476. [Google Scholar] [CrossRef]
- Konjikusic, M.J.; Gray, R.S.; Wallingford, J.B. The developmental biology of kinesins. Dev. Biol. 2021, 469, 26–36. [Google Scholar] [CrossRef]
- Mann, B.J.; Wadsworth, P. Kinesin-5 Regulation and Function in Mitosis. Trends Cell Biol. 2019, 29, 66–79. [Google Scholar] [CrossRef]
- Rath, O.; Kozielski, F. Kinesins and cancer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef]
- Wei, D.; Rui, B.; Qingquan, F.; Chen, C.; Ping, H.Y.; Xiaoling, S.; Hao, W.; Jun, G. KIF11 promotes cell proliferation via ERBB2/PI3K/AKT signaling pathway in gallbladder cancer. Int. J. Biol. Sci. 2021, 17, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, G.; Cui, S.; Du, G. Upregulation of KIF11 in TP53 Mutant Glioma Promotes Tumor Stemness and Drug Resistance. Cell Mol. Neurobiol. 2021, 42, 1477–1485. [Google Scholar] [CrossRef]
- Ling, J.; Wang, Y.; Ma, L.; Zheng, Y.; Tang, H.; Meng, L.; Zhang, L. KIF11, a plus end-directed kinesin, as a key gene in benzo(a)pyrene-induced non-small cell lung cancer. Environ. Toxicol. Pharmacol. 2022, 89, 103775. [Google Scholar] [CrossRef] [PubMed]
- Hansson, K.; Radke, K.; Aaltonen, K.; Saarela, J.; Mañas, A.; Sjölund, J.; Smith, E.M.; Pietras, K.; Påhlman, S.; Wennerberg, K.; et al. Therapeutic targeting of KSP in preclinical models of high-risk neuroblastoma. Sci. Transl. Med. 2020, 12, eaba4434. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Saez, I.; Skoufias, D.A. Eg5 targeting agents: From new anti-mitotic based inhibitor discovery to cancer therapy and resistance. Biochem. Pharmacol. 2021, 184, 114364. [Google Scholar] [CrossRef]
- Jin, Q.; Huang, F.; Wang, X.; Zhu, H.; Xian, Y.; Li, J.; Zhang, S.; Ni, Q. High Eg5 expression predicts poor prognosis in breast cancer. Oncotarget 2017, 8, 62208–62216. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Chen, W.R.; Yang, L.C.; Wang, J.; Sun, J.Y.; Zhang, W.W.; He, Z.Y.; Wu, S.G. KIF11 Functions as an Oncogene and Is Associated with Poor Outcomes from Breast Cancer. Cancer Res. Treat. 2019, 51, 1207–1221. [Google Scholar] [CrossRef]
- Guido, B.C.; Ramos, L.M.; Nolasco, D.O.; Nobrega, C.C.; Andrade, B.Y.; Pic-Taylor, A.; Neto, B.A.; Corrêa, J.R. Impact of kinesin Eg5 inhibition by 3,4-dihydropyrimidin-2(1H)-one derivatives on various breast cancer cell features. BMC Cancer 2015, 15, 283. [Google Scholar] [CrossRef] [Green Version]
- De Iuliis, F.; Taglieri, L.; Salerno, G.; Giuffrida, A.; Milana, B.; Giantulli, S.; Carradori, S.; Silvestri, I.; Scarpa, S. The kinesin Eg5 inhibitor K858 induces apoptosis but also survivin-related chemoresistance in breast cancer cells. Investig. New Drugs 2016, 34, 399–406. [Google Scholar] [CrossRef]
- Planas-Silva, M.D.; Filatova, I.S. Estrogen-dependent regulation of Eg5 in breast cancer cells. Anti-Cancer Drugs 2007, 18, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Huang, M.; Guo, J.; Zhang, H.; Wang, D.; Liu, T.; Liu, H.; Chen, S.; Gao, P.; Mu, K. The Role of CHK1 Varies with the Status of Oestrogen-receptor and Progesterone-receptor in the Targeted Therapy for Breast Cancer. Int. J. Biol. Sci. 2020, 16, 1388–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talapatra, S.K.; Tham, C.L.; Guglielmi, P.; Cirilli, R.; Chandrasekaran, B.; Karpoormath, R.; Carradori, S.; Kozielski, F. Crystal structure of the Eg5-K858 complex and implications for structure-based design of thiadiazole-containing inhibitors. Eur. J. Med. Chem. 2018, 156, 641–651. [Google Scholar] [CrossRef]
