
Consequential Impact of Particulate Matter Linked Inter-Fibrillar Mitochondrial Dysfunction in Rat Myocardium Subjected to Ischemia Reperfusion Injury


 ,
,  ,
,  and
and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Animal Experimental Groups
2.3. Assessment of Cardiac Hemodynamics and Injury Parameters
2.4. Assessment of Mitochondrial Electron Transport Chain Enzyme Activities and Oxidative Stress
2.5. Gene Expression Studies
2.6. MtDNA Copy Number Estimation
2.7. Analysis of In Vitro Protein Translation Capacity of Mitochondria
2.8. Inductively Coupled Mass Spectrophotometry Analysis
2.9. Statistical Analysis
3. Results
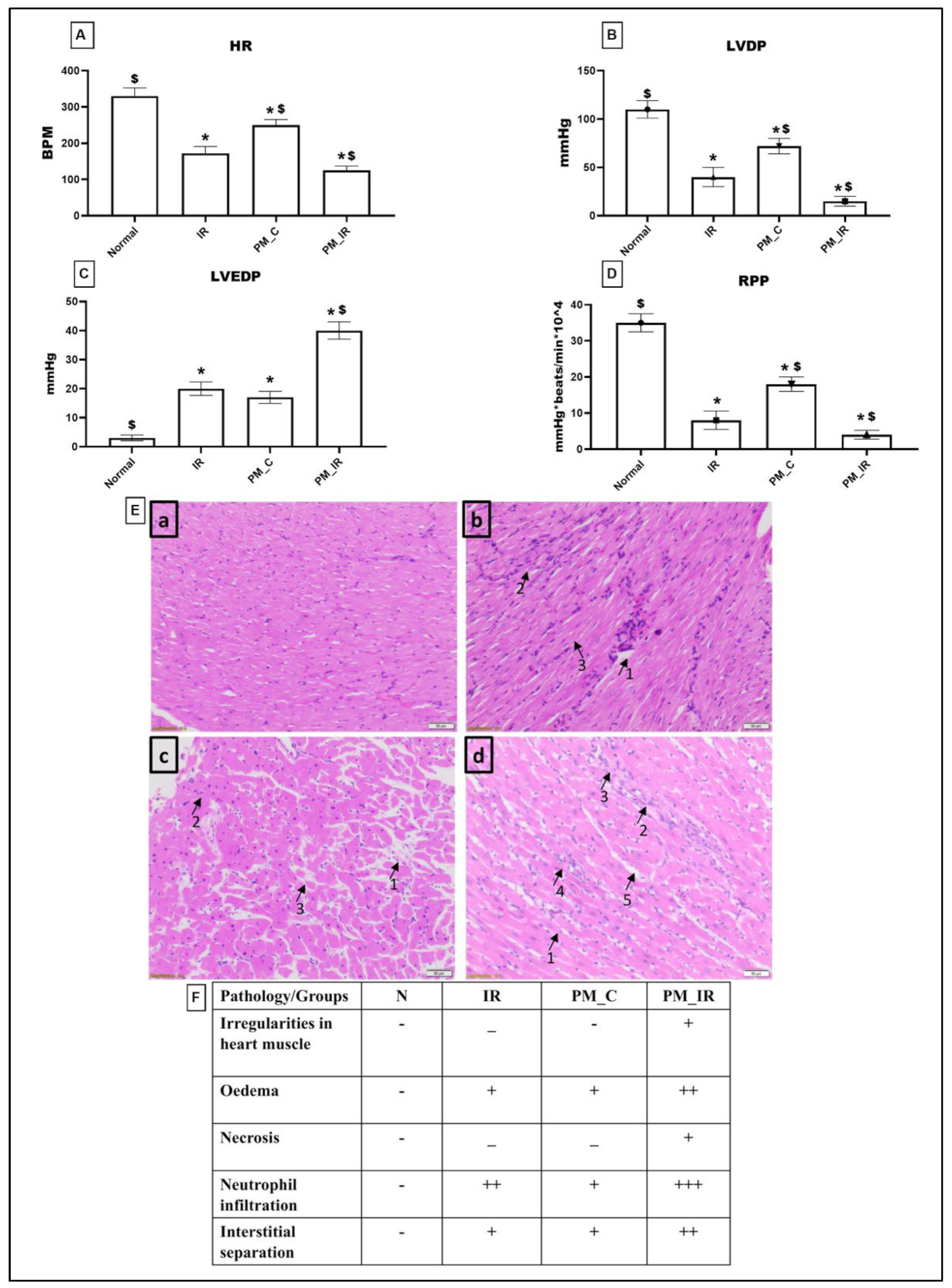
3.1. Administration of PM2.5 Aggravated Cardiac Injury and Compromised Cardiac Hemodynamics in Isolated Rat Myocardium
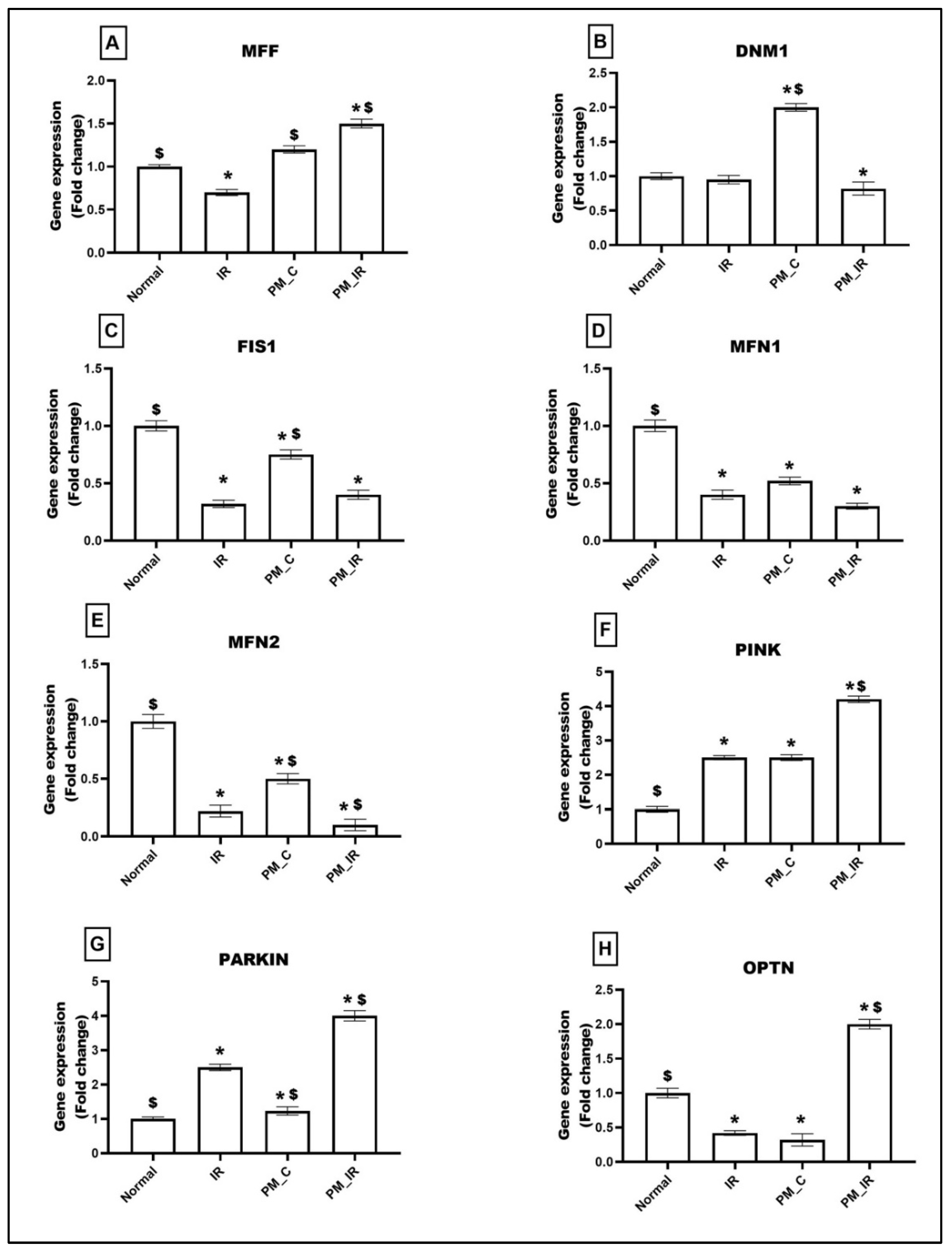
3.2. Administration of PM2.5 Altered the Mitochondrial Dynamics and Increased Mitophagy in Ischemia Reperfused Myocardium
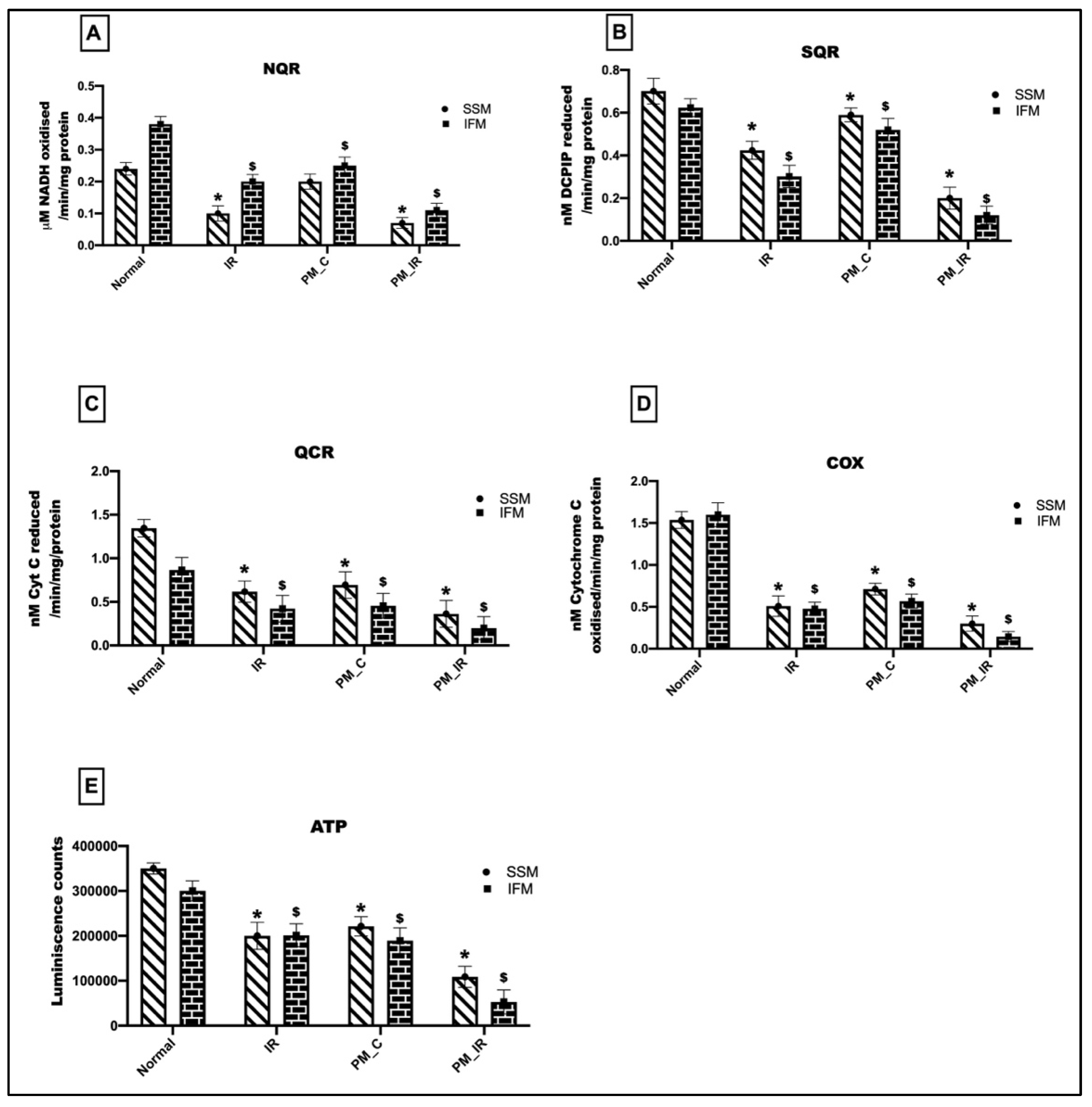
3.3. Administration of PM2.5 Altered the Mitochondrial Bioenergetics Function and Elevated Oxidative Stress in the IFM Fraction of Ischemia Reperfused Myocardium
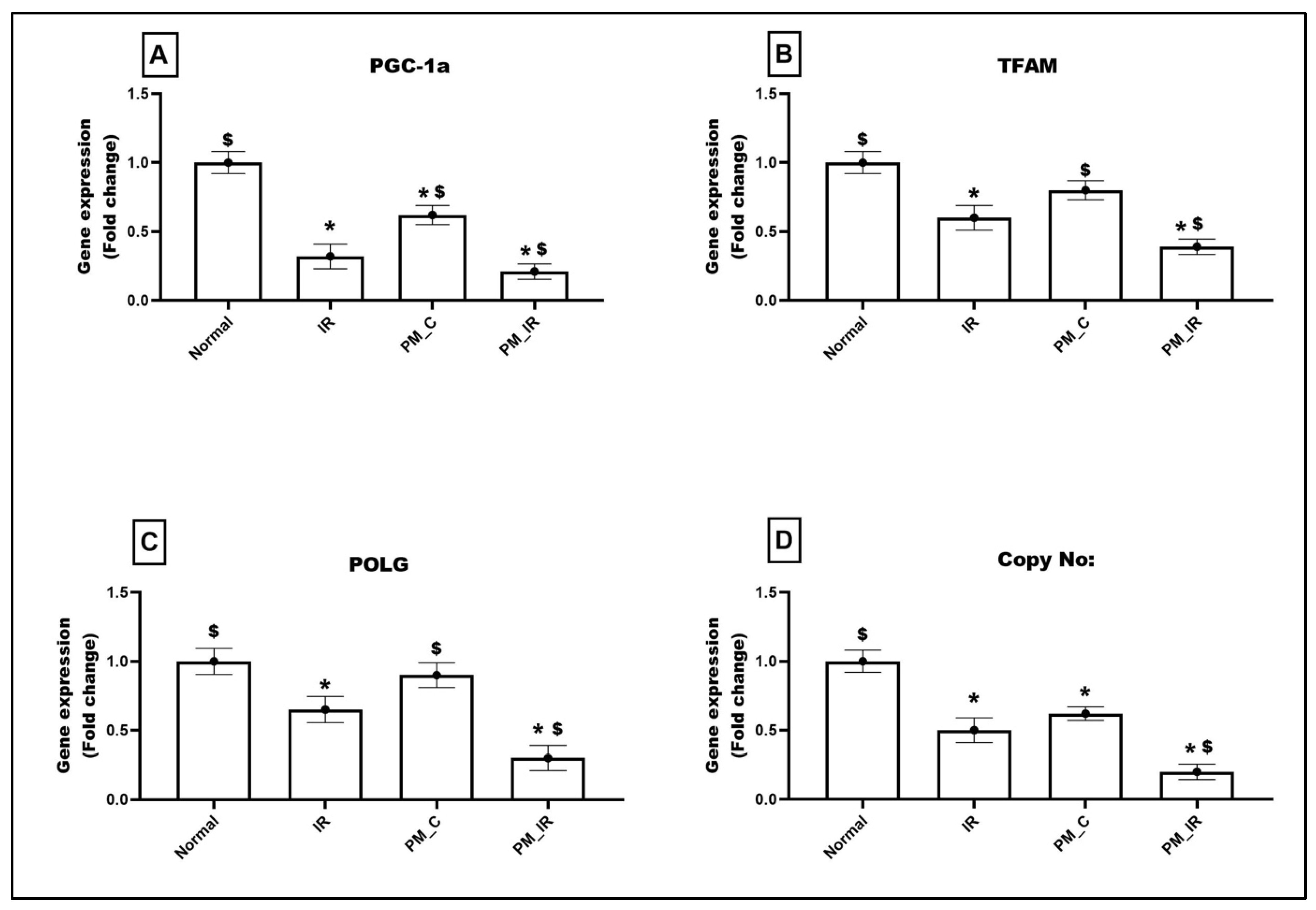
3.4. Administration of PM2.5 Altered the Expression of Mitochondrial Regulatory Genes and Reduced the Mitochondrial DNA Copy Number in Ischemia Reperfused Myocardium
3.5. Administration of PM2.5 Altered the Expression of Mitochondrial DNA Maintenance Genes in Ischemia Reperfused Myocardium
3.6. Administration of PM2.5 Decreased the In Vitro Protein Translation Capacity in the IFM Fraction of Ischemia Reperfused Myocardium
3.7. ICPMS Data Reveals the Deposition of Metal Components from PM2.5 in the Mitochondria of Hearts Administered with PM2.5
