
Side-by-Side Comparison of Post-Entry Quarantine and High Throughput Sequencing Methods for Virus and Viroid Diagnosis

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material Preparation and Nucleic Acid Extraction
2.2. Routine Diagnostics Assays
2.3. Library Construction and Sequencing
- Sequencing provider 1 (SP1)
- Sequencing provider 2 (SP2)
2.4. Bioinformatics Analyses
3. Results
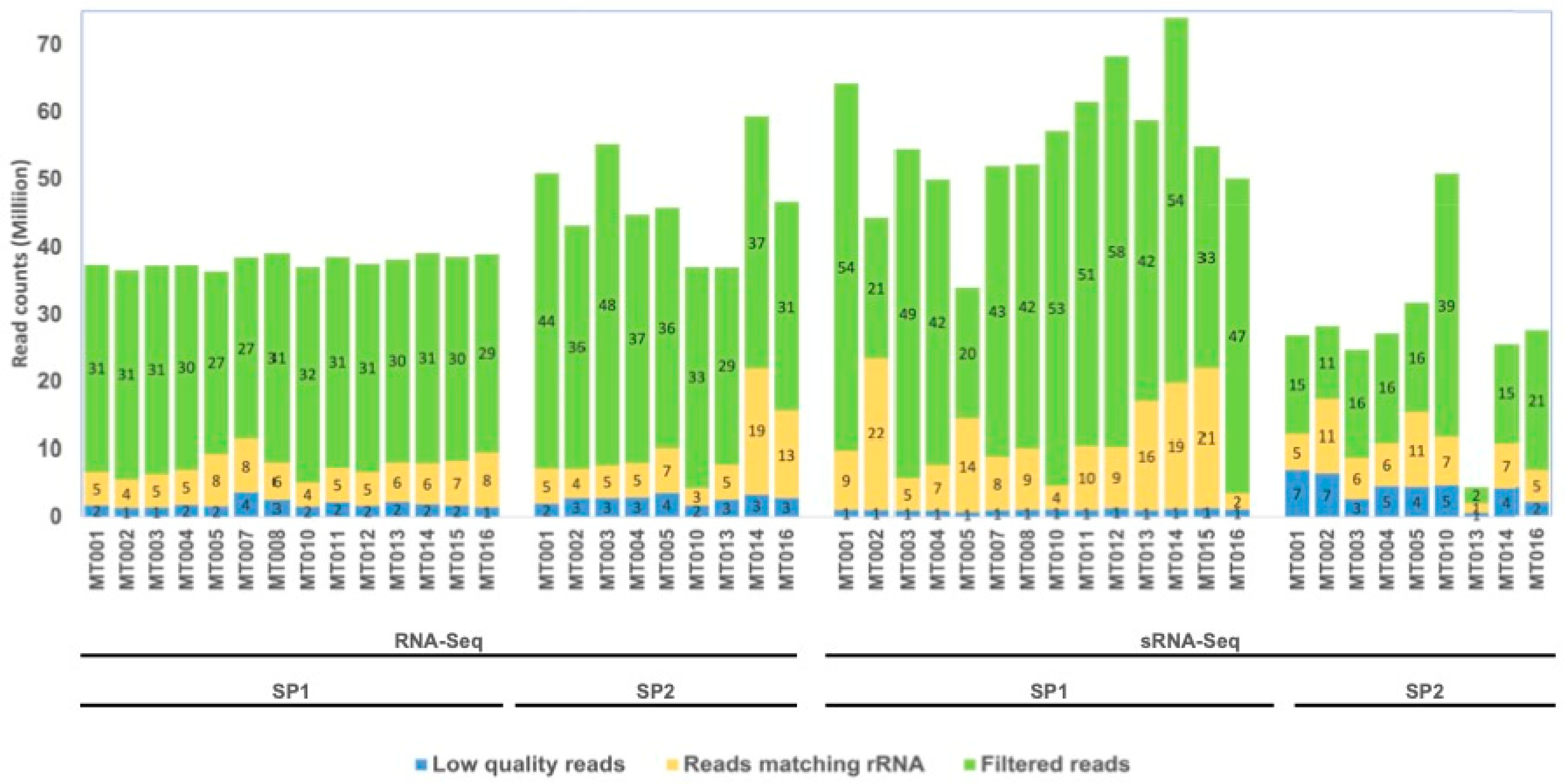
3.1. General Sequencing Statistics
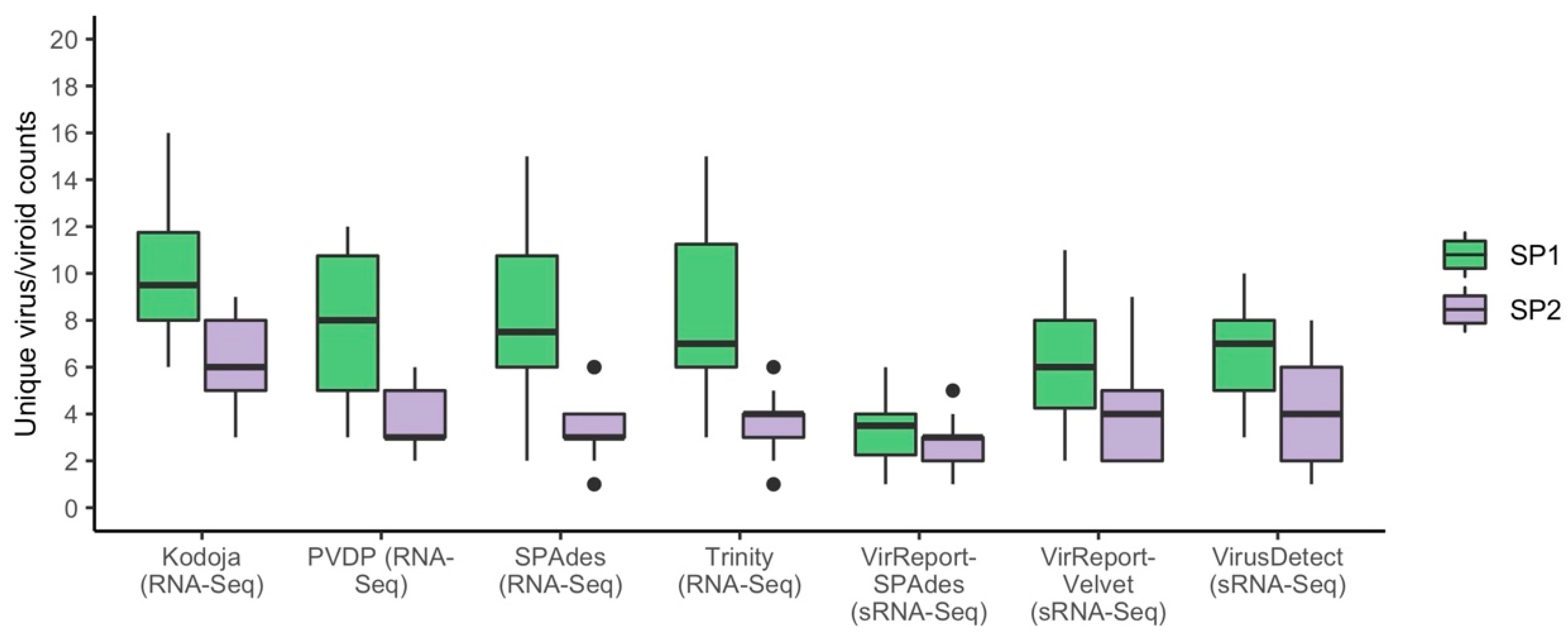
3.2. Small RNA HTS Data Generates Fewer and Targeted De Novo Assembled Scaffolds
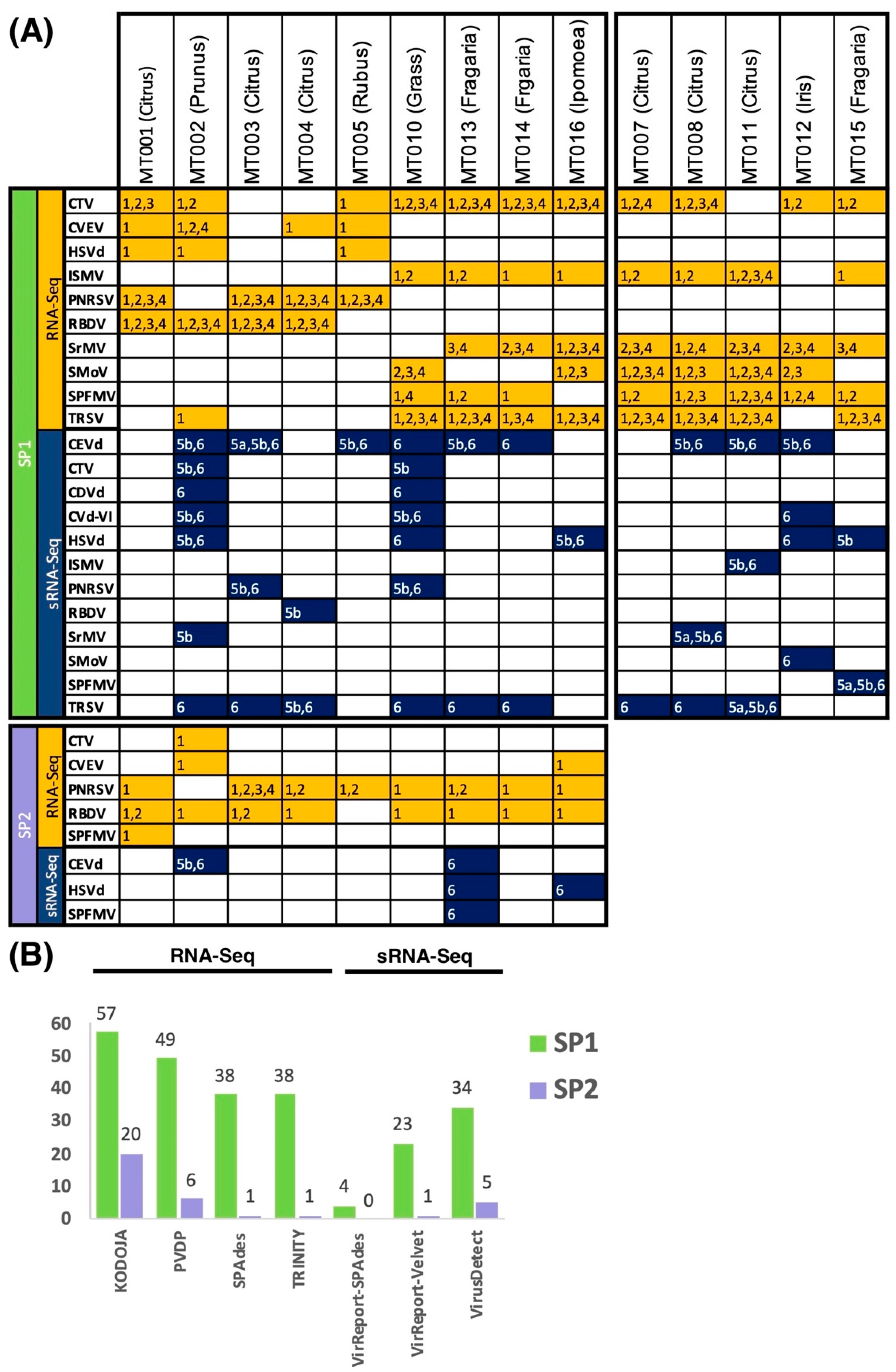
3.3. Evaluating Sensitivity of Detection of Plant Virus and Viroids
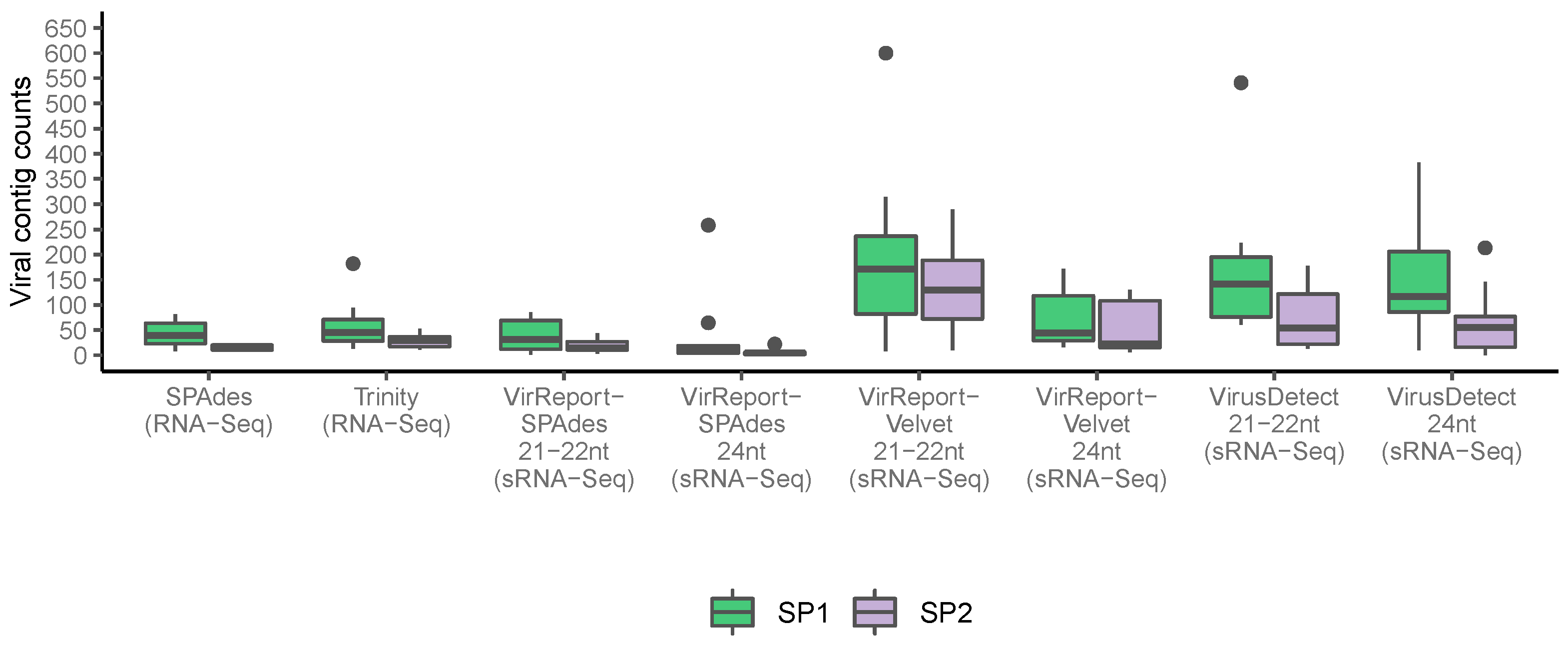
3.4. Viral Abundance Highly Variable
3.5. Detection of Cross-Sample Contamination Events
3.6. Subsampling Reduces False Discovery Rate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations. Preparing to Use High-Throughput Sequencing (HTS) Technologies as a Diagnostic Tool for Phytosanitary Purposes. In Commission on Phytosanitary Measures Recommendation; FAO: Rome, Italy, 2019; p. 6. [Google Scholar]
- Loebenstein, G. Plant virus diseases: Economic aspects. In Encyclopedia of Virology, 3rd ed.; Mahy, B.W.J., Van Regenmortel, M.H.V., Eds.; Academic Press: Oxford, UK, 2008; pp. 197–201. [Google Scholar]