- Marconi, G.D.; Carradori, S.; Ricci, A.; Guglielmi, P.; Cataldi, A.; Zara, S. Kinesin Eg5 Targeting Inhibitors as a New Strategy for Gastric Adenocarcinoma Treatment. Molecules 2019, 24, 3948. [Google Scholar] [CrossRef] [Green Version]
- Ricci, A.; Gallorini, M.; Del Bufalo, D.; Cataldi, A.; D’Agostino, I.; Carradori, S.; Zara, S. Negative Modulation of the Angiogenic Cascade Induced by Allosteric Kinesin Eg5 Inhibitors in a Gastric Adenocarcinoma In Vitro Model. Molecules 2022, 27, 957. [Google Scholar] [CrossRef] [PubMed]
- De Monte, C.; Carradori, S.; Secci, D.; D’Ascenzio, M.; Guglielmi, P.; Mollica, A.; Morrone, S.; Scarpa, S.; Aglianò, A.M.; Giantulli, S.; et al. Synthesis and pharmacological screening of a large library of 1,3,4-thiadiazolines as innovative therapeutic tools for the treatment of prostate cancer and melanoma. Eur. J. Med. Chem. 2015, 105, 245–262. [Google Scholar] [CrossRef] [PubMed]
- De Colli, M.; Tortorella, P.; Marconi, G.D.; Agamennone, M.; Campestre, C.; Tauro, M.; Cataldi, A.; Zara, S. In vitro comparison of new bisphosphonic acids and zoledronate effects on human gingival fibroblasts viability, inflammation and matrix turnover. Clin. Oral Investig. 2016, 20, 2013–2021. [Google Scholar] [CrossRef]
- Scully, O.J.; Bay, B.H.; Yip, G.; Yu, Y. Breast cancer metastasis. Cancer Genom. Proteom. 2012, 9, 311–320. [Google Scholar]
- Novikov, N.M.; Zolotaryova, S.Y.; Gautreau, A.M.; Denisov, E.V. Mutational drivers of cancer cell migration and invasion. Br. J. Cancer 2021, 124, 102–114. [Google Scholar] [CrossRef]
- Sun, X.D.; Shi, X.J.; Sun, X.O.; Luo, Y.G.; Wu, X.J.; Yao, C.F.; Yu, H.Y.; Li, D.W.; Liu, M.; Zhou, J. Dimethylenastron suppresses human pancreatic cancer cell migration and invasion in vitro via allosteric inhibition of mitotic kinesin Eg5. Acta Pharmacol. Sin. 2011, 32, 1543–1548. [Google Scholar] [CrossRef] [Green Version]
- Nicolai, A.; Taurone, S.; Carradori, S.; Artico, M.; Greco, A.; Costi, R.; Scarpa, S. The kinesin Eg5 inhibitor K858 exerts antiproliferative and proapoptotic effects and attenuates the invasive potential of head and neck squamous carcinoma cells. Investig. New Drugs 2022, 40, 556. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Adhikari, N.; Banerjee, S.; Amin, S.A.; Jha, T. Matrix metalloproteinase-9 (MMP-9) and its inhibitors in cancer: A minireview. Eur. J. Med. Chem. 2020, 194, 112260. [Google Scholar] [CrossRef] [PubMed]
- Taglieri, L.; Rubinacci, G.; Giuffrida, A.; Carradori, S.; Scarpa, S. The kinesin Eg5 inhibitor K858 induces apoptosis and reverses the malignant invasive phenotype in human glioblastoma cells. Investig. New Drugs 2018, 36, 28–35. [Google Scholar] [CrossRef]
- Takahra, T.; Smart, D.E.; Oakley, F.; Mann, D.A. Induction of myofibroblast MMP-9 transcription in three-dimensional collagen I gel cultures: Regulation by NF-kappaB, AP-1 and Sp. Int. J. Biochem. Cell Biol. 2004, 36, 353–363. [Google Scholar] [CrossRef]
- Wu, L.; Zhao, K.Q.; Wang, W.; Cui, L.N.; Hu, L.L.; Jiang, X.X.; Shuai, J.; Sun, Y.P. Nuclear receptor coactivator 6 promotes HTR-8/SVneo cell invasion and migration by activating NF-κB-mediated MMP9 transcription. Cell Prolif. 2020, 53, e12876. [Google Scholar] [CrossRef]
