
EGR1 Is Implicated in Right Ventricular Cardiac Remodeling Associated with Pulmonary Hypertension

 ,
,  , ,
, ,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Ethics Statement
2.3. Sample Acquisition
2.4. Microarray
2.5. Biocomputational Analyses
2.6. Identification of Transcription Factor Binding Sites (TFBS)
2.7. Immunohistochemistry
2.8. Statistical Analyses
3. Results
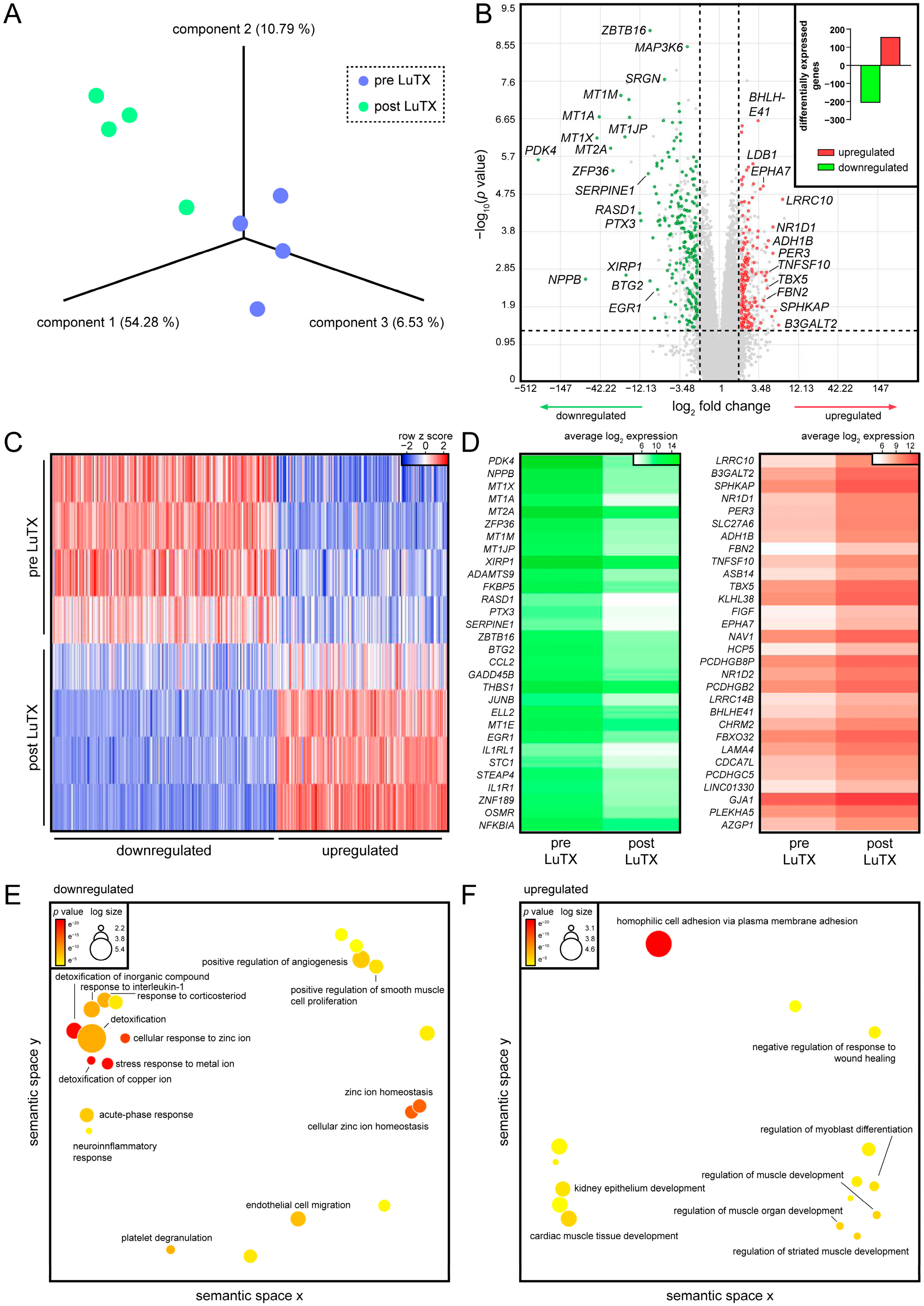
3.1. Recovered Hearts of Lung-Transplanted iPAH Patients Displayed Decreased Inflammatory Processes and Increased Cardiac Muscle Development
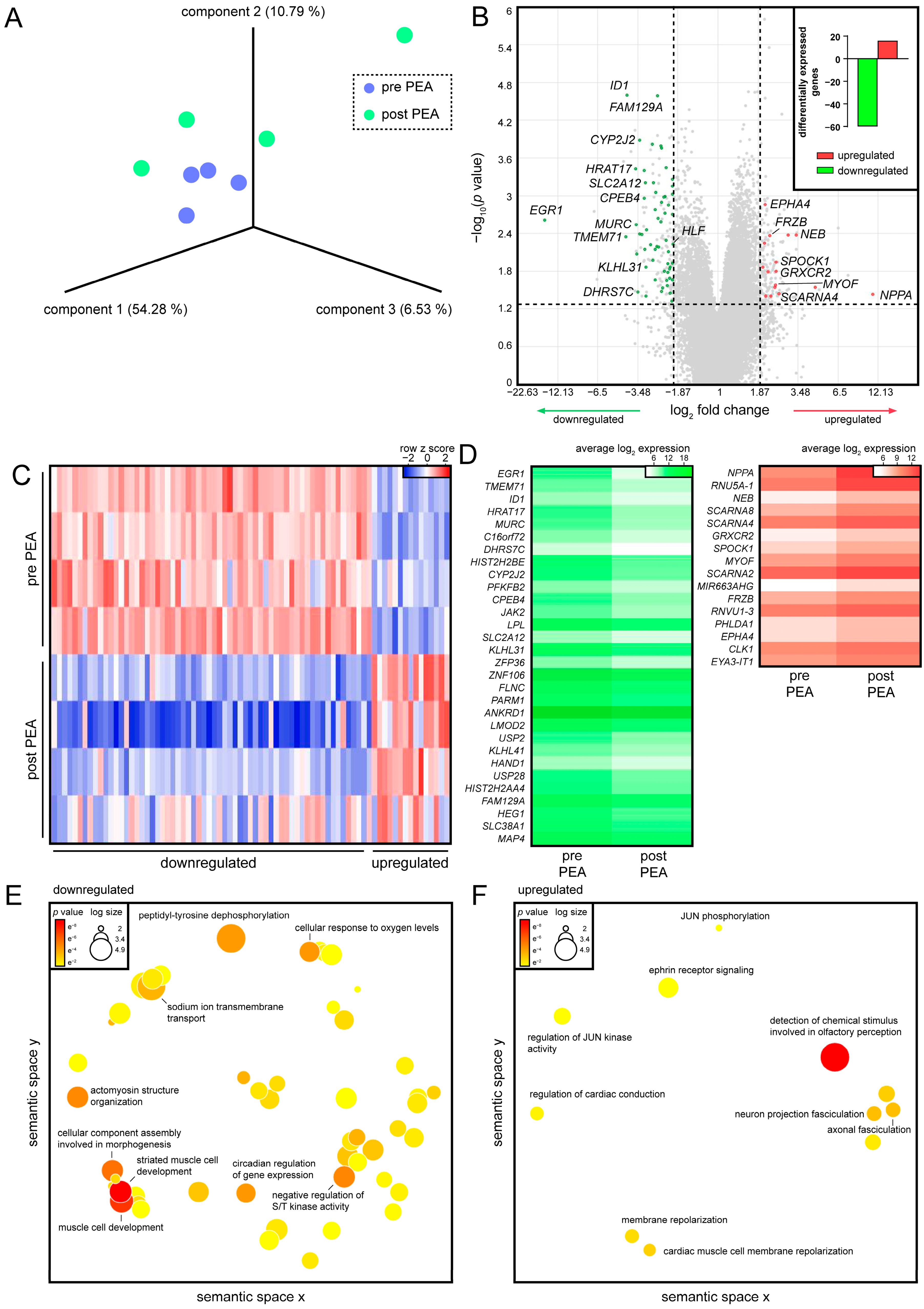
3.2. PEA of CTEPH Patients Promotes Cardiac Conduction in Regenerated RVs
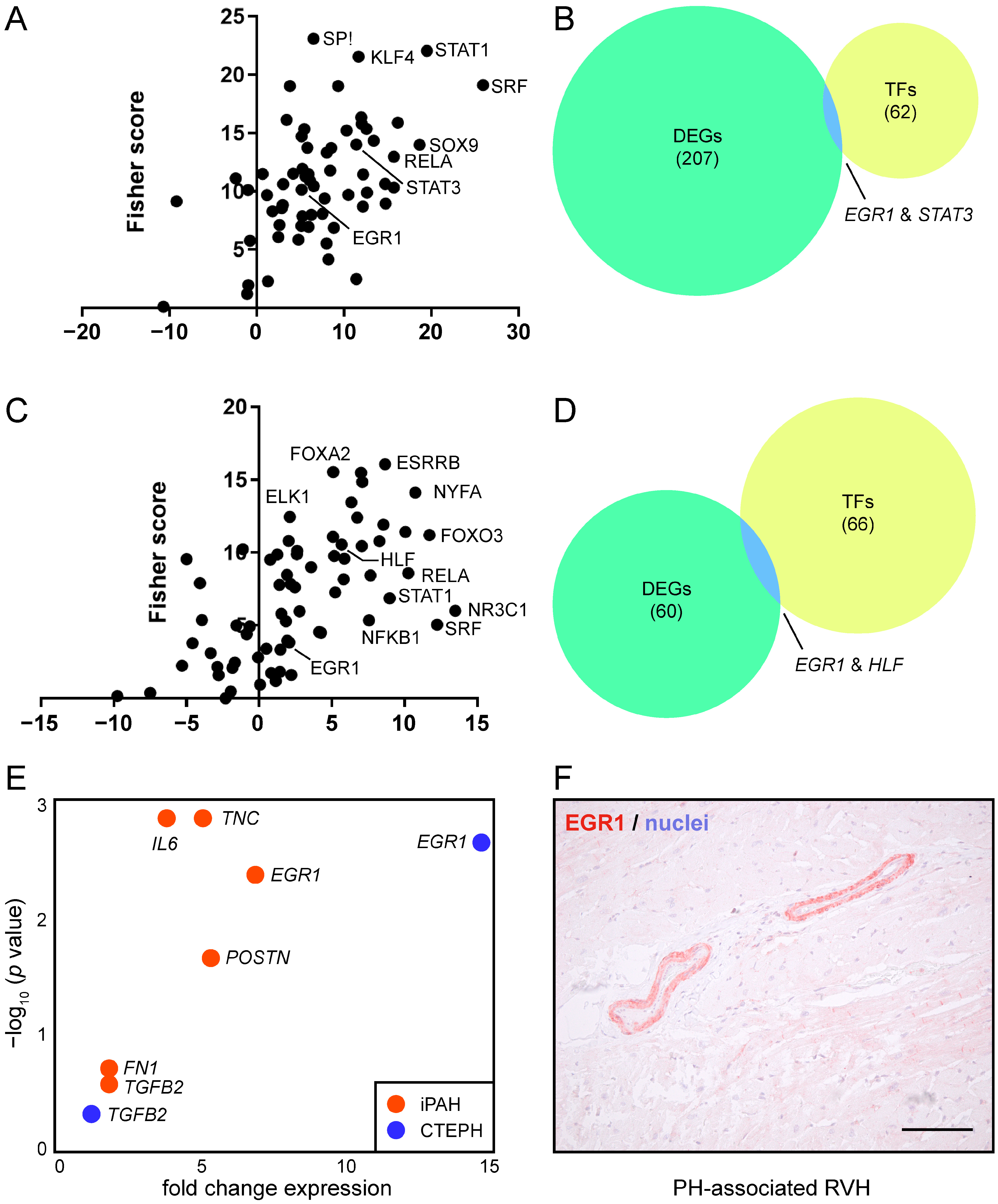
3.3. EGR1 Is Implicated in Reverse Remodeling of RVH
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruffenach, G.; Hong, J.; Vaillancourt, M.; Medzikovic, L.; Eghbali, M. Pulmonary hypertension secondary to pulmonary fibrosis: Clinical data, histopathology and molecular insights. Respir. Res. 2020, 21, 303. [Google Scholar] [CrossRef]
- Ruaro, B.; Salton, F.; Baratella, E.; Confalonieri, P.; Geri, P.; Pozzan, R.; Torregiani, C.; Bulla, R.; Confalonieri, M.; Matucci-Cerinic, M.; et al. An Overview of Different Techniques for Improving the Treatment of Pulmonary Hypertension Secondary in Systemic Sclerosis Patients. Diagnostics 2022, 12, 616. [Google Scholar] [CrossRef]
- Delcroix, M.; Torbicki, A.; Gopalan, D.; Sitbon, O.; Klok, F.A.; Lang, I.; Jenkins, D.; Kim, N.H.; Humbert, M.; Jais, X.; et al. ERS statement on chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2021, 57, 2002828. [Google Scholar] [CrossRef]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar]
- Simonneau, G.; Hoeper, M.M. The revised definition of pulmonary hypertension: Exploring the impact on patient management. Eur. Heart J. Suppl. 2019, 21 (Suppl. K), K4–K8. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Simonneau, G.; Galie, N.; Rubin, L.J.; Langleben, D.; Seeger, W.; Domenighetti, G.; Gibbs, S.; Lebrec, D.; Speich, R.; Beghetti, M.; et al. Clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2004, 43 (Suppl. S12), 5S–12S. [Google Scholar] [CrossRef]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. S25), D34–D41. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef] [Green Version]
- Farber, H.W.; Miller, D.P.; Poms, A.D.; Badesch, D.B.; Frost, A.E.; Muros-Le Rouzic, E.; Romero, A.J.; Benton, W.W.; Elliott, C.G.; McGoon, M.D.; et al. Five-Year outcomes of patients enrolled in the REVEAL Registry. Chest 2015, 148, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Rol, N.; Timmer, E.M.; Faes, T.J.; Vonk Noordegraaf, A.; Grünberg, K.; Bogaard, H.J.; Westerhof, N. Vascular narrowing in pulmonary arterial hypertension is heterogeneous: Rethinking resistance. Physiol. Rep. 2017, 5, e13159. [Google Scholar] [CrossRef] [Green Version]
- van de Veerdonk, M.C.; Kind, T.; Marcus, J.T.; Mauritz, G.J.; Heymans, M.W.; Bogaard, H.J.; Boonstra, A.; Marques, K.M.; Westerhof, N.; Vonk-Noordegraaf, A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J. Am. Coll. Cardiol. 2011, 58, 2511–2519. [Google Scholar] [CrossRef] [Green Version]
- Bing, R.; Dweck, M.R. Myocardial fibrosis: Why image, how to image and clinical implications. Heart 2019, 105, 1832–1840. [Google Scholar] [CrossRef] [Green Version]
