Transcriptome Analysis of Developing Wheat Grains at Rapid Expanding Phase Reveals Dynamic Gene Expression Patterns

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. RNA Extraction and Sequencing
2.3. Identification of Differentially Expressed Genes
2.4. k-Means Cluster Analysis
2.5. Genome-Wide Identification of Transcription Factor Family
2.6. Comparison of Relative Expression Levels among the Three Homoeologs of Triads
3. Results
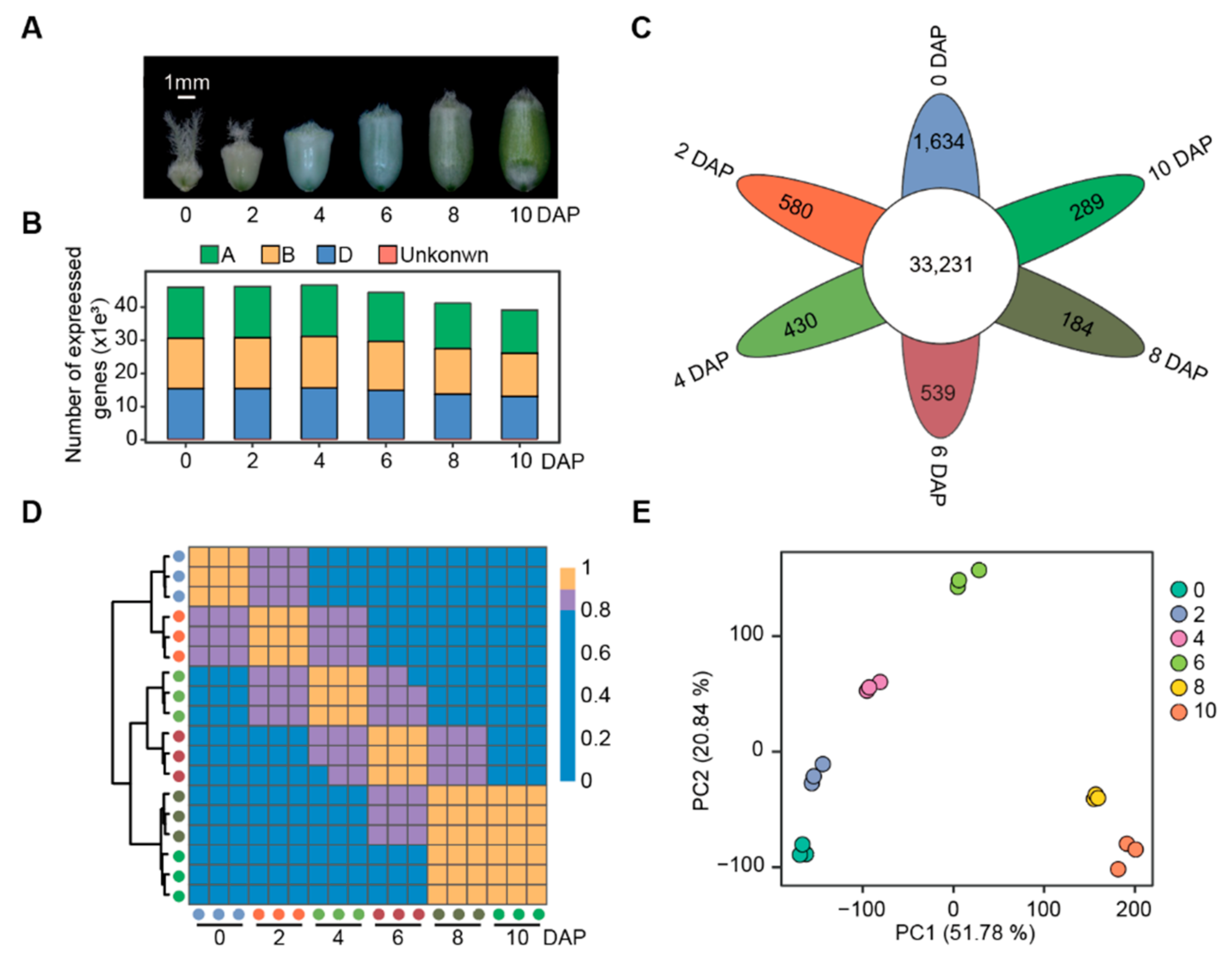
3.1. Transcriptome Analysis of Wheat Grains at Expanding Phase
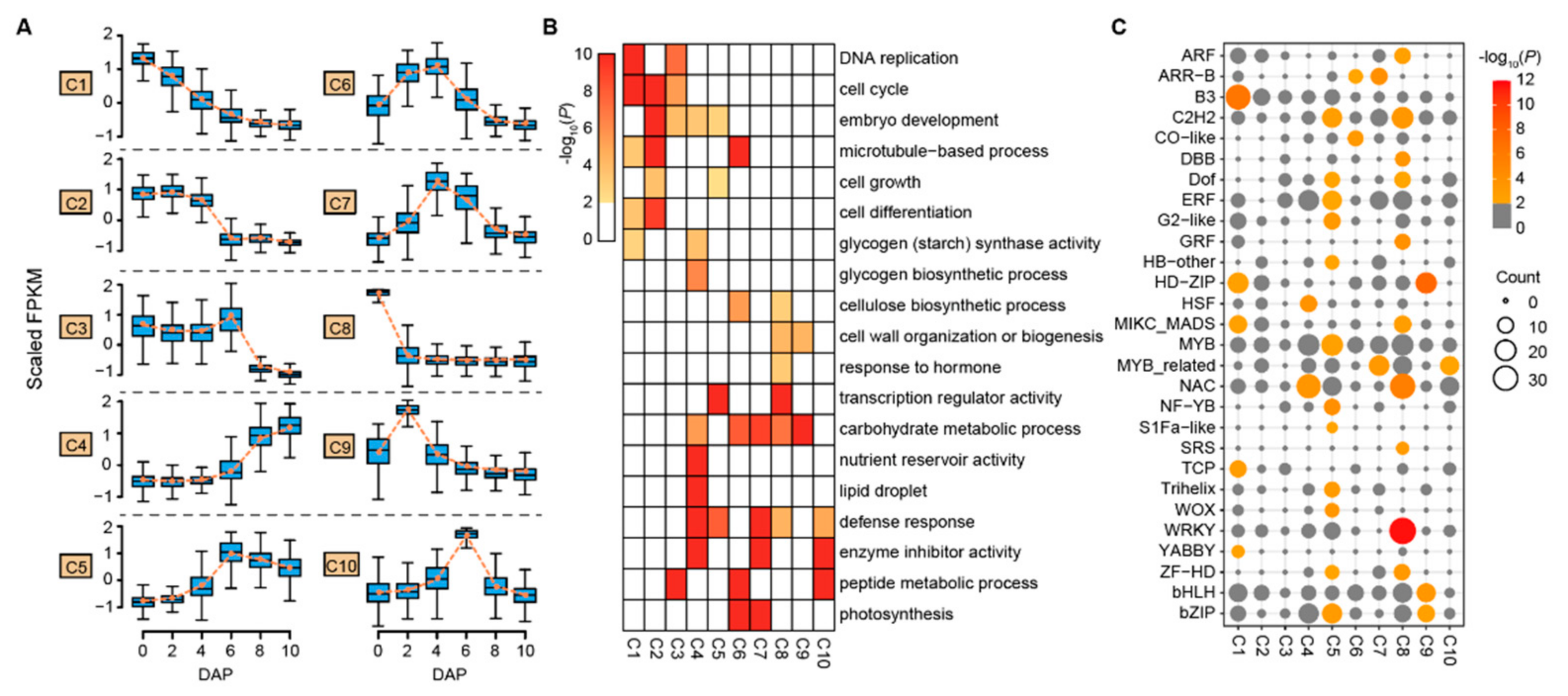
3.2. Expression Clusters and Cellular Function of Differentially Expressed Genes
3.3. Expression Profiles of Key Genes Involved in Cell Division and Starch Biosynthesis
3.4. Differentially Expressed Transcription Factors
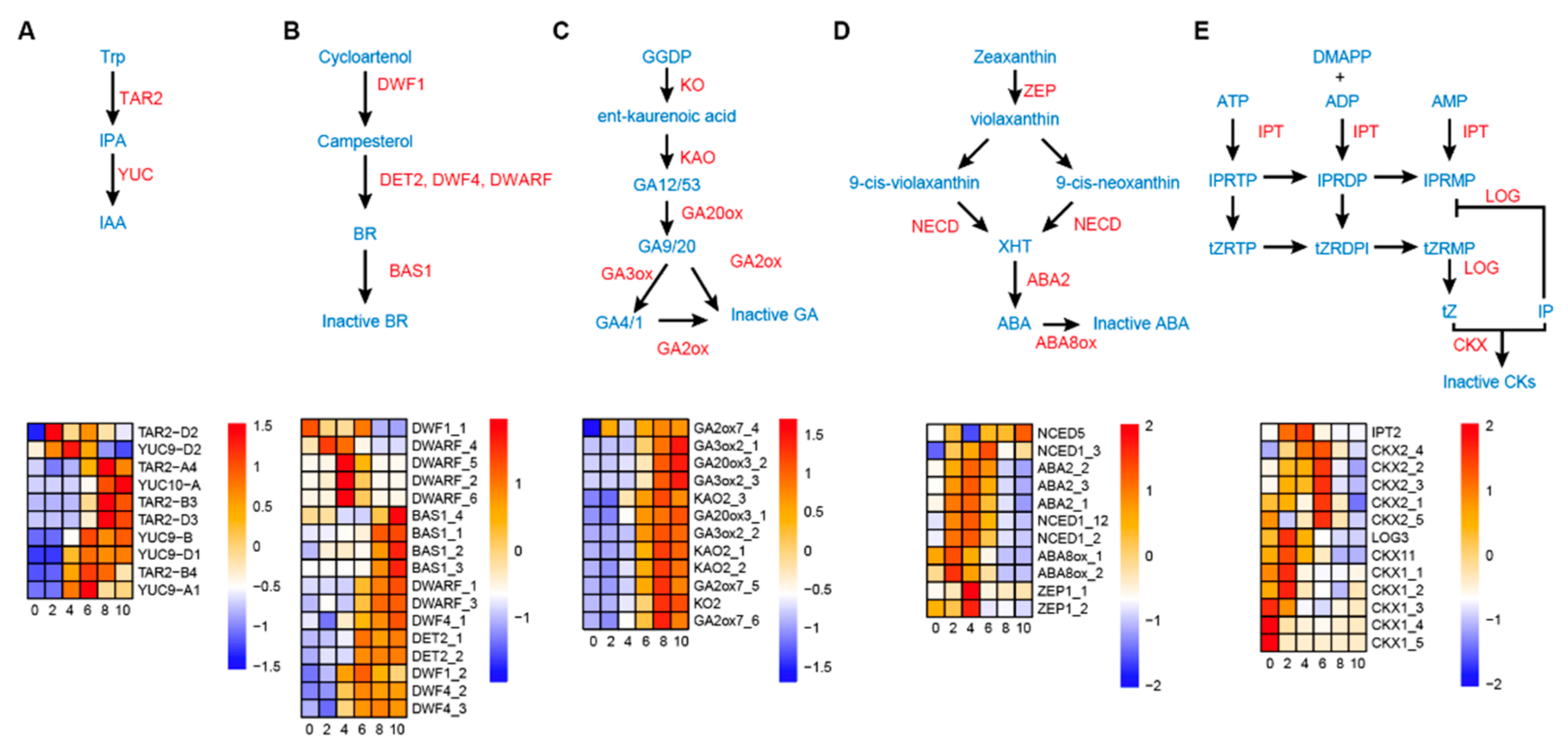
3.5. Dynamic Hormone Metabolism during the Wheat Grain Expanding Phase
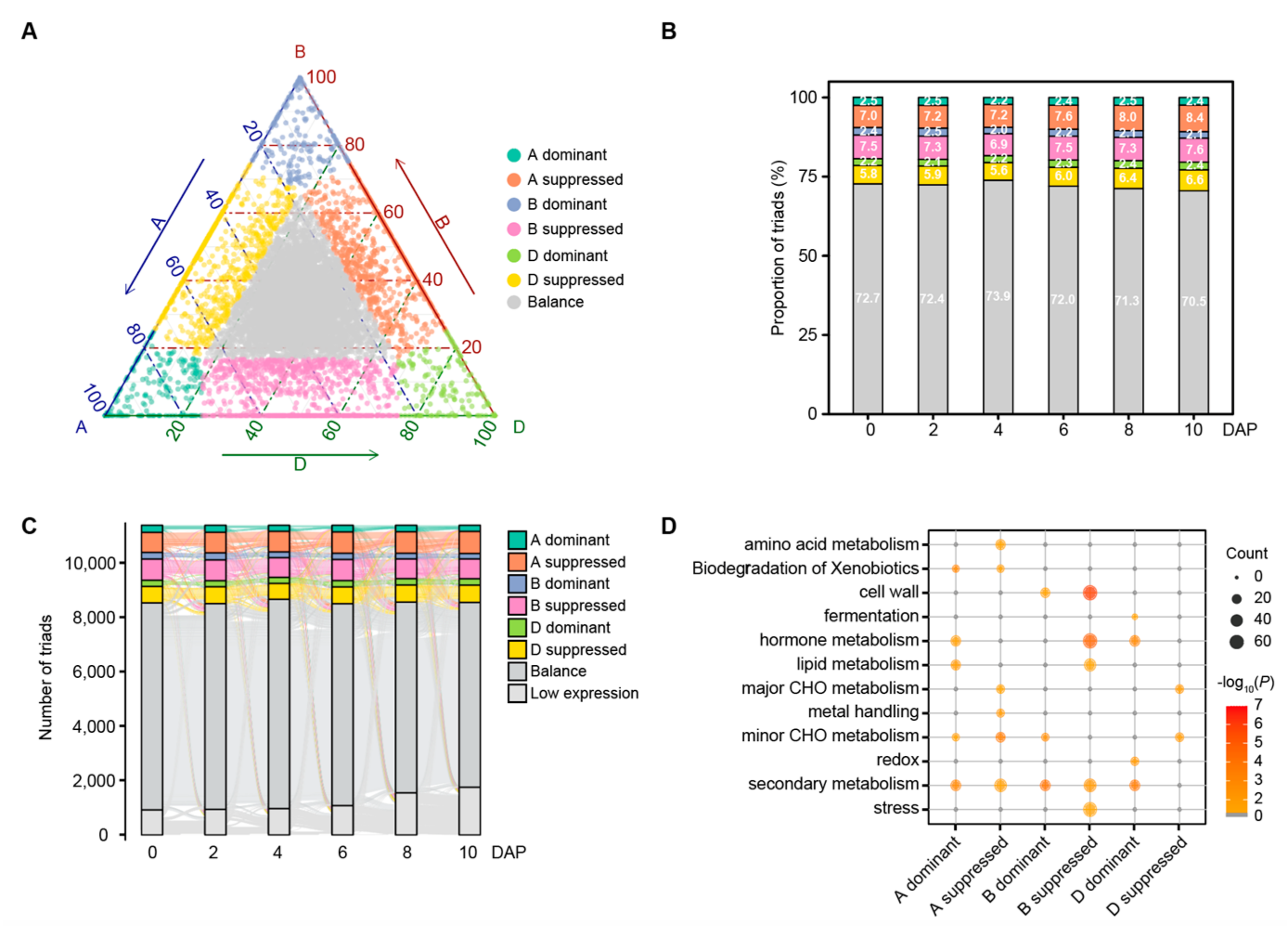
3.6. Homoeolog Expression Bias during Wheat Grain Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pfeifer, M.; Kugler, K.G.; Sandve, S.R.; Zhan, B.; Rudi, H.; Hvidsten, T.R.; Mayer, K.F.X.; Olsen, O.-A. International Wheat Genome Sequencing Consortium Genome interplay in the grain transcriptome of hexaploid bread wheat. Science 2014, 345, 1250091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mechin, V.; Thevenot, C.; Le Guilloux, M.; Prioul, J.L.; Damerval, C. Developmental analysis of maize endosperm proteome suggests a pivotal role for pyruvate orthophosphate dikinase. Plant Physiol. 