- Yenmis, G.; Yaprak Sarac, E.; Besli, N.; Soydas, T.; Tastan, C.; Dilek Kancagi, D.; Yilanci, M.; Senol, K.; Karagulle, O.O.; Ekmekci, C.G.; et al. Anti-cancer effect of metformin on the metastasis and invasion of primary breast cancer cells through mediating NF-kB activity. Acta Histochem. 2021, 123, 151709. [Google Scholar] [CrossRef] [PubMed]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)-key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Prior, C.; Perez-Gracia, J.L.; Garcia-Donas, J.; Rodriguez-Antona, C.; Guruceaga, E.; Esteban, E.; Suarez, C.; Castellano, D.; del Alba, A.G.; Lozano, M.D.; et al. Identification of tissue microRNAs predictive of sunitinib activity in patients with metastatic renal cell carcinoma. PLoS ONE 2014, 9, e86263. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Jadhav, U.; Chigurupati, S.; Lakka, S.S.; Mohanam, S. Inhibition of matrix metalloproteinase-9 reduces in vitro invasion and angiogenesis in human microvascular endothelial cells. Int. J. Oncol. 2004, 25, 1407–1414. [Google Scholar] [CrossRef]
- Exertier, P.; Javerzat, S.; Wang, B.; Franco, M.; Herbert, J.; Platonova, N.; Winandy, M.; Pujol, N.; Nivelles, O.; Ormenese, S.; et al. Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5. Oncotarget 2013, 4, 2302–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed. Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, K.M.; Jakovljevic, J.; Woolford, J.L., Jr.; Saunders, W.S. Kinesin molecular motor Eg5 functions during polypeptide synthesis. Mol. Biol. Cell 2011, 22, 3420–3430. [Google Scholar] [CrossRef] [PubMed]
- Wakana, Y.; Villeneuve, J.; van Galen, J.; Cruz-Garcia, D.; Tagaya, M.; Malhotra, V. Kinesin-5/Eg5 is important for transport of CARTS from the trans-Golgi network to the cell surface. J. Cell Biol. 2013, 202, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giantulli, S.; De Iuliis, F.; Taglieri, L.; Carradori, S.; Menichelli, G.; Morrone, S.; Scarpa, S.; Silvestri, I. Growth arrest and apoptosis induced by kinesin Eg5 inhibitor K858 and by its 1,3,4-thiadiazoline analogue in tumor cells. Anti-Cancer Drugs 2018, 29, 674–681. [Google Scholar] [CrossRef]
- Jungwirth, G.; Yu, T.; Cao, J.; Eddine, M.A.; Moustafa, M.; Warta, R.; Debus, J.; Unterberg, A.; Abdollahi, A.; Herold-Mende, C. KIF11 inhibitors filanesib and ispinesib inhibit meningioma growth in vitro and in vivo. Cancer Lett. 2021, 506, 1–10. [Google Scholar] [CrossRef]
- Leizerman, I.; Avunie-Masala, R.; Elkabets, M.; Fich, A.; Gheber, L. Differential effects of monastrol in two human cell lines. Cell Mol. Life Sci. 2004, 61, 2060–2070. [Google Scholar] [CrossRef]
- Asraf, H.; Avunie-Masala, R.; Hershfinkel, M.; Gheber, L. Mitotic slippage and expression of survivin are linked to differential sensitivity of human cancer cell-lines to the Kinesin-5 inhibitor monastrol. PLoS ONE 2015, 10, e0129255. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, A.; Cataldi, A.; Carradori, S.; Zara, S. Kinesin Eg5 Selective Inhibition by Newly Synthesized Molecules as an Alternative Approach to Counteract Breast Cancer Progression: An In Vitro Study. Biology 2022, 11, 1450. https://doi.org/10.3390/biology11101450
Ricci A, Cataldi A, Carradori S, Zara S. Kinesin Eg5 Selective Inhibition by Newly Synthesized Molecules as an Alternative Approach to Counteract Breast Cancer Progression: An In Vitro Study. Biology. 2022; 11(10):1450. https://doi.org/10.3390/biology11101450
Chicago/Turabian StyleRicci, Alessia, Amelia Cataldi, Simone Carradori, and Susi Zara. 2022. "Kinesin Eg5 Selective Inhibition by Newly Synthesized Molecules as an Alternative Approach to Counteract Breast Cancer Progression: An In Vitro Study" Biology 11, no. 10: 1450. https://doi.org/10.3390/biology11101450