

A Systems Biology and LASSO-Based Approach to Decipher the Transcriptome–Interactome Signature for Predicting Non-Small Cell Lung Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
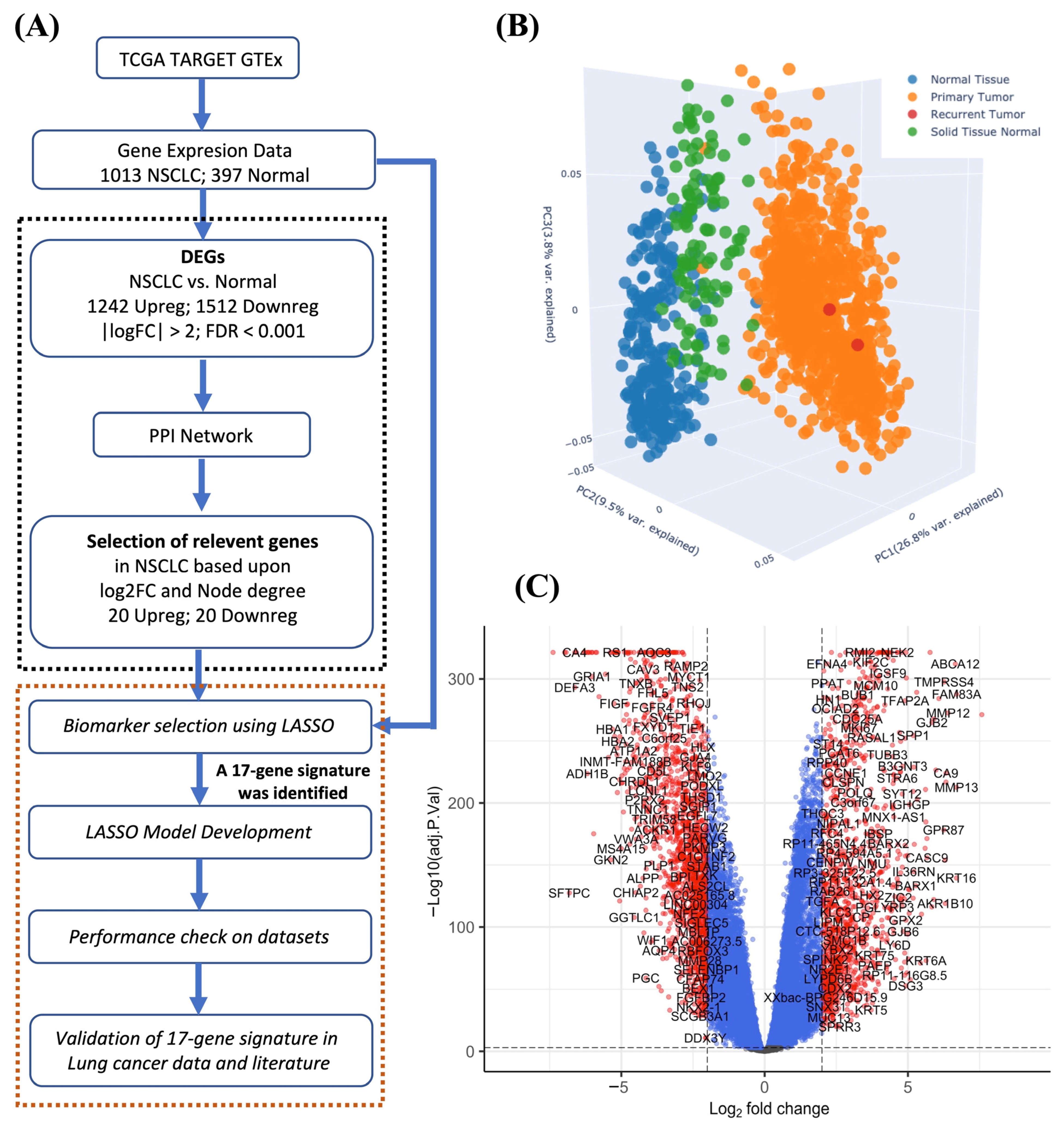
2.1. Identification of DEGs
2.2. Construction of the Interaction Network
2.3. Identification of Biologically Important Nodes in the Network
2.4. Training and Testing Dataset
2.5. Construction of the LASSO Model
2.6. Performance of the Models
- (1)
- Sensitivity, also called the recall or true positive rate, which indicates the percentage of correctly predicted cancer samples.