- Rybicki, E.P. A Top Ten list for economically important plant viruses. Arch. Virol. 2014, 160, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Gucek, T.; Trdan, S.; Jakse, J.; Javornik, B.; Matousek, J.; Radisek, S. Diagnostic techniques for viroids. Plant Pathol. 2016, 66, 339–358. [Google Scholar] [CrossRef]
- Mehetre, G.; Leo, V.; Singh, G.; Sorokan, A.; Maksimov, I.; Yadav, M.; Upadhyaya, K.; Hashem, A.; Alsaleh, A.; Dawoud, T.; et al. Current Developments and Challenges in Plant Viral Diagnostics: A Systematic Review. Viruses 2021, 13, 412. [Google Scholar] [CrossRef] [PubMed]
- Whattam, M.; Dinsdale, A.; Elliott, C. Evolution of Plant Virus Diagnostics Used in Australian Post Entry Quarantine. Plants 2021, 10, 1430. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Baizan-Edge, A.; MacFarlane, S.; Torrance, L. Viral Diagnostics in Plants Using Next Generation Sequencing: Computational Analysis in Practice. Front. Plant Sci. 2017, 8, 1770. [Google Scholar] [CrossRef]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High Throughput Sequencing for Plant Virus Detection and Discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef]
- Visser, M.; Bester, R.; Burger, J.T.; Maree, H.J. Next-generation sequencing for virus detection: Covering all the bases. Virol. J. 2016, 13, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, E.E.; Coletta-Filho, H.D.; Nouri, S.; Falk, B.W.; Nerva, L.; Oliveira, T.S.; Dorta, S.O.; Machado, M.A. Deep Sequencing Analysis of RNAs from Citrus Plants Grown in a Citrus Sudden Death-Affected Area Reveals Diverse Known and Putative Novel Viruses. Viruses 2017, 9, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, A.; Andrusch, A.; Belka, A.; Wylezich, C.; Höper, D.; Pohlmann, A.; Petersen, T.N.; Lucas, P.; Blanchard, Y.; Papa, A.; et al. Proficiency Testing of Virus Diagnostics Based on Bioinformatics Analysis of Simulated In Silico High-Throughput Sequencing Data Sets. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaafar, Y.Z.A.; Ziebell, H. Comparative study on three viral enrichment approaches based on RNA extraction for plant virus/viroid detection using high-throughput sequencing. PLoS ONE 2020, 15, e0237951. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Komínek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus Detection by High-Throughput Sequencing of Small RNAs: Large-Scale Performance Testing of Sequence Analysis Strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Kutnjak, D.; Tamisier, L.; Adams, I.; Boonham, N.; Candresse, T.; Chiumenti, M.; De