- Kasimir, M.T.; Seebacher, G.; Jaksch, P.; Winkler, G.; Schmid, K.; Marta, G.M.; Simon, P.; Klepetko, W. Reverse cardiac remodelling in patients with primary pulmonary hypertension after isolated lung transplantation. Eur. J. Cardiothorac. Surg. 2004, 26, 776–781. [Google Scholar] [CrossRef]
- Sarashina, T.; Nakamura, K.; Akagi, S.; Oto, T.; Oe, H.; Ejiri, K.; Nakagawa, K.; Nishii, N.; Matsubara, H.; Kobayashi, M.; et al. Reverse Right Ventricular Remodeling After Lung Transplantation in Patients With Pulmonary Arterial Hypertension Under Combination Therapy of Targeted Medical Drugs. Circ. J. 2017, 81, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Ruaro, B.; Baratella, E.; Caforio, G.; Confalonieri, P.; Wade, B.; Marrocchio, C.; Geri, P.; Pozzan, R.; Andrisano, A.G.; Cova, M.A.; et al. Chronic Thromboembolic Pulmonary Hypertension: An Update. Diagnostics 2022, 12, 235. [Google Scholar] [CrossRef]
- Sharma, M.; Levine, D.J. Revisiting a Distinct Entity in Pulmonary Vascular Disease: Chronic Thromboembolic Pulmonary Hypertension (CTEPH). Medicina 2021, 57, 355. [Google Scholar] [CrossRef]
- Kramm, T.; Wilkens, H.; Fuge, J.; Schafers, H.J.; Guth, S.; Wiedenroth, C.B.; Weingard, B.; Huscher, D.; Pittrow, D.; Cebotari, S.; et al. Incidence and characteristics of chronic thromboembolic pulmonary hypertension in Germany. Clin. Res. Cardiol. 2018, 107, 548–553. [Google Scholar] [CrossRef] [Green Version]
- Pengo, V.; Lensing, A.W.; Prins, M.H.; Marchiori, A.; Davidson, B.L.; Tiozzo, F.; Albanese, P.; Biasiolo, A.; Pegoraro, C.; Iliceto, S.; et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N. Engl. J. Med. 2004, 350, 2257–2264. [Google Scholar] [CrossRef] [Green Version]
- Pepke-Zaba, J.; Delcroix, M.; Lang, I.; Mayer, E.; Jansa, P.; Ambroz, D.; Treacy, C.; D’Armini, A.M.; Morsolini, M.; Snijder, R.; et al. Chronic thromboembolic pulmonary hypertension (CTEPH): Results from an international prospective registry. Circulation 2011, 124, 1973–1981. [Google Scholar] [CrossRef] [Green Version]
- Lang, I.M.; Pesavento, R.; Bonderman, D.; Yuan, J.X. Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: A current understanding. Eur. Respir. J. 2013, 41, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Morris, T.A.; Marsh, J.J.; Chiles, P.G.; Magana, M.M.; Liang, N.C.; Soler, X.; Desantis, D.J.; Ngo, D.; Woods, V.L., Jr. High prevalence of dysfibrinogenemia among patients with chronic thromboembolic pulmonary hypertension. Blood 2009, 114, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Lang, I. Chronic thromboembolic pulmonary hypertension: A distinct disease entity. Eur. Respir. Rev. 2015, 24, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Quarck, R.; Wynants, M.; Verbeken, E.; Meyns, B.; Delcroix, M. Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2015, 46, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Reesink, H.J.; Marcus, J.T.; Tulevski, I.I.; Jamieson, S.; Kloek, J.J.; Noordegraaf, A.V.; Bresser, P. Reverse right ventricular remodeling after pulmonary endarterectomy in patients with chronic thromboembolic pulmonary hypertension: Utility of magnetic resonance imaging to demonstrate restoration of the right ventricle. J. Thorac. Cardiovasc. Surg. 2007, 133, 58–64. [Google Scholar] [CrossRef] [Green Version]