- (2)
- Specificity, which indicates the percentage of correctly predicted normal samples.
- (3)
- Accuracy is the percentage of correct predictions overall.
- (4)
- Positive predictive value (PPV), also called the precision.
- (5)
- Negative predictive value (NPV)
- (6)
- Area Under the Curve (AUC). The performance was tested at various thresholds using the receiver operating characteristics (ROC) to plot a graph of the true positive rate (sensitivity on the y-axis) versus the false positive rate (1 – specificity on the x-axis). The higher the mean AUC-ROC values, the better the model was for distinguishing between lung cancer and normal samples. In addition, we used precision–recall (PRC), which is a plot of the precision (positive predictive value on the y-axis) versus the recall (sensitivity or true positive rate on the x-axis) for all possible thresholds. The larger the value of AUC-PRC, the better the model’s performance. If the positive and negative data were imbalanced, the PRC curve was preferred for checking the model’s performance.
2.7. Functional Enrichment of Key Genes Obtained by the LASSO Model
3. Results
3.1. Identification of DEGs
3.2. Identification of the Relevant Interacting Genes
3.3. Development of the LASSO Model
3.4. Performance of the LASSO Model on Independent Datasets
3.5. Comparative Analysis of Logistic Regression Models
3.6. Interaction Network and Functional Enrichment Analysis of Genes from the LASSO Model
3.7. Validation of the 17-Gene Signature in Lung Cancer Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Hendriks, L.E.L. Targeted therapies for unresectable stage III non-small cell lung cancer. Mediastinum 2021, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Beckett, P.; Tata, L.J.; Hubbard, R.B. Risk factors and survival outcome for non-elective referral in non-small cell lung cancer patients--analysis based on the National Lung Cancer Audit. Lung Cancer 2014, 83, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Taylor-Stokes, G.; Roughley, A. Symptom burden and quality of life in advanced non-small cell lung cancer patients in France and Germany. Lung Cancer 2013, 81, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.S.; Wong, W.; Ravelo, A.; Miller, P.J.E.; Schwartzberg, L.S. Effectiveness outcomes and health related quality of life impact of disease progression in patients with advanced nonsquamous NSCLC treated in real-world community oncology settings: Results from a prospective medical record registry study. Health Qual. Life Outcomes 2017, 15, 160. [Google Scholar] [CrossRef] [Green Version]
- Grant, M.; Sun, V.; Fujinami, R.; Sidhu, R.; Otis-Green, S.; Juarez, G.; Klein, L.; Ferrell, B. Family caregiver burden, skills preparedness, and quality of life in non-small cell lung cancer. Oncol. Nurs. Forum 2013, 40, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Gibson, G.J.; Loddenkemper, R.; Lundbäck, B.; Sibille, Y. Respiratory health and disease in Europe: The new European Lung White Book. Eur. Respir. J. 2013, 42, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Soda, H.; Tomita, H.; Kohno, S.; Oka, M. Limitation of annual screening chest radiography for the diagnosis of lung cancer. A retrospective study. Cancer 1993, 72, 2341–2346. [Google Scholar] [CrossRef]