Jonghe, K.; Kreuze, J.; Lefebvre, M.; Silva, G.; et al. A Primer on the Analysis of High-Throughput Sequencing Data for Detection of Plant Viruses. Microorganisms 2021, 9, 841. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Islas, C.; Rowhani, A. Comparison of Next-Generation Sequencing Versus Biological Indexing for the Optimal Detection of Viral Pathogens in Grapevine. Phytopathology 2015, 105, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Santala, J.; Valkonen, J.P.T. Sensitivity of Small RNA-Based Detection of Plant Viruses. Front. Microbiol. 2018, 9, 939. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.R.; Constable, F.; Tzanetakis, I.E. Quarantine regulations and the impact of modern detection methods. Ann. Rev. Phytopathol. 2016, 54, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Gaafar, Y.Z.A.; Westenberg, M.; Botermans, M.; László, K.; De Jonghe, K.; Foucart, Y.; Ferretti, L.; Kutnjak, D.; Pecman, A.; Mehle, N.; et al. Interlaboratory Comparison Study on Ribodepleted Total RNA High-Throughput Sequencing for Plant Virus Diagnostics and Bioinformatic Competence. Pathogens 2021, 10, 1174. [Google Scholar] [CrossRef]
- Höper, D.; Mettenleiter, T.; Beer, M. Metagenomic approaches to identifying infectious agents. Rev. Sci. Tech. 2016, 35, 83–93. [Google Scholar] [CrossRef]