- D’Armini, A.M.; Zanotti, G.; Ghio, S.; Magrini, G.; Pozzi, M.; Scelsi, L.; Meloni, G.; Klersy, C.; Vigano, M. Reverse right ventricular remodeling after pulmonary endarterectomy. J. Thorac. Cardiovasc. Surg. 2007, 133, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Fukui, S.; Ogo, T.; Morita, Y.; Tsuji, A.; Tateishi, E.; Ozaki, K.; Sanda, Y.; Fukuda, T.; Yasuda, S.; Ogawa, H.; et al. Right ventricular reverse remodelling after balloon pulmonary angioplasty. Eur. Respir. J. 2014, 43, 1394–1402. [Google Scholar] [CrossRef] [Green Version]
- Condliffe, R.; Kiely, D.G.; Gibbs, J.S.; Corris, P.A.; Peacock, A.J.; Jenkins, D.P.; Hodgkins, D.; Goldsmith, K.; Hughes, R.J.; Sheares, K.; et al. Improved outcomes in medically and surgically treated chronic thromboembolic pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2008, 177, 1122–1127. [Google Scholar] [CrossRef]
- Havis, E.; Duprez, D. EGR1 Transcription Factor is a Multifaceted Regulator of Matrix Production in Tendons and Other Connective Tissues. Int. J. Mol. Sci. 2020, 21, 1664. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, M.G.; Kowalski, P.S.; Bartelds, B.; Borgdorff, M.A.; van der Feen, D.; Sietsma, H.; Molema, G.; Kamps, J.A.; Berger, R.M. A critical role for Egr-1 during vascular remodelling in pulmonary arterial hypertension. Cardiovasc. Res. 2014, 103, 573–584. [Google Scholar] [CrossRef]
- van der Feen, D.E.; Dickinson, M.G.; Bartelds, B.; Borgdorff, M.A.; Sietsma, H.; Levy, M.; Berger, R.M. Egr-1 identifies neointimal remodeling and relates to progression in human pulmonary arterial hypertension. J. Heart Lung Transplant. 2016, 35, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. S25), D42–D50. [Google Scholar] [CrossRef] [Green Version]
- Bogaard, H.J.; Abe, K.; Vonk Noordegraaf, A.; Voelkel, N.F. The right ventricle under pressure: Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009, 135, 794–804. [Google Scholar] [CrossRef] [Green Version]
- Galie, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.L.; Barbera, J.A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J.S.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2009, 30, 2493–2537. [Google Scholar]
- Thistlethwaite, P.A.; Mo, M.; Madani, M.M.; Deutsch, R.; Blanchard, D.; Kapelanski, D.P.; Jamieson, S.W. Operative classification of thromboembolic disease determines outcome after pulmonary endarterectomy. J. Thorac. Cardiovasc. Surg. 2002, 124, 1203–1211. [Google Scholar] [CrossRef] [Green Version]
- Cooper, L.T.; Baughman, K.L.; Feldman, A.M.; Frustaci, A.; Jessup, M.; Kuhl, U.; Levine, G.N.; Narula, J.; Starling, R.C.; Towbin, J.; et al. The role of endomyocardial biopsy in the management of cardiovascular disease: A scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. Eur. Heart J. 2007, 28, 3076–3093. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Kwon, A.T.; Arenillas, D.J.; Worsley Hunt, R.; Wasserman, W.W. oPOSSUM-3: Advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 2012, 2, 987–1002. [Google Scholar] [CrossRef]