2007, 143, 1203–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Liu, Z.; Xu, H.; Zhang, X.; Zhao, A.; Liang, F.; Xin, M.; Peng, H.; Yao, Y.; Sun, Q.; et al. Transcriptome analysis reveals potential mechanisms for different grain size between natural and resynthesized allohexaploid wheats with near-identical AABB genomes. BMC Plant Biol. 2018, 18, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saulnier, L.; Guillon, F.; Chateigner-Boutin, A.-L. Cell wall deposition and metabolism in wheat grain. J. Cereal Sci. 2012, 56, 91–108. [Google Scholar] [CrossRef]
- Capron, D.; Mouzeyar, S.; Boulaflous, A.; Girousse, C.; Rustenholz, C.; Laugier, C.; Paux, E.; Bouzidi, M.F. Transcriptional profile analysis of E3 ligase and hormone-related genes expressed during wheat grain development. BMC Plant Biol. 2012, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Shewry, P.R.; Mitchell, R.A.; Tosi, P.; Wan, Y.; Underwood, C.; Lovegrove, A.; Freeman, J.; Toole, G.A.; Mills, E.C.; Ward, J.L. An integrated study of grain development of wheat (cv. Hereward). J. Cereal Sci. 2012, 56, 21–30. [Google Scholar] [CrossRef]
- Locascio, A.; Roig-Villanova, I.; Bernardi, J.; Varotto, S. Current perspectives on the hormonal control of seed development in Arabidopsis and maize: A focus on auxin. Front. Plant Sci. 2014, 5, 412. [Google Scholar] [CrossRef] [Green Version]
- Basunia, M.; Nonhebel, H.M. Hormonal regulation of cereal endosperm development with a focus on rice (Oryza sativa). Funct. Plant Biol. 2019, 46, 493–506. [Google Scholar] [CrossRef]
- Rijavec, T.; Dermastia, M. Cytokinins and their Function in Developing Seeds. Acta Chim. Slov. 2010, 57, 617–629. [Google Scholar]