- Vivek, A.; Zahra, S.; Kumar, S. From current knowledge to best practice: A primer on viral diagnostics using deep sequencing of virus-derived small interfering RNAs (vsiRNAs) in infected plants. Methods 2019, 183, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Tamisier, L.; Haegeman, A.; Foucart, Y.; Fouillien, N.; Al Rwahnih, M.; Buzkan, N.; Candresse, T.; Chiumenti, M.; De Jonghe, K.; Lefebvre, M.; et al. Semi-artificial datasets as a resource for validation of bioinformatics pipelines for plant virus detection. Peer Community J. 2021, 1, e53. [Google Scholar] [CrossRef]
- Cao, M.; Zhang, S.; Li, M.; Liu, Y.; Dong, P.; Li, S.; Kuang, M.; Li, R.; Zhou, Y. Discovery of Four Novel Viruses Associated with Flower Yellowing Disease of Green Sichuan Pepper (Zanthoxylum armatum) by Virome Analysis. Viruses 2019, 11, 696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maachi, A.; Torre, C.; Sempere, R.; Hernando, Y.; Aranda, M.; Donaire, L. Use of High-Throughput Sequencing and Two RNA Input Methods to Identify Viruses Infecting Tomato Crops. Microorganisms 2021, 9, 1043. [Google Scholar] [CrossRef]
- Krueger, F. Trim_Galore: A Wrapper Tool Around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 25 January 2022).