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lee, B.J.; Kim, B.; Lee, K. Air pollution exposure and cardiovascular disease. Toxicol. Res. 2014, 30, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Waidyatillake, N.T.; Campbell, P.T.; Vicendese, D.; Dharmage, S.C.; Curto, A.; Stevenson, M. Particulate Matter and Premature Mortality: A Bayesian Meta-Analysis. Int. J. Environ. Res. Public Health 2021, 18, 7655. [Google Scholar] [CrossRef] [PubMed]
- Pardo, M.; Qiu, X.; Zimmermann, R.; Rudich, Y. Particulate Matter Toxicity Is Nrf2 and Mitochondria Dependent: The Roles of Metals and Polycyclic Aromatic Hydrocarbons. Chem. Res. Toxicol. 2020, 33, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhang, J.; Ning, R.; Du, Z.; Liu, J.; Batibawa, J.W.; Duan, J.; Sun, Z. The critical role of endothelial function in fine particulate matter-induced atherosclerosis. Part. Fibre Toxicol. 2020, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lu, W.; Wei, Z.; Zhang, H. Air Pollution and Cardiac Arrhythmias: From Epidemiological and Clinical Evidences to Cellular Electrophysiological Mechanisms. Front. Cardiovasc. Med. 2021, 8, 736151. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Sivakumar, B.; Kurian, G.A. Diesel particulate matter exposure deteriorates cardiovascular health and increases the sensitivity of rat heart towards ischemia reperfusion injury via suppressing mitochondrial bioenergetics function. Chem. Biol. Interact. 2022, 351, 109769. [Google Scholar] [CrossRef]