- Yang, J.; Zhang, J.; Huang, Z.; Wang, Z.; Zhu, Q.; Liu, L. Correlation of cytokinin levels in the endosperms and roots with cell number and cell division activity during endosperm development in rice. Ann. Bot. 2002, 90, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Friml, J.; Vieten, A.; Sauer, M.; Weijers, D.; Schwarz, H.; Hamann, T.; Offringa, R.; Jurgens, G. Efflux-dependent auxin gradients establish the apical-basal axis of Arabidopsis. Nature 2003, 426, 147–153. [Google Scholar] [CrossRef]
- Li, N.; Xu, R.; Li, Y. Molecular Networks of Seed Size Control in Plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef]
- Cheng, Z.J.; Zhao, X.Y.; Shao, X.X.; Wang, F.; Zhou, C.; Liu, Y.G.; Zhang, Y.; Zhang, X.S. Abscisic Acid Regulates Early Seed Development in Arabidopsis by ABI5-Mediated Transcription of SHORT HYPOCOTYL UNDER BLUE1. Plant Cell 2014, 26, 1053–1068. [Google Scholar] [CrossRef] [Green Version]
- Marcussen, T.; Sandve, S.R.; Heier, L.; Spannagl, M.; Pfeifer, M.; Jakobsen, K.S.; Wulff, B.B.H.; Steuernagel, B.; Mayer, K.F.X.; Olsen, O.-A.; et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science 2014, 345, 1250092. [Google Scholar] [CrossRef]
- Ramirez-Gonzalez, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361, 6403. [Google Scholar] [CrossRef] [Green Version]
- Leach, L.J.; Belfield, E.J.; Jiang, C.; Brown, C.; Mithani, A.; Harberd, N.P. Patterns of homoeologous gene expression shown by RNA sequencing in hexaploid bread wheat. BMC Genom. 2014, 15, 276. [Google Scholar] [CrossRef] [Green Version]
- Powell, J.J.; Fitzgerald, T.L.; Stiller, J.; Berkman, P.J.; Gardiner, D.M.; Manners, J.M.; Henry, R.J.; Kazan, K. The defence-associated transcriptome of hexaploid wheat displays homoeolog expression and induction bias. Plant Biotechnol. J. 2017, 15, 533–543. [Google Scholar] [CrossRef]