- Prorok, P.C.; Andriole, G.L.; Bresalier, R.S.; Buys, S.S.; Chia, D.; Crawford, E.D.; Fogel, R.; Gelmann, E.P.; Gilbert, F.; Hasson, M.A.; et al. Design of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control. Clin. Trials 2000, 21, 273S–309S. [Google Scholar] [CrossRef]
- Prabhakar, B.; Shende, P.; Augustine, S. Current trends and emerging diagnostic techniques for lung cancer. Biomed. Pharmacother. 2018, 106, 1586–1599. [Google Scholar] [CrossRef]
- Rodríguez, J.; Avila, J.; Rolfo, C.; Ruíz-Patiño, A.; Russo, A.; Ricaurte, L.; Ordóñez-Reyes, C.; Arrieta, O.; Zatarain-Barrón, Z.L.; Recondo, G.; et al. When Tissue is an Issue the Liquid Biopsy is Nonissue: A Review. Oncol. Ther. 2021, 9, 89–110. [Google Scholar] [CrossRef]
- Goebel, C.; Louden, C.L.; McKenna, R., Jr.; Onugha, O.; Wachtel, A.; Long, T. Diagnosis of Non-small Cell Lung Cancer for Early Stage Asymptomatic Patients. Cancer Genom. Proteom. 2019, 16, 229–244. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, Q.; Yuan, S.; Xie, W.; Liu, Y.; Xiang, Y.; Wu, N.; Wu, L.; Ma, X.; Cai, T.; et al. Genetic predisposition to lung cancer: Comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies. Sci. Rep. 2017, 7, 8371. [Google Scholar] [CrossRef] [Green Version]
- Walser, T.; Cui, X.; Yanagawa, J.; Lee, J.M.; Heinrich, E.; Lee, G.; Sharma, S.; Dubinett, S.M. Smoking and lung cancer: The role of inflammation. Proc. Am. Thorac. Soc. 2008, 5, 811–815. [Google Scholar] [CrossRef] [Green Version]
- Dietel, M.; Bubendorf, L.; Dingemans, A.M.; Dooms, C.; Elmberger, G.; Garcia, R.C.; Kerr, K.M.; Lim, E.; Lopez-Rios, F.; Thunnissen, E.; et al. Diagnostic procedures for non-small-cell lung cancer (NSCLC): Recommendations of the European Expert Group. Thorax 2016, 71, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, F. Integrated Network Analysis Reveals FOXM1 and MYBL2 as Key Regulators of Cell Proliferation in Non-small Cell Lung Cancer. Front. Oncol. 2019, 9, 1011. [Google Scholar] [CrossRef]
- Giannos, P.; Kechagias, K.S.; Gal, A. Identification of Prognostic Gene Biomarkers in Non-Small Cell Lung Cancer Progression by Integrated Bioinformatics Analysis. Biology 2021, 10, 1200. [Google Scholar] [CrossRef]
- Feng, Z.; Qu, J.; Liu, X.; Liang, J.; Li, Y.; Jiang, J.; Zhang, H.; Tian, H. Integrated bioinformatics analysis of differentially expressed genes and immune cell infiltration characteristics in Esophageal Squamous cell carcinoma. Sci. Rep. 2021, 11, 16696. [Google Scholar] [CrossRef]
- Giannos, P.; Kechagias, K.S.; Bowden, S.; Tabassum, N.; Paraskevaidi, M.; Kyrgiou, M. PCNA in Cervical Intraepithelial Neoplasia and Cervical Cancer: An Interaction Network Analysis of Differentially Expressed Genes. Front. Oncol. 2021, 11, 779042. [Google Scholar] [CrossRef]
- Cassidy, A.; Duffy, S.W.; Myles, J.P.; Liloglou, T.; Field, J.K. Lung cancer risk prediction: A tool for early detection. Int. J. Cancer 2007, 120, 1–6. [Google Scholar] [CrossRef]