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, B.; Megerle, A.; Bekos, C.; Janik, S.; Szerafin, T.; Birner, P.; Schiefer, A.I.; Mildner, M.; Lang, I.; Skoro-Sajer, N.; et al. Local and systemic RAGE axis changes in pulmonary hypertension: CTEPH and iPAH. PLoS ONE 2014, 9, e106440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Zhao, X.; Zhang, J.; Li, Y.; Sheng, P.; Ma, C.; Zhang, L.; Hao, X.; Zheng, X.; Xing, Y.; et al. Dacomitinib, a new pan-EGFR inhibitor, is effective in attenuating pulmonary vascular remodeling and pulmonary hypertension. Eur. J. Pharmacol. 2019, 850, 97–108. [Google Scholar] [CrossRef]
- Overbeek, M.J.; Boonstra, A.; Voskuyl, A.E.; Vonk, M.C.; Vonk-Noordegraaf, A.; van Berkel, M.P.; Mooi, W.J.; Dijkmans, B.A.; Hondema, L.S.; Smit, E.F.; et al. Platelet-derived growth factor receptor-beta and epidermal growth factor receptor in pulmonary vasculature of systemic sclerosis-associated pulmonary arterial hypertension versus idiopathic pulmonary arterial hypertension and pulmonary veno-occlusive disease: A case-control study. Arthritis Res. Ther. 2011, 13, R61. [Google Scholar] [PubMed] [Green Version]
- Laggner, M.; Hacker, P.; Oberndorfer, F.; Bauer, J.; Raunegger, T.; Gerges, C.; Szerafin, T.; Thanner, J.; Lang, I.; Skoro-Sajer, N.; et al. The Roles of S100A4 and the EGF/EGFR Signaling Axis in Pulmonary Hypertension with Right Ventricular Hypertrophy. Biology 2022, 11, 18. [Google Scholar] [CrossRef]
- Saygin, D.; Tabib, T.; Bittar, H.E.T.; Valenzi, E.; Sembrat, J.; Chan, S.Y.; Rojas, M.; Lafyatis, R. Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension. Pulm. Circ. 2020, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gaskill, C.; Marriott, S.; Pratap, S.; Menon, S.; Hedges, L.K.; Fessel, J.P.; Kropski, J.A.; Ames, D.; Wheeler, L.; Loyd, J.E.; et al. Shared gene expression patterns in mesenchymal progenitors derived from lung and epidermis in pulmonary arterial hypertension: Identifying key pathways in pulmonary vascular disease. Pulm. Circ. 2016, 6, 483–497. [Google Scholar] [CrossRef] [Green Version]
- West, J.D.; Austin, E.D.; Gaskill, C.; Marriott, S.; Baskir, R.; Bilousova, G.; Jean, J.C.; Hemnes, A.R.; Menon, S.; Bloodworth, N.C.; et al. Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2014, 307, C415–C430. [Google Scholar] [CrossRef] [Green Version]
- Franco, V. Right ventricular remodeling in pulmonary hypertension. Heart Fail. Clin. 2012, 8, 403–412. [Google Scholar] [CrossRef]
- Olaniyi, K.S.; Olatunji, L.A. Preventive effects of l-glutamine on gestational fructose-induced cardiac hypertrophy: Involvement of pyruvate dehydrogenase kinase-4. Appl. Physiol. Nutr. Metab. 2019, 44, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Zhu, Z.Y.; Zhou, X.; Xie, M.L. Chrysanthemum morifolium extract improves hypertension-induced cardiac hypertrophy in rats by reduction of blood pressure and inhibition of myocardial hypoxia inducible factor-1alpha expression. Pharm. Biol. 2016, 54, 2895–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.Y.; Gao, T.; Huang, Y.; Xue, J.; Xie, M.L. Apigenin ameliorates hypertension-induced cardiac hypertrophy and down-regulates cardiac hypoxia inducible factor-lalpha in rats. Food Funct. 2016, 7, 1992–1998. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gu, J.; Xu, Z.; Zhang, Z.; Bai, T.; Xu, J.; Cai, J.; Barnes, G.; Liu, Q.J.; Freedman, J.H.; et al. Zinc rescues obesity-induced cardiac hypertrophy via stimulating metallothionein to suppress oxidative stress-activated BCL10/CARD9/p38 MAPK pathway. J. Cell. Mol. Med. 2017, 21, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Li, X.; Hein, D.W.; Xiang, X.; Marshall, J.P.; Prabhu, S.D.; Cai, L. Metallothionein suppresses angiotensin II-induced nicotinamide adenine dinucleotide phosphate oxidase activation, nitrosative stress, apoptosis, and pathological remodeling in the diabetic heart. J. Am. Coll. Cardiol. 2008, 52, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Dong, F.; Li, Q.; Sreejayan, N.; Nunn, J.M.; Ren, J. Metallothionein prevents high-fat diet induced cardiac contractile dysfunction: Role of peroxisome proliferator activated receptor gamma coactivator 1alpha and mitochondrial biogenesis. Diabetes 2007, 56, 2201–2212. [Google Scholar] [CrossRef] [Green Version]
- Havlenova, T.; Skaroupkova, P.; Miklovic, M.; Behounek, M.; Chmel, M.; Jarkovska, D.; Sviglerova, J.; Stengl, M.; Kolar, M.; Novotny, J.; et al. Right versus left ventricular remodeling in heart failure due to chronic volume overload. Sci. Rep. 2021, 11, 17136. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Shah, S.J.; Strande, J.L.; Baldridge, A.S.; Flevaris, P.; Puckelwartz, M.J.; McNally, E.M.; Rasmussen-Torvik, L.J.; Lee, D.C.; Carr, J.C.; et al. Identification of Cardiac Fibrosis in Young Adults With a Homozygous Frameshift Variant in SERPINE1. JAMA Cardiol. 2021, 6, 841–846. [Google Scholar] [CrossRef]
- Brody, M.J.; Feng, L.; Grimes, A.C.; Hacker, T.A.; Olson, T.M.; Kamp, T.J.; Balijepalli, R.C.; Lee, Y. LRRC10 is required to maintain cardiac function in response to pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H269–H278. [Google Scholar] [CrossRef]
- Brody, M.J.; Lee, Y. The Role of Leucine-Rich Repeat Containing Protein 10 (LRRC10) in Dilated Cardiomyopathy. Front. Physiol. 2016, 7, 337. [Google Scholar] [CrossRef] [Green Version]
- Brody, M.J.; Hacker, T.A.; Patel, J.R.; Feng, L.; Sadoshima, J.; Tevosian, S.G.; Balijepalli, R.C.; Moss, R.L.; Lee, Y. Ablation of the cardiac-specific gene leucine-rich repeat containing 10 (Lrrc10) results in dilated cardiomyopathy. PLoS ONE 2012, 7, e51621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koleini, N.; Nickel, B.E.; Nagalingam, R.S.; Landry, N.M.; Fandrich, R.R.; Cheung, D.Y.C.; Dixon, I.M.; Czubryt, M.P.; Jassal, D.S.; Cattini, P.A.; et al. Elimination of endogenous high molecular weight FGF2 prevents pressure-overload-induced systolic dysfunction, linked to increased FGFR1 activity and NR1D1 expression. Cell Tissue Res. 2021, 385, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Hirt, M.N.; Sorensen, N.A.; Bartholdt, L.M.; Boeddinghaus, J.; Schaaf, S.; Eder, A.; Vollert, I.; Stohr, A.; Schulze, T.; Witten, A.; et al. Increased afterload induces pathological cardiac hypertrophy: A new in vitro model. Basic Res. Cardiol. 