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yuxin, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2017, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Andrews, S.; Babraham, I. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2013. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 January 2022).
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, P.; Rivillas, A.; Tejada, D.; Giraldo, S.; Restrepo, A.; Ospina, M.; Cadavid, S.; Gallo, Y.; Marín, M. PVDP: A portable open source pipeline for detection of plant viruses in RNAseq data. A case study on potato viruses in Antioquia (Colombia). Physiol. Mol. Plant Pathol. 2021, 113, 101604. [Google Scholar] [CrossRef]
- Baizan-Edge, A.; Cock, P.; Macfarlane, S.; McGavin, W.; Torrance, L.; Jones, S. Kodoja: A workflow for virus detection in plants using k-mer analysis of RNA-sequencing data. J. Gen. Virol. 2019, 100, 533–542. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA Sequence Assembly Program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Barrero, R.A.; Napier, K.R.; Cunnington, J.; Liefting, L.; Keenan, S.; Frampton, R.A.; Szabó, T.O.; Bulman, S.; Hunter, A.; Ward, L.; et al. An internet-based bioinformatics toolkit for plant biosecurity diagnosis and surveillance of viruses and viroids. BMC Bioinform. 2017, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling genomes and mini-metagenomes from highly chimeric reads. In Proceedings of the Annual international conference on research in computational molecular biology, Beijing, China, 7–10 April 2013; Springer: Berlin/Heidelberg, Germany, 2013; pp. 158–170. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Liu, D. Algorithms for efficiently collapsing reads with Unique Molecular Identifiers. PeerJ 2019, 7, e8275. [Google Scholar] [CrossRef] [PubMed]
- Pawitan, Y.; Michiels, S.; Koscielny, S.; Gusnanto, A.; Ploner, A. False discovery rate, sensitivity and sample size for microarray studies. Bioinformatics 2005, 21, 3017–3024. [Google Scholar] [CrossRef] [PubMed]
- Li, H. seqtk, Toolkit for Processing Sequences in FASTA/Q Formats. Available online: https://github.com/lh3/seqtk (accessed on 25 January 2022).
- Ho, T.; Broome, J.C.; Buhler, J.P.; O’Donovan, W.; Tian, T.; Diaz-Lara, A.; Martin, R.R.; Tzanetakis, I.E. Characterization of endogenous Rubus yellow net virus in raspberries. bioRxiv 2021. [Google Scholar] [CrossRef]
- Effects of Index Misassignment on Multiplexing and Downstream Analysis (White Paper). Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/whitepapers/index-hopping-white-paper-770-2017-004.pdf (accessed on 9 November 2021).