- Gray, E.P.; Teare, M.D.; Stevens, J.; Archer, R. Risk Prediction Models for Lung Cancer: A Systematic Review. Clin. Lung Cancer 2016, 17, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.S.; Mayya, A.M. A new tool to predict lung cancer based on risk factors. Heliyon 2020, 6, e03402. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.C.; Wang, Y.H.; Yang, H.C.; Bai, K.J.; Wang, H.H.; Li, Y.J. Artificial Intelligence-Based Prediction of Lung Cancer Risk Using Nonimaging Electronic Medical Records: Deep Learning Approach. J. Med. Internet Res. 2021, 23, e26256. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Ma, X.; Song, P. A prognostic model of non small cell lung cancer based on TCGA and ImmPort databases. Sci. Rep. 2022, 12, 437. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Li, J.; Li, G.; Poon, S.; Harrington, P.B. Analysis and Modeling for Big Data in Cancer Research. BioMed Res. Int. 2017, 2017, 1972097. [Google Scholar] [CrossRef] [Green Version]
- Shait Mohammed, M.R.; Zamzami, M.; Choudhry, H.; Ahmed, F.; Ateeq, B.; Khan, M.I. The Histone H3K27me3 Demethylases KDM6A/B Resist Anoikis and Transcriptionally Regulate Stemness-Related Genes. Front. Cell Dev. Biol. 2022, 10, 780176. [Google Scholar] [CrossRef]
- Chin, L.; Hahn, W.C.; Getz, G.; Meyerson, M. Making sense of cancer genomic data. Genes Dev. 2011, 25, 534–555. [Google Scholar] [CrossRef] [Green Version]
- Karimi, M.R.; Karimi, A.H.; Abolmaali, S.; Sadeghi, M.; Schmitz, U. Prospects and challenges of cancer systems medicine: From genes to disease networks. Brief. Bioinform. 2022, 23, bbab343. [Google Scholar] [CrossRef]
- Ahmed, F.; Kumar, M.; Raghava, G.P. Prediction of polyadenylation signals in human DNA sequences using nucleotide frequencies. Silico Biol. 2009, 9, 135–148. [Google Scholar] [CrossRef]
- Ahmed, F.; Raghava, G.P. Designing of highly effective complementary and mismatch siRNAs for silencing a gene. PLoS ONE 2011, 6, e23443. [Google Scholar] [CrossRef]
- Ahmed, F.; Senthil-Kumar, M.; Dai, X.; Ramu, V.S.; Lee, S.; Mysore, K.S.; Zhao, P.X. pssRNAit: A Web Server for Designing Effective and Specific Plant siRNAs with Genome-Wide Off-Target Assessment. Plant Physiol. 2020, 184, 65–81. [Google Scholar] [CrossRef]
- Ahmed, F.; Ansari, H.R.; Raghava, G.P. Prediction of guide strand of microRNAs from its sequence and secondary structure. BMC Bioinform. 2009, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, F.; Kaundal, R.; Raghava, G.P. PHDcleav: A SVM based method for predicting human Dicer cleavage sites using sequence and secondary structure of miRNA precursors. BMC Bioinform. 2013, 14 (Suppl. 14), S9. [Google Scholar] [CrossRef] [Green Version]
- Elemento, O.; Leslie, C.; Lundin, J.; Tourassi, G. Artificial intelligence in cancer research, diagnosis and therapy. Nat. Rev. Cancer 2021, 21, 747–752. [Google Scholar] [CrossRef]
- Arjmand, B.; Hamidpour, S.K.; Tayanloo-Beik, A.; Goodarzi, P.; Aghayan, H.R.; Adibi, H.; Larijani, B. Machine Learning: A New Prospect in Multi-Omics Data Analysis of Cancer. Front. Genet. 2022, 13, 824451. [Google Scholar] [CrossRef]
- Liu, R.; Rizzo, S.; Whipple, S.; Pal, N.; Pineda, A.L.; Lu, M.; Arnieri, B.; Lu, Y.; Capra, W.; Copping, R.; et al. Evaluating eligibility criteria of oncology trials using real-world data and AI. Nature 2021, 592, 629–633. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Network, C.G.A.R. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Carithers, L.J.; Ardlie, K.; Barcus, M.; Branton, P.A.; Britton, A.; Buia, S.A.; Compton, C.C.; DeLuca, D.S.; Peter-Demchok, J.; Gelfand, E.T.; et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv. Biobank. 2015, 13, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2018. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 10 November 2021).