- Sivakumar, B.; Kurian, G.A. Inhalation of PM2.5 from diesel exhaust promote impairment of mitochondrial bioenergetics and dysregulate mitochondrial quality in rat heart: Implications in isoproterenol-induced myocardial infarction model. Inhal. Toxicol. 2022, 34, 107–119. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Thai, P.N.; Lu, S.; Pu, J.; Bers, D.M. Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes. J. Mol. Cell. Cardiol. 2019, 136, 72–84. [Google Scholar] [CrossRef]
- Hollander, J.M.; Thapa, D.; Shepherd, D.L. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: Influence of cardiac pathologies. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1–H14. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Mutlu, G.M. Particulate Matter Air Pollution: Effects on the Cardiovascular System. Front. Endocrinol. 2018, 9, 680. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Ji, A.; Li, D.; Shi, L.; Gao, M.; Lv, N.; Zhang, Y.; Zhang, R.; Chen, R.; Chen, W.; et al. Mitochondria damage in ambient particulate matter induced cardiotoxicity: Roles of PPAR alpha/PGC-1 alpha signaling. Environ. Pollut. 2021, 288, 117792. [Google Scholar] [CrossRef]
- Magnani, N.D.; Marchini, T.; Calabró, V.; Alvarez, S.; Evelson, P. Role of Mitochondria in the Redox Signaling Network and Its Outcomes in High Impact Inflammatory Syndromes. Front. Endocrinol. 2020, 11, 568305. [Google Scholar] [CrossRef]
- Huang, J.; Li, R.; Wang, C. The Role of Mitochondrial Quality Control in Cardiac Ischemia/Reperfusion Injury. Oxidative Med. Cell. Longev. 2021, 2021, 5543452. [Google Scholar] [CrossRef]
- Sivakumar, B.; Kurian, G.A. PM2.5 Exposure Lowers Mitochondrial Endurance During Cardiac Recovery in a Rat Model of Myocardial Infarction. Cardiovasc. Toxicol. 2022, 22, 545–557. [Google Scholar] [CrossRef]