2012, 107, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, T.; Ueyama, T.; Isodono, K.; Tagawa, M.; Takehara, N.; Kawashima, T.; Harada, K.; Takahashi, T.; Shioi, T.; Matsubara, H.; et al. MURC, a muscle-restricted coiled-coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol. Cell. Biol. 2008, 28, 3424–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, T.; Naito, D.; Nakanishi, N.; Hayashi, Y.K.; Taniguchi, T.; Miyagawa, K.; Hamaoka, T.; Maruyama, N.; Matoba, S.; Ikeda, K.; et al. MURC/Cavin-4 facilitates recruitment of ERK to caveolae and concentric cardiac hypertrophy induced by alpha1-adrenergic receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 3811–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riechert, E.; Kmietczyk, V.; Stein, F.; Schwarzl, T.; Sekaran, T.; Jurgensen, L.; Kamuf-Schenk, V.; Varma, E.; Hofmann, C.; Rettel, M.; et al. Identification of dynamic RNA-binding proteins uncovers a Cpeb4-controlled regulatory cascade during pathological cell growth of cardiomyocytes. Cell Rep. 2021, 35, 109100. [Google Scholar] [CrossRef]
- Fu, M.; Luo, F.; Wang, E.; Jiang, Y.; Liu, S.; Peng, J.; Liu, B. Magnolol Attenuates Right Ventricular Hypertrophy and Fibrosis in Hypoxia-Induced Pulmonary Arterial Hypertensive Rats Through Inhibition of the JAK2/STAT3 Signaling Pathway. Front. Pharmacol. 2021, 12, 755077. [Google Scholar] [CrossRef]
- Gupta, V.A.; Beggs, A.H. Kelch proteins: Emerging roles in skeletal muscle development and diseases. Skelet. Muscle 2014, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Ohara, M.; Saito, Y.; Watanabe, M.; Mizutani, S.; Kobayashi, M.; Iida, A.; Nishino, I.; Fujigasaki, H. An adult nemaline myopathy patient with respiratory and heart failure harboring a novel NEB variant. Eneurologicalsci 2020, 21, 100268. [Google Scholar] [CrossRef]
- Zhang, Y.; Storey, K.B. Expression of nuclear factor of activated T cells (NFAT) and downstream muscle-specific proteins in ground squirrel skeletal and heart muscle during hibernation. Mol. Cell. Biochem. 2016, 412, 27–40. [Google Scholar] [CrossRef]
- Hwang, D.M.; Dempsey, A.A.; Lee, C.Y.; Liew, C.C. Identification of differentially expressed genes in cardiac hypertrophy by analysis of expressed sequence tags. Genomics 2000, 66, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.A.; Perry, G.; Mori, T.; Hayashi, T.; Oparil, S.; Chen, Y.F. Pressure-independent enhancement of cardiac hypertrophy in atrial natriuretic peptide-deficient mice. Clin. Exp. Pharmacol. Physiol. 2003, 30, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Chen, Y.F.; Feng, J.A.; Hayashi, T.; Oparil, S.; Perry, G.J. Volume overload results in exaggerated cardiac hypertrophy in the atrial natriuretic peptide knockout mouse. Cardiovasc. Res. 2004, 61, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Twinn, D.S.; Blackmore, H.L.; Siggens, L.; Giussani, D.A.; Cross, C.M.; Foo, R.; Ozanne, S.E. The programming of cardiac hypertrophy in the offspring by maternal obesity is associated with hyperinsulinemia, AKT, ERK, and mTOR activation. Endocrinology 2012, 153, 5961–5971. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Song, M.J.; Lee, H.A.; Kang, S.H.; Kim, M.; Yang, E.K.; Lee do, Y.; Ro, S.; Cho, J.M.; Kim, I. Histone deacetylase inhibitor, CG200745, attenuates cardiac hypertrophy and fibrosis in DOCA-induced hypertensive rats. Korean J. Physiol. Pharmacol. 2016, 20, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, M.W. WNT signaling in adult cardiac hypertrophy and remodeling: Lessons learned from cardiac development. Circ. Res. 2010, 107, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Person, A.D.; Garriock, R.J.; Krieg, P.A.; Runyan, R.B.; Klewer, S.E. Frzb modulates Wnt-9a-mediated beta-catenin signaling during avian atrioventricular cardiac cushion development. Dev. Biol. 2005, 278, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Lyn, D.; Liu, X.; Bennett, N.A.; Emmett, N.L. Gene expression profile in mouse myocardium after ischemia. Physiol. Genom. 2000, 2, 93–100. [Google Scholar] [CrossRef]