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. A Publ. Protein Soc. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Killcoyne, S.; Carter, G.W.; Smith, J.; Boyle, J. Cytoscape: A community-based framework for network modeling. Methods Mol. Biol. 2009, 563, 219–239. [Google Scholar] [CrossRef]
- Sanchez-Palencia, A.; Gomez-Morales, M.; Gomez-Capilla, J.A.; Pedraza, V.; Boyero, L.; Rosell, R.; Fárez-Vidal, M.E. Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. Int. J. Cancer 2011, 129, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.Y.; Juan, C.C.; Hisa, J.Y.; Su, L.J.; Lee, Y.C.; Chou, H.Y.; Chen, J.M.; Wu, Y.C.; Chiu, S.C.; Hsu, C.P.; et al. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 2012, 103, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.P.; Tsai, M.H.; Lee, J.M.; Hsu, C.P.; Chen, P.C.; Lin, C.W.; Shih, J.Y.; Yang, P.C.; Hsiao, C.K.; Lai, L.C.; et al. Identification of a novel biomarker, SEMA5A, for non-small cell lung carcinoma in nonsmoking women. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2590–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W3221. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, N. Cancer survival data emphasise importance of early diagnosis. Bmj 2019, 364, l408. [Google Scholar] [CrossRef]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Blandin Knight, S.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef]
- Goncalves, S.; Fong, P.C.; Blokhina, M. Artificial intelligence for early diagnosis of lung cancer through incidental nodule detection in low- and middle-income countries-acceleration during the COVID-19 pandemic but here to stay. Am. J. Cancer Res. 2022, 12, 1–16. [Google Scholar]
- Joshi, S.; Pandit, S.V.; Shukla, P.K.; Almalki, A.H.; Othman, N.A.; Alharbi, A.; Alhassan, M. Analysis of Smart Lung Tumour Detector and Stage Classifier Using Deep Learning Techniques with Internet of Things. Comput. Intell. Neurosci. 2022, 2022, 4608145. [Google Scholar] [CrossRef]
- Mao, Q.; Zhang, L.; Zhang, Y.; Dong, G.; Yang, Y.; Xia, W.; Chen, B.; Ma, W.; Hu, J.; Jiang, F.; et al. A network-based signature to predict the survival of non-smoking lung adenocarcinoma. Cancer Manag. Res. 2018, 10, 2683–2693. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lu, D.; Liu, X.; Jiang, J.; Feng, S.; Dong, X.; Shi, X.; Wu, H.; Xiong, G.; Cai, K. Survival-related risk score of lung adenocarcinoma identified by weight gene co-expression network analysis. Oncol. Lett. 2019, 18, 4441–4448. [Google Scholar] [CrossRef] [Green Version]
- Batada, N.N.; Hurst, L.D.; Tyers, M. Evolutionary and physiological importance of hub proteins. PLoS Comput. Biol. 2006, 2, e88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.L.; Qu, L.W.; Ma, L.; Zhou, Y.C.; Wang, G.Z.; Zhao, X.C.; Zhang, C.; Zhang, Y.F.; Wang, M.; Zhang, M.Y.; et al. Genome-wide identification of transcription factors that are critical to non-small cell lung cancer. Cancer Lett. 2018, 434, 132–143. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, H.; Fan, L.; Fang, M.; He, X.; Lu, B.; Pang, Z. CLEC4s as Potential Therapeutic Targets in Hepatocellular Carcinoma Microenvironment. Front. Cell Dev. Biol. 2021, 9, 681372. [Google Scholar] [CrossRef]
- Zheng, Q.; Min, S.; Zhou, Q. Identification of potential diagnostic and prognostic biomarkers for LUAD based on TCGA and GEO databases. Biosci. Rep. 2021, 41, BSR20204370. [Google Scholar] [CrossRef]