- Holmuhamedov, E.L.; Oberlin, A.; Short, K.; Terzic, A.; Jahangir, A. Cardiac subsarcolemmal and interfibrillar mitochondria display distinct responsiveness to protection by diazoxide. PLoS ONE 2012, 7, e44667. [Google Scholar] [CrossRef]
- Shanmugam, K.; Ravindran, S.; Kurian, G.A.; Rajesh, M. Fisetin Confers Cardioprotection against Myocardial Ischemia Reperfusion Injury by Suppressing Mitochondrial Oxidative Stress and Mitochondrial Dysfunction and Inhibiting Glycogen Synthase Kinase 3β Activity. Oxidative Med. Cell. Longev. 2018, 2018, 9173436. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, H.; Li, H.; Guo, W.; An, Z.; Zeng, X.; Li, W.; Li, H.; Song, J.; Wu, W. Amelioration of PM2.5-induced lung toxicity in rats by nutritional supplementation with fish oil and Vitamin E. Respir. Res. 2019, 20, 76. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.E. Inhaled efficacious dose translation from rodent to human: A retrospective analysis of clinical standards for respiratory diseases. Pharmacol. Ther. 2017, 178, 141–147. [Google Scholar] [CrossRef]
- Selley, L.; Schuster, L.; Marbach, H.; Forsthuber, T.; Forbes, B.; Gant, T.W.; Sandström, T.; Camiña, N.; Athersuch, T.J.; Mudway, I.; et al. Brake dust exposure exacerbates inflammation and transiently compromises phagocytosis in macrophages. Met. Integr. Biometal Sci. 2020, 12, 371–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, J.W.; Tandler, B.; Hoppel, C.L. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 1977, 252, 8731–8739. [Google Scholar] [CrossRef] [PubMed]