- Khachigian, L.M. Early growth response-1 in cardiovascular pathobiology. Circ. Res. 2006, 98, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Rayner, B.S.; Figtree, G.A.; Sabaretnam, T.; Shang, P.; Mazhar, J.; Weaver, J.C.; Lay, W.N.; Witting, P.K.; Hunyor, S.N.; Grieve, S.M.; et al. Selective inhibition of the master regulator transcription factor Egr-1 with catalytic oligonucleotides reduces myocardial injury and improves left ventricular systolic function in a preclinical model of myocardial infarction. J. Am. Heart Assoc. 2013, 2, e000023. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.C.; Chang, Y.T.; Chen, C.C. Early growth response 1 is an early signal inducing Cav3.2 T-type calcium channels during cardiac hypertrophy. Cardiovasc. Res. 2013, 100, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buitrago, M.; Lorenz, K.; Maass, A.H.; Oberdorf-Maass, S.; Keller, U.; Schmitteckert, E.M.; Ivashchenko, Y.; Lohse, M.J.; Engelhardt, S. The transcriptional repressor Nab1 is a specific regulator of pathological cardiac hypertrophy. Nat. Med. 2005, 11, 837–844. [Google Scholar] [CrossRef]
- Khachigian, L.M. Early Growth Response-1, an Integrative Sensor in Cardiovascular and Inflammatory Disease. J. Am. Heart Assoc. 2021, 10, e023539. [Google Scholar] [CrossRef] [PubMed]
- Khachigian, L.M.; Lindner, V.; Williams, A.J.; Collins, T. Egr-1-induced endothelial gene expression: A common theme in vascular injury. Science 1996, 271, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Silverman, E.S.; Khachigian, L.M.; Lindner, V.; Williams, A.J.; Collins, T. Inducible PDGF A-chain transcription in smooth muscle cells is mediated by Egr-1 displacement of Sp1 and Sp3. Am. J. Physiol. 1997, 273, H1415–H1426. [Google Scholar] [CrossRef]
- Santiago, F.S.; Lowe, H.C.; Kavurma, M.M.; Chesterman, C.N.; Baker, A.; Atkins, D.G.; Khachigian, L.M. New DNA enzyme targeting Egr-1 mRNA inhibits vascular smooth muscle proliferation and regrowth after injury. Nat. Med. 1999, 5, 1264–1269. [Google Scholar] [CrossRef]
- Ohtani, K.; Egashira, K.; Usui, M.; Ishibashi, M.; Hiasa, K.I.; Zhao, Q.; Aoki, M.; Kaneda, Y.; Morishita, R.; Takeshita, A. Inhibition of neointimal hyperplasia after balloon injury by cis-element ‘decoy’ of early growth response gene-1 in hypercholesterolemic rabbits. Gene. Ther. 2004, 11, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Wang, C.Y.; Hwang, D.W.; Sakaguchi, T.; Olson, K.E.; Yoshikawa, Y.; Minamoto, K.; Mazer, S.P.; Yan, S.F.; Pinsky, D.J. Transcriptional control of cardiac allograft vasculopathy by early growth response gene-1 (Egr-1). Circ. Res. 2002, 91, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Gitenay, D.; Baron, V.T. Is EGR1 a potential target for prostate cancer therapy? Future Oncol. 2009, 5, 993–1003. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Gender | F | F | F | M |
| Age at BLTX (years) | 27 | 38 | 39 | 40 |