- Mei, L.; Huang, C.; Wang, A.; Zhang, X. Association between ADRB2, IL33, and IL2RB gene polymorphisms and lung cancer risk in a Chinese Han population. Int. Immunopharmacol. 2019, 77, 105930. [Google Scholar] [CrossRef]
- Tian, Z.Q.; Li, Z.H.; Wen, S.W.; Zhang, Y.F.; Li, Y.; Cheng, J.G.; Wang, G.Y. Identification of Commonly Dysregulated Genes in Non-small-cell Lung Cancer by Integrated Analysis of Microarray Data and qRT-PCR Validation. Lung 2015, 193, 583–592. [Google Scholar] [CrossRef]
- Treweek, T.M.; Meehan, S.; Ecroyd, H.; Carver, J.A. Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell Mol. Life Sci. 2015, 72, 429–451. [Google Scholar] [CrossRef] [PubMed]
- Kamradt, M.C.; Lu, M.; Werner, M.E.; Kwan, T.; Chen, F.; Strohecker, A.; Oshita, S.; Wilkinson, J.C.; Yu, C.; Oliver, P.G.; et al. The small heat shock protein alpha B-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. J. Biol. Chem. 2005, 280, 11059–11066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Zhang, D.W.; Lin, H.; Xiong, L.; Liu, Y.; Li, Q.D.; Ma, J.; Cao, Q.; Chen, R.J.; Zhu, J.; et al. Alpha B-crystallin is a new prognostic marker for laryngeal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2012, 31, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.K.; Lui, P.C.; Tan, P.H.; Yamaguchi, R.; Moriya, T.; Yu, A.M.; Shao, M.M.; Hliang, T.; Wong, S.I.; Tse, G.M. Increased alpha-B-crystallin expression in mammary metaplastic carcinomas. Histopathology 2011, 59, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Lloyd, A.J.; Mundy, J.; Deva, R.; Lampe, G.; Hawley, C.; Boyle, G.; Griffin, R.; Thompson, C.; Shah, P. Is alpha-B crystallin an independent marker for prognosis in lung cancer? Heart Lung Circ. 2013, 22, 759–766. [Google Scholar] [CrossRef]
- Xu, G.; Dang, C. CMTM5 is downregulated and suppresses tumour growth in hepatocellular carcinoma through regulating PI3K-AKT signalling. Cancer Cell Int. 2017, 17, 113. [Google Scholar] [CrossRef]
- He, J.; Wu, M.; Xiong, L.; Gong, Y.; Yu, R.; Peng, W.; Li, L.; Li, L.; Tian, S.; Wang, Y.; et al. BTB/POZ zinc finger protein ZBTB16 inhibits breast cancer proliferation and metastasis through upregulating ZBTB28 and antagonizing BCL6/ZBTB27. Clin. Epigenetics 2020, 12, 82. [Google Scholar] [CrossRef]
- Jin, Y.; Pan, Y.; Zheng, S.; Liu, Y.; Xu, J.; Peng, Y.; Zhang, Z.; Wang, Y.; Xiong, Y.; Xu, L.; et al. Inactivation of EGLN3 hydroxylase facilitates Erk3 degradation via autophagy and impedes lung cancer growth. Oncogene 2022, 41, 1752–1766. [Google Scholar] [CrossRef]
- Qiu, H.L.; Deng, S.Z.; Li, C.; Tian, Z.N.; Song, X.Q.; Yao, G.D.; Geng, J.S. High expression of KIF14 is associated with poor prognosis in patients with epithelial ovarian cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 239–245. [Google Scholar]
- Arora, A.; Agarwal, D.; Abdel-Fatah, T.M.; Lu, H.; Croteau, D.L.; Moseley, P.; Aleskandarany, M.A.; Green, A.R.; Ball, G.; Rakha, E.A.; et al. RECQL4 helicase has oncogenic potential in sporadic breast cancers. J. Pathol. 2016, 238, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Alzahrani, F.A.; Ahmed, F.; Sharma, M.; Rehan, M.; Mahfuz, M.; Baeshen, M.N.; Hawsawi, Y.; Almatrafi, A.; Alsagaby, S.A.; Kamal, M.A.; et al. Investigating the pathogenic SNPs in BLM helicase and their biological consequences by computational approach. Sci. Rep. 2020, 10, 12377. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef]