- Ashar, F.N.; Moes, A.; Moore, A.Z.; Grove, M.L.; Chaves, P.; Coresh, J.; Newman, A.B.; Matteini, A.M.; Bandeen-Roche, K.; Boerwinkle, E.; et al. Association of mitochondrial DNA levels with frailty and all-cause mortality. J. Mol. Med. 2015, 93, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Silva, P.; Acín-Pérez, R.; Fernández-Vizarra, E.; Pérez-Martos, A.; Enriquez, J.A. In Vivo and in organello analyses of mitochondrial translation. Methods Cell Biol. 2007, 80, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Khosravipour, M.; Safari-Faramani, R.; Rajati, F.; Omidi, F. The long-term effect of exposure to respirable particulate matter on the incidence of myocardial infarction: A systematic review and meta-analysis study. Environ. Sci. Pollut. Res. Int. 2022, 29, 42347–42371. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.; Thomson, A.L.; Carter, R.; Stott, H.R.; Shaw, C.A.; Hadoke, P.W.; Newby, D.E.; Miller, M.R.; Gray, G.A. Pulmonary diesel particulate increases susceptibility to myocardial ischemia/reperfusion injury via activation of sensory TRPV1 and β1 adrenoreceptors. Part. Fibre Toxicol. 2014, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Tong, F.; Zhang, H. Pulmonary Exposure to Particulate Matter (PM2.5) Affects the Sensitivity to Myocardial Ischemia/Reperfusion Injury Through Farnesoid-X-Receptor-Induced Autophagy. Cell. Physiol. Biochem. 2018, 46, 1493–1507. [Google Scholar] [CrossRef]
- Lukyanenko, V.; Chikando, A.; Lederer, W.J. Mitochondria in cardiomyocyte Ca2+ signaling. Int. J. Biochem. Cell Biol. 2009, 41, 1957–1971. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxidative Med. Cell. Longev. 2016, 2016, 1656450. [Google Scholar] [CrossRef]
- Mahalakshmi, A.; Kurian, G.A. Evaluating the impact of diabetes and diabetic cardiomyopathy rat heart on the outcome of ischemia-reperfusion associated oxidative stress. Free. Radic. Biol. Med. 2018, 118, 35–43. [Google Scholar] [CrossRef]