- Li, Q.; Zhao, X.; Zhang, W.; Wang, L.; Wang, J.; Xu, D.; Mei, Z.; Liu, Q.; Du, S.; Li, Z.; et al. Reliable multiplex sequencing with rare index mis-assignment on DNB-based NGS platform. BMC Genom. 2019, 20, 215. [Google Scholar] [CrossRef] [Green Version]
- MacConaill, L.E.; Burns, R.T.; Nag, A.; Coleman, H.A.; Slevin, M.K.; Giorda, K.; Light, M.; Lai, K.; Jarosz, M.; McNeill, M.S.; et al. Unique, dual-indexed sequencing adapters with UMIs effectively eliminate index cross-talk and significantly improve sensitivity of massively parallel sequencing. BMC Genom. 2018, 19, 30. [Google Scholar] [CrossRef] [Green Version]
- Vigne, E.; Garcia, S.; Komar, V.; Lemaire, O.; Hily, J.-M. Comparison of Serological and Molecular Methods with High-Throughput Sequencing for the Detection and Quantification of Grapevine Fanleaf Virus in Vineyard Samples. Front. Microbiol. 2018, 9, 2726. [Google Scholar] [CrossRef] [Green Version]
- Costello, M.; Fleharty, M.; Abreu, J.; Farjoun, Y.; Ferriera, S.; Holmes, L.; Granger, B.; Green, L.; Howd, T.; Mason, T.; et al. Characterization and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genom. 2018, 19, 332. [Google Scholar] [CrossRef]
- Sinha, R.; Stanley, G.; Gulati, G.S.; Ezran, C.; Travaglini, K.J.; Wei, E.; Chan, C.K.F.; Nabhan, A.N.; Su, T.; Morganti, R.M.; et al. Index switching causes “spreading-of-signal” among multiplexed samples in Illumina HiSeq 4000 DNA sequencing. bioRxiv 2017, 125724. [Google Scholar] [CrossRef]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Adkar-Purushothama, C.R.; Bolduc, F.; Bru, P.; Perreault, J.-P. Insights into Potato Spindle Tuber Viroid Quasi-Species from Infection to Disease. Front. Microbiol. 2020, 11, 1235. [Google Scholar] [CrossRef]
- Rogers, S.O.; Bendich, A.J. Ribosomal RNA genes in plants: Variability in copy number and in the intergenic spacer. Plant Mol. Biol. 1987, 9, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Commodity | Species | Positive Detections in PEQ |
|---|---|---|---|
| MT001 | Citrus | Citrus Troyer × Frost-Lisbon | CEVd |
| MT002 | Prunus | Prunus persica | PNRSV |
| MT003 | Citrus | Citrus × aurantiifolia (Christm.) Swingle | CTV, CVEV, CDVd, HSVd |
| MT004 | Citrus | Citrus medica L. | CEVd, CTV, HSVd |
| MT005 | Raspberry | Rubus idaeus | RBDV |
| MT007 | Citrus | Citrus sp. | CDVd, HSVd |
| MT008 | Citrus | Citrus sinensis | CDVd, HSVd |
| MT010 | Ornamental grass | Miscanthus sinensis ‘Morning light’ | Novel potyvirus (MsiMV) |
| MT011 | Citrus | Citrus medica L. | CTV, CVd-VI, HSVd |
| MT012 | Iris | Iris sp. ‘Crimson colossus’ | ISMV, TRSV |
| MT013 | Strawberry | Fragaria vesca ‘Alpine’ | SMoV |
| MT014 | Strawberry | Fragaria vesca ‘UC4’ | SMoV |
| MT015 | Strawberry. | Fragaria sp. | SMoV |
| MT016 | Sweet potato | Ipomoea batatas | SPFMV |
| Sequencing Provider | Sequencing Technology | Software | Sensitivity (%) |
|---|---|---|---|
| SP1 | RNA-Seq | Kodoja | 100 (24/24) |
| PVDP | 95.8 (23/24) | ||
| SPAdes | 95.8 (23/24) | ||
| Trinity | 95.8 (23/24) | ||
| sRNA-Seq | VirusDetect | 100 (24/24) | |