- Chen, Y.; Chu, C.W.; Chen, M.I.C.; Cook, A.R. The utility of LASSO-based models for real time forecasts of endemic infectious diseases: A cross country comparison. J. Biomed. Inform. 2018, 81, 16–30. [Google Scholar] [CrossRef]
- Meng, Z.; Wang, M.; Guo, S.; Zhou, Y.; Zheng, M.; Liu, M.; Chen, Y.; Yang, Z.; Zhao, B.; Ying, B. Development and Validation of a LASSO Prediction Model for Better Identification of Ischemic Stroke: A Case-Control Study in China. Front. Aging Neurosci. 2021, 13, 630437. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Type | Disease Class | Training Dataset (TR) | Test Dataset 1 (TD1) |
|---|---|---|---|
| Primary tumor | Lung cancer (positive = 1) | 802 | 209 |
| Recurrent tumor | 2 | 0 | |
| Normal solid tissue | Normal lung (negative = 0) | 91 | 18 |
| Normal tissue (GTEx) | 233 | 55 | |
| Total | 1128 | 282 |
| Upregulate Genes | Downregulated Genes | ||||
|---|---|---|---|---|---|
| Log2FC | Degree | Name | Log2FC | Degree | Name |
| 4.76 | 698 | SOX2 | −5.16 | 279 | GPR17 |
| 4.33 | 804 | CDC20 | −5.06 | 297 | ZBTB16 |
| 4.19 | 1143 | ANLN | −4.13 | 278 | CMTM5 |
| 4.08 | 1063 | KIF20A | −3.66 | 723 | ACTC1 |
| 3.73 | 1834 | KIF14 | −3.55 | 294 | USHBP1 |
| 3.51 | 817 | AURKB | −2.87 | 429 | TRIM63 |
| 3.29 | 550 | MKI67 | −2.84 | 404 | ADRB2 |
| 3.24 | 635 | CDK1 | −2.82 | 715 | LRRK2 |
| 3.14 | 577 | RAD51 | −2.70 | 270 | NR4A1 |
| 3.12 | 1207 | MCM2 | −2.70 | 764 | MEOX2 |
| 3.10 | 695 | PLK1 | −2.69 | 843 | CAV1 |
| 2.86 | 529 | CDKN2A | −2.59 | 315 | CLEC4D |
| 2.62 | 1032 | KIF23 | −2.46 | 411 | CLEC4E |
| 2.58 | 934 | ECT2 | −2.43 | 455 | GPR182 |
| 2.50 | 986 | PRC1 | −2.41 | 433 | SYNE3 |
| 2.50 | 1465 | RECQL4 | −2.33 | 342 | CRYAB |
| 2.37 | 553 | KRT31 | −2.27 | 294 | KANK2 |
| 2.35 | 1354 | EGLN3 | −2.19 | 297 | ALB |
| 2.31 | 849 | CDH1 | −2.09 | 367 | LMO2 |
| 2.14 | 1189 | AGR2 | −2.07 | 348 | HECW2 |
| Threshold | Accuracy | Specificity | Sensitivity | TN | TP | FN | FP | NPV | PPV |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.741 | 0.000 | 1.000 | 0 | 209 | 0 | 73 | NA | 0.741 |
| 0.1 | 0.982 | 0.945 | 0.995 | 69 | 208 | 1 | 4 | 0.986 | 0.981 |
| 0.2 | 0.982 | 0.945 | 0.995 | 69 | 208 | 1 | 4 | 0.986 | 0.981 |
| 0.3 | 0.982 | 0.945 | 0.995 | 69 | 208 | 1 | 4 | 0.986 | 0.981 |
| 0.4 | 0.982 | 0.945 | 0.995 | 69 | 208 | 1 | 4 | 0.986 | 0.981 |
| 0.5 | 0.986 | 0.959 | 0.995 | 70 | 208 | 1 | 3 | 0.986 | 0.986 |
| 0.6 | 0.982 | 0.959 | 0.990 | 70 | 207 | 2 | 3 | 0.972 | 0.986 |
| 0.7 | 0.986 | 0.973 | 0.990 | 71 | 207 | 2 | 2 | 0.973 | 0.990 |
| 0.8 | 0.982 | 0.973 | 0.986 | 71 | 206 | 3 | 2 | 0.959 | 0.990 |
| 0.9 | 0.986 | 1.000 | 0.981 | 73 | 205 | 4 | 0 | 0.948 | 1.000 |
| 1 | 0.259 | 1.000 | 0.000 | 73 | 0 | 209 | 0 | 0.259 | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, F.; Khan, A.A.; Ansari, H.R.; Haque, A. A Systems Biology and LASSO-Based Approach to Decipher the Transcriptome–Interactome Signature for Predicting Non-Small Cell Lung Cancer. Biology 2022, 11, 1752. https://doi.org/10.3390/biology11121752
Ahmed F, Khan AA, Ansari HR, Haque A. A Systems Biology and LASSO-Based Approach to Decipher the Transcriptome–Interactome Signature for Predicting Non-Small Cell Lung Cancer. Biology. 2022; 11(12):1752. https://doi.org/10.3390/biology11121752
Chicago/Turabian StyleAhmed, Firoz, Abdul Arif Khan, Hifzur Rahman Ansari, and Absarul Haque. 2022. "A Systems Biology and LASSO-Based Approach to Decipher the Transcriptome–Interactome Signature for Predicting Non-Small Cell Lung Cancer" Biology 11, no. 12: 1752. https://doi.org/10.3390/biology11121752