- Alissa, E.M.; Ferns, G.A. Heavy metal poisoning and cardiovascular disease. J. Toxicol. 2011, 2011, 870125. [Google Scholar] [CrossRef]
- Farahani, V.J.; Pirhadi, M.; Sioutas, C. Are standardized diesel exhaust particles (DEP) representative of ambient particles in air pollution toxicological studies? Sci. Total Environ. 2021, 788, 147854. [Google Scholar] [CrossRef]
- Bai, L.; Zhao, Y.; Zhao, L.; Zhang, M.; Cai, Z.; Yung, K.; Dong, C.; Li, R. Ambient air PM2.5 exposure induces heart injury and cardiac hypertrophy in rats through regulation of miR-208a/b, α/β-MHC, and GATA4. Environ. Toxicol. Pharmacol. 2021, 85, 103653. [Google Scholar] [CrossRef]
- Vincent, R.; Kumarathasan, P.; Goegan, P.; Bjarnason, S.G.; Guénette, J.; Bérubé, D.; Adamson, I.Y.; Desjardins, S.; Burnett, R.T.; Miller, F.J.; et al. Inhalation Toxicology of Urban Ambient Particulate Matter: Acute Cardiovascular Effects in Rats; Health Effects Institute: Boston, MA, USA, 2001; pp. 5–62. [Google Scholar]
- Fischbacher, A.; von Sonntag, C.; Schmidt, T.C. Hydroxyl radical yields in the Fenton process under various pH, ligand concentrations and hydrogen peroxide/Fe(II) ratios. Chemosphere 2017, 182, 738–744. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leung, M.C.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a target of environmental toxicants. Toxicol. Sci. 2013, 134, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Tinkov, A.A.; Nguyen, T.T.; Santamaria, A.; Bowman, A.B.; Buha Djordjevic, A.; Paoliello, M.; Skalny, A.V.; Aschner, M. Sirtuins as molecular targets, mediators, and protective agents in metal-induced toxicity. Arch. Toxicol. 