| VirReport-SPAdes | 100 (24/24) | ||
| VirReport-Velvet | 100 (24/24) | ||
| SP2 | RNA-Seq | Kodoja | 100 (14/14) |
| PVDP | 100 (14/14) | ||
| SPAdes | 100 (14/14) | ||
| Trinity | 92.9 (13/14) | ||
| sRNA-Seq | VirusDetect | 100 (14/14) | |
| VirReport-SPAdes | 78.6 (11/14) | ||
| VirReport-Velvet | 100 (14/14) |
| Sequencing Technology | Subsampling | Viruses | Viroids | ||
|---|---|---|---|---|---|
| Sensitivity (%) | False Discovery Rate (%) | Sensitivity (%) | False Discovery Rate (%) | ||
| RNA-Seq SP1 (Kodoja) | 1 M | 100 (13/13) | 66.7 (26) | 72.7 (8/11) | 0 |
| 2.5 M | 100 (13/13) | 71.7 (33) | 72.7 (8/11) | 0 | |
| 4 M | 100 (13/13) | 73.5 (36) | 81.8 (9/11) | 0 | |
| 5 M | 100 (13/13) | 75.9 (41) | 81.8 (9/11) | 0 | |
| 10 M | 100 (13/13) | 78.3 (47) | 100 (11/11) | 8.3 (1) | |
| All reads | 100 (13/13) | 80.6 (54) | 100 (11/11) | 21.4 (3) | |
| sRNA-Seq SP1 (VirReport-Velvet) | 1 M | 92.3 (12/13) | 7.7 (1) | 100 (11/11) | 8.3 (1) |
| 2.5 M | 100 (13/13) | 7.1 (1) | 100 (11/11) | 15.3 (2) | |
| 4 M | 100 (13/13) | 13.3 (2) | 100 (11/11) | 21.4 (3) | |
| All reads | 100 (13/13) | 48 (12) | 100 (11/11) | 52.1 (11) | |
| RNA-Seq SP2 (Kodoja) | 1 M | 100 (9/9) | 25 (3) | 80 (4/5) | 0 |
| 2.5 M | 100 (9/9) | 35.7 (5) | 100 (5/5) | 0 | |
| 4 M | 100 (9/9) | 43.8 (7) | 100 (5/5) | 0 | |
| 5 M | 100 (9/9) | 50.0 (9) | 100 (5/5) | 0 | |
| 10 M | 100 (9/9) | 57.1 (12) | 100 (5/5) | 0 | |
| All reads | 100 (9/9) | 69.9 (20) | 100 (5/5) | 0 | |
| sRNA-Seq SP2 (VirReport-Velvet) | 1 M | 100 (9/9) | 0 | 80 (4/5) | 0 |
| 2.5 M | 100 (9/9) | 0 | 100 (5/5) | 16.7 (1) | |
| 4 M | 100 (9/9) | 0 | 100 (5/5) | 16.7 (1) | |
| All reads | 100 (9/9) | 0 | 100 (5/5) | 16.7 (1) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gauthier, M.-E.A.; Lelwala, R.V.; Elliott, C.E.; Windell, C.; Fiorito, S.; Dinsdale, A.; Whattam, M.; Pattemore, J.; Barrero, R.A. Side-by-Side Comparison of Post-Entry Quarantine and High Throughput Sequencing Methods for Virus and Viroid Diagnosis. Biology 2022, 11, 263. https://doi.org/10.3390/biology11020263
Gauthier M-EA, Lelwala RV, Elliott CE, Windell C, Fiorito S, Dinsdale A, Whattam M, Pattemore J, Barrero RA. Side-by-Side Comparison of Post-Entry Quarantine and High Throughput Sequencing Methods for Virus and Viroid Diagnosis. Biology. 2022; 11(2):263. https://doi.org/10.3390/biology11020263
Chicago/Turabian StyleGauthier, Marie-Emilie A., Ruvini V. Lelwala, Candace E. Elliott, Craig Windell, Sonia Fiorito, Adrian Dinsdale, Mark Whattam, Julie Pattemore, and Roberto A. Barrero. 2022. "Side-by-Side Comparison of Post-Entry Quarantine and High Throughput Sequencing Methods for Virus and Viroid Diagnosis" Biology 11, no. 2: 263. https://doi.org/10.3390/biology11020263
APA StyleGauthier, M.-E. A., Lelwala, R. V., Elliott, C. E., Windell, C., Fiorito, S., Dinsdale, A., Whattam, M., Pattemore, J., & Barrero, R. A. (2022). Side-by-Side Comparison of Post-Entry Quarantine and High Throughput Sequencing Methods for Virus and Viroid Diagnosis. Biology, 11(2), 263. https://doi.org/10.3390/biology11020263