- Liu, Z.; Xin, M.; Qin, J.; Peng, H.; Ni, Z.; Yao, Y.; Sun, Q. Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.). BMC Plant Biol. 2015, 15, 152. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Niknafs, Y.S.; Pandian, B.; Iyer, H.K.; Chinnaiyan, A.M.; Iyer, M.K. TACO produces robust multisample transcriptome assemblies from RNA-seq. Nat. Methods 2017, 14, 68–70. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, J.; Tang, L.; Zhao, Y.; Gu, X.; Gao, G.; Luo, J. PlantTFDB 2.0: Update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011, 39, D1114–D1117. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Dante, R.A.; Larkins, B.A.; Sabelli, P.A. Cell cycle control and seed development. Front. Plant Sci. 2014, 5, 493. [Google Scholar] [CrossRef] [Green Version]
- Inze, D.; De Veylder, L. Cell cycle regulation in plant development. Annu. Rev. Genet. 2006, 40, 77–105. [Google Scholar] [CrossRef]
- Guo, J.; Wang, F.; Song, J.; Sun, W.; Zhang, X.S. The expression of Orysa;CycB1;1 is essential for endosperm formation and causes embryo enlargement in rice. Planta 2010, 231, 293–303. [Google Scholar] [CrossRef]
- De Veylder, L.; Beemster, G.T.; Beeckman, T.; Inze, D. CKS1At overexpression in Arabidopsis thaliana inhibits growth by reducing meristem size and inhibiting cell-cycle progression. Plant J. 2001, 25, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Opanowicz, M.; Hands, P.; Betts, D.; Parker, M.L.; Toole, G.A.; Mills, E.N.; Doonan, J.H.; Drea, S. Endosperm development in Brachypodium distachyon. J. Exp. Bot. 2011, 62, 735–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.; Smith, A.M. Starch biosynthesis. Plant Cell 1995, 7, 971–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, S.; Hiratsuka, K.; Chua, N.H. Transcription factors in plant growth and development. Curr. Opin. Genet. Dev. 1994, 4, 642–646. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Day, R.C.; Herridge, R.P.; Ambrose, B.A.; Macknight, R.C. Transcriptome analysis of proliferating Arabidopsis endosperm reveals biological implications for the control of syncytial division, cytokinin signaling, and gene expression regulation. Plant Physiol. 2008, 148, 1964–1984. [Google Scholar] [CrossRef] [Green Version]
- Borrill, P.; Adamski, N.; Uauy, C. Genomics as the key to unlocking the polyploid potential of wheat. New Phytol. 2015, 208, 1008–1022. [Google Scholar] [CrossRef]
- Xu, X.; E, Z.; Zhang, D.; Yun, Q.; Zhou, Y.; Niu, B.; Chen, C. OsYUC11-mediated auxin biosynthesis is essential for endosperm development of rice. Plant Physiol. 2021, 185, 934–950. [Google Scholar] [CrossRef]
- Olsen, O.A. Nuclear endosperm development in cereals and Arabidopsis thaliana. Plant Cell 2004, 16, S214–S227. [Google Scholar] [CrossRef] [Green Version]
- Jia, M.; Li, Y.; Wang, Z.; Tao, S.; Sun, G.; Kong, X.; Wang, K.; Ye, X.; Liu, S.; Geng, S.; et al. TaIAA21 represses TaARF25-mediated expression of TaERFs required for grain size and weight development in wheat. Plant J. 2021, 108, 1754–1767. [Google Scholar] [CrossRef]
- Xu, J.-J.; Zhang, X.-F.; Xue, H.-W. Rice aleurone layer specific OsNF-YB1 regulates grain filling and endosperm development by interacting with an ERF transcription factor. J. Exp. Bot. 2016, 67, 6399–6411. [Google Scholar] [CrossRef]
- Gao, Y.; An, K.; Guo, W.; Chen, Y.; Zhang, R.; Zhang, X.; Chang, S.; Rossi, V.; Jin, F.; Cao, X.; et al. The endosperm-specific transcription factor TaNAC019 regulates glutenin and starch accumulation and its elite allele improves wheat grain quality. Plant Cell 2021, 33, 603–622. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, J.; Ji, C.; Wu, Y.; Messing, J. NAC-type transcription factors regulate accumulation of starch and protein in maize seeds. Proc. Natl. Acad. Sci. USA 2019, 116, 11223–11228. [Google Scholar] [CrossRef] [Green Version]