2021, 95, 2263–2278. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Duarte-Hospital, C.; Tête, A.; Brial, F.; Benoit, L.; Koual, M.; Tomkiewicz, C.; Kim, M.J.; Blanc, E.B.; Coumoul, X.; Bortoli, S. Mitochondrial Dysfunction as a Hallmark of Environmental Injury. Cells 2021, 11, 110. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer | |
|---|---|---|---|
| 1 | PGC-1α | 5′-GAGGGACGAATACCGCAGAG-3′ | 5′-CTCTCAGTTCTGTCCGCGTT-3′ |
| 2 | Dnm1 | 5′-TTGCCCTCTTCAACACTGAGC-3′ | 5′-ATGAAGCTGTCAGAGCCGTT-3′ |
| 3 | Parkin | 5′-AGTTTGTCCACGACGCTCAA-3′ | 5′-CAGAAAACGAACCCACAGCC-3′ |
| 4 | MFN1 | 5′-TGACTTGGACTACTCGTGCG-3′ | 5′-GGCACAGTCGAGCAAAAGTG-3′ |
| 5 | MFN2 | 5′-CTCTGTGCTGGTTGACGAGT-3′ | 5′-TCGAGGGACCAGCATGTCTA-3′ |
| 6 | DRP1 | 5′-TGGAAAGAGCTCAGTGCTGG-3′ | 5′-TCAACTCCATTTTCTTCTCCTGT-3′ |
| 7 | MFF | 5′-GAAAACACCTCCACGTGTGC-3′ | 5′-CTGCTCGGATCTCTTCGCTT-3′ |
| 8 | FIS | 5′-CCAGAGATGAAGCTGCAAGGA-3′ | 5′-TTCCTTGAGCCGGTAGTTGC-3′ |
| 9 | PINK1 | 5′-TGTATGAAGCCACCATGCCC-3′ | 5′-TCTGCTCCCTTTGAGACGAC-3′ |
| 10 | TFAM | 5′-GTTGCTGTCGCTTGTGAGTG-3′ | 5′-GTCTTTGAGTCCCCCATCCC-3′ |
| 11 | β-actin | 5′-GTGTGGTCAGCCCTGTAGTT-3′ | 5′-CCTAGAAGCATTTGCGGTGC-3′ |
| 12 | POLG1 | 5′-CTTTGGGCTCCAGCTTGACT-3′ | 5′-TGGAGAAAATGCTTGGCACG-3′ |
| 13 | Casp9-F | 5’-GAGGATATTCAGCGGGCAGG-3’ | 5’-GCAGGAGATGAAGCGAGGAA-3’ |
| 14 | Casp3-F | 5’-CGGACCTGTGGACCTGAAAA-3’ | 5’-TAACCGGGTGCGGTAGAGTA-3’ |
| 15 | Casp7-F | 5’-TTCGACGGAAGACGGAGTTG-3’ | 5’-CCGGACATCCATACCTGTCG-3’ |
| 16 | PARP-F | 5’-ACCACGCACAATGCCTATGA-3’ | 5’-AGCAGTCTCCGGTTGTGAAG-3’ |
| 17 | Glutaredoxin1-F | 5’-AGCATGGCTCAGGACTTTGT-3’ | 5’-TTGAATCGCATTGGTGTTGT-3’ |