| Type of LuTX | BLTX size reduced: resection of ML and lingula | Lobar TX: RLL and LUL | Lobar TX: RLL and LUL | Size-reduced BLTX: ML resection |
| Preop WHO-FC | 3 | 3 | 3 | 3 |
| Postop WHO-FC | 1 | 1 | 0.5 | 2 |
| Preop 6-MWD (m) | 300 | 160 | - | - |
| Preop PH-specific treatments | Double-therapy | Triple-therapy | Double-therapy | Double-therapy |
| Postop cardiological medication (s) | Bisoprolol | Bisoprolol | Ramipril | Nitrendipin, ivabradin, molsidomin |
| Pre- and Postoperative Hemodynamics | ||||
| Preop PAPsys (mmHg) | 103 | 180 | 168 | 168 |
| Postop PAPsys (mmHg) | No TR signal * | No TR signal * | No TR signal * | 46 |
| Patient ID | 5 | 6 | 7 | 8 |
|---|---|---|---|---|
| Gender | M | F | F | M |
| Age at PEA (years) | 54 | 61 | 66 | 50 |
| PA:AA ratio | 1.06 | 1.33 | 1.46 | 0.97 |
| History of VTE | PE 9 months prior to PEA | PE 3 months prior to PEA | DVT and PE 3 years prior to PEA | DVT and PE 1 year prior to PEA |
| Preop PH-specific medications | None | None | LTOT | Riociguat |
| Postop PH-specific medications | None | None | None | None |
| CAD or stenosis | Yes | No | LAD stenosis type B1: 70–90% * | No |
| Concomitant surgery | CABG 2-vessel surgery | No | No | No |
| Comorbidities | Chronic bronchitis | History of ileus, hysterectomy | Heterozygote prothrombin SNP G20210A, psoriasis arthritis | Arterial hypertension, asthma |
| UCSD classification of surgical specimens | 2 | 3 | 3 | 3 |
| Preop WHO-FC | 3 | 3 | 3 | 3 |
| Postop WHO-FC | 1 | 2 | 1 | 1 |
| Pre- and Postoperative Hemodynamics | ||||
| Preop PAP (s/d/m) (mmHg) | 75/24/40 | 64/24/40 | PAPm 26 | 55/22/36 |
| Postop PAP (s/d/m) (mmHg) | 22/11/16 | 62/23/33 | 32/13/20 | 38/17/25 |
| Preop PVR (WU) | 4.19 | 9.61 | 6.95 | 5.95 |
| Postop PVR (WU) | 1.24 | 4.31 | 1.76 | 2.82 |
| Preop CI (L/min/m2) | 2.9 | 3.1 | 2.8 | 2.1 |
| Postop CI (L/min/m2) | 3.1 | 2.2 | 3.8 | 3.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laggner, M.; Oberndorfer, F.; Golabi, B.; Bauer, J.; Zuckermann, A.; Hacker, P.; Lang, I.; Skoro-Sajer, N.; Gerges, C.; Taghavi, S.; et al. EGR1 Is Implicated in Right Ventricular Cardiac Remodeling Associated with Pulmonary Hypertension. Biology 2022, 11, 677. https://doi.org/10.3390/biology11050677
Laggner M, Oberndorfer F, Golabi B, Bauer J, Zuckermann A, Hacker P, Lang I, Skoro-Sajer N, Gerges C, Taghavi S, et al. EGR1 Is Implicated in Right Ventricular Cardiac Remodeling Associated with Pulmonary Hypertension. Biology. 2022; 11(5):677. https://doi.org/10.3390/biology11050677
Chicago/Turabian StyleLaggner, Maria, Felicitas Oberndorfer, Bahar Golabi, Jonas Bauer, Andreas Zuckermann, Philipp Hacker, Irene Lang, Nika Skoro-Sajer, Christian Gerges, Shahrokh Taghavi, and et al. 2022. "EGR1 Is Implicated in Right Ventricular Cardiac Remodeling Associated with Pulmonary Hypertension" Biology 11, no. 5: 677. https://doi.org/10.3390/biology11050677
APA StyleLaggner, M., Oberndorfer, F., Golabi, B., Bauer, J., Zuckermann, A., Hacker, P., Lang, I., Skoro-Sajer, N., Gerges, C., Taghavi, S., Jaksch, P., Mildner, M., Ankersmit, H. J., & Moser, B. (2022). EGR1 Is Implicated in Right Ventricular Cardiac Remodeling Associated with Pulmonary Hypertension. Biology, 11(5), 677. https://doi.org/10.3390/biology11050677