- Renak, D.; Gibalova, A.; Solcova, K.; Honys, D. A new link between stress response and nucleolar function during pollen development in Arabidopsis mediated by AtREN1 protein. Plant Cell Environ. 2014, 37, 670–683. [Google Scholar] [CrossRef]
- Kotak, S.; Vierling, E.; Baumlein, H.; von Koskull-Doring, P. A novel transcriptional cascade regulating expression of heat stress proteins during seed development of Arabidopsis. Plant Cell 2007, 19, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tong, J.; Bai, A.; Liu, C.; Xiao, L.; Xue, H. Phytohormone dynamics in developing endosperm influence rice grain shape and quality. J. Integr. Plant Biol. 2020, 62, 1625–1637. [Google Scholar] [CrossRef]
- Sun, X.; Shantharaj, D.; Kang, X.; Ni, M. Transcriptional and hormonal signaling control of Arabidopsis seed development. Curr. Opin. Plant Biol. 2010, 13, 611–620. [Google Scholar] [CrossRef]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin synthesized by the YUCCA flavin Monooxygenases is essential for embryogenesis and leaf formation in Arabidopsis. Plant Cell 2007, 19, 2430–2439. [Google Scholar] [CrossRef] [Green Version]
- McAdam, E.L.; Meitzel, T.; Quittenden, L.J.; Davidson, S.E.; Dalmais, M.; Bendahmane, A.I.; Thompson, R.; Smith, J.J.; Nichols, D.S.; Urquhart, S.; et al. Evidence that auxin is required for normal seed size and starch synthesis in pea. New Phytol. 2017, 216, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.S.; Bayer, E.M. Auxin transport-feedback models of patterning in plants. Plant Cell Environ. 2009, 32, 1258–1271. [Google Scholar] [CrossRef] [Green Version]