| 18 | Peroxiredoxin 6-F | 5’-CCTGGAGCAAGGACATCAAT-3’ | 5’-GTTTCTTGTCAGGGCCAAAA-3’ |
| 19 | Peroxiredoxin 3- F | 5’-CCAGAGTCCCCTACGATCAA-3’ | 5’-TGGCCACCTTTAACCTGAAC-3’ |
| 20 | GUF 1 | 5’-GGTGTCTTGGAGCCAGGG-3’ | 5’-CCTCTTTCTCGCTCCACTTGT-3’ |
| 21 | LRPPRC | 5’-CAACAACATTGCTCTGGCCC-3’ | 5’-TCCACCTTGCCTGAATCCAC-3’ |
| 22 | SLC25A4 | 5’-GGATCTTCCCAGCGTGAGTT-3’ | 5’-TGCACATTCTTGGGGTCTGG-3’ |
| Metals | Normal | PM_C |
|---|---|---|
| Li | BDL | BDL |
| Be | BDL | BDL |
| B | BDL | 0.01 |
| Na | 0.03 | 0.06 |
| Mg | BDL | BDL |
| Al | BDL | BDL |
| P | BDL | BDL |
| K | 0.04 | 0.06 |
| Ca | 0.01 | 0.02 |
| Cr | BDL | BDL |
| Mn | 0.03 | 0.04 |
| Fe | 0.05 | 0.09 |
| Co | BDL | BDL |
| Ni | BDL | BDL |
| Cu | 0.02 | 0.03 |
| Zn | 0.01 | 0.01 |
| As | BDL | BDL |
| Cd | BDL | BDL |
| Pb | BDL | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivakumar, B.; AlAsmari, A.F.; Ali, N.; Waseem, M.; Kurian, G.A. Consequential Impact of Particulate Matter Linked Inter-Fibrillar Mitochondrial Dysfunction in Rat Myocardium Subjected to Ischemia Reperfusion Injury. Biology 2022, 11, 1811. https://doi.org/10.3390/biology11121811
Sivakumar B, AlAsmari AF, Ali N, Waseem M, Kurian GA. Consequential Impact of Particulate Matter Linked Inter-Fibrillar Mitochondrial Dysfunction in Rat Myocardium Subjected to Ischemia Reperfusion Injury. Biology. 2022; 11(12):1811. https://doi.org/10.3390/biology11121811
Chicago/Turabian StyleSivakumar, Bhavana, Abdullah F. AlAsmari, Nemat Ali, Mohammad Waseem, and Gino A. Kurian. 2022. "Consequential Impact of Particulate Matter Linked Inter-Fibrillar Mitochondrial Dysfunction in Rat Myocardium Subjected to Ischemia Reperfusion Injury" Biology 11, no. 12: 1811. https://doi.org/10.3390/biology11121811