- Ugartechea-Chirino, Y.; Swarup, R.; Swarup, K.; Peret, B.; Whitworth, M.; Bennett, M.; Bougourd, S. The AUX1 LAX family of auxin influx carriers is required for the establishment of embryonic root cell organization in Arabidopsis thaliana. Ann. Bot. 2010, 105, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Li, G.; Qu, D.; Li, X.; Wang, Y. Into the Seed: Auxin Controls Seed Development and Grain Yield. Int. J. Mol. Sci. 2020, 21, 1662. [Google Scholar] [CrossRef] [Green Version]
- Bethke, P.C.; Schuurink, R.; Jones, R.L. Hormonal signalling in cereal aleurone. J. Exp. Bot. 1997, 48, 1337–1356. [Google Scholar] [CrossRef] [Green Version]
- Feldman, M.; Levy, A.A.; Fahima, T.; Korol, A. Genomic asymmetry in allopolyploid plants: Wheat as a model. J. Exp. Bot. 2012, 63, 5045–5059. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, J.; Wang, Z.; Liu, S.; Kong, X.; Wang, F.; Sun, G.; Geng, S.; Mao, L.; Zhou, P.; Li, A. Transcriptome Analysis of Developing Wheat Grains at Rapid Expanding Phase Reveals Dynamic Gene Expression Patterns. Biology 2022, 11, 281. https://doi.org/10.3390/biology11020281
Guan J, Wang Z, Liu S, Kong X, Wang F, Sun G, Geng S, Mao L, Zhou P, Li A. Transcriptome Analysis of Developing Wheat Grains at Rapid Expanding Phase Reveals Dynamic Gene Expression Patterns. Biology. 2022; 11(2):281. https://doi.org/10.3390/biology11020281
Chicago/Turabian StyleGuan, Jiantao, Zhenyu Wang, Shaoshuai Liu, Xingchen Kong, Fang Wang, Guoliang Sun, Shuaifeng Geng, Long Mao, Peng Zhou, and Aili Li. 2022. "Transcriptome Analysis of Developing Wheat Grains at Rapid Expanding Phase Reveals Dynamic Gene Expression Patterns" Biology 11, no. 2: 281. https://doi.org/10.3390/biology11020281