
Transcriptome Analysis of Plenodomus tracheiphilus Infecting Rough Lemon (Citrus jambhiri Lush.) Indicates a Multifaceted Strategy during Host Pathogenesis

 , ,
, ,  , , ,
, , ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Inoculum
2.2. DNA Extraction and Real-Time Confirmation of Infected Plants
2.3. RNA Extraction and Library Preparation and Sequencing
2.4. Fungus Transcriptome Recovery
3. Results
3.1. Analysis of Plenodomus tracheiphilus Infection on Rough Lemon Leaves and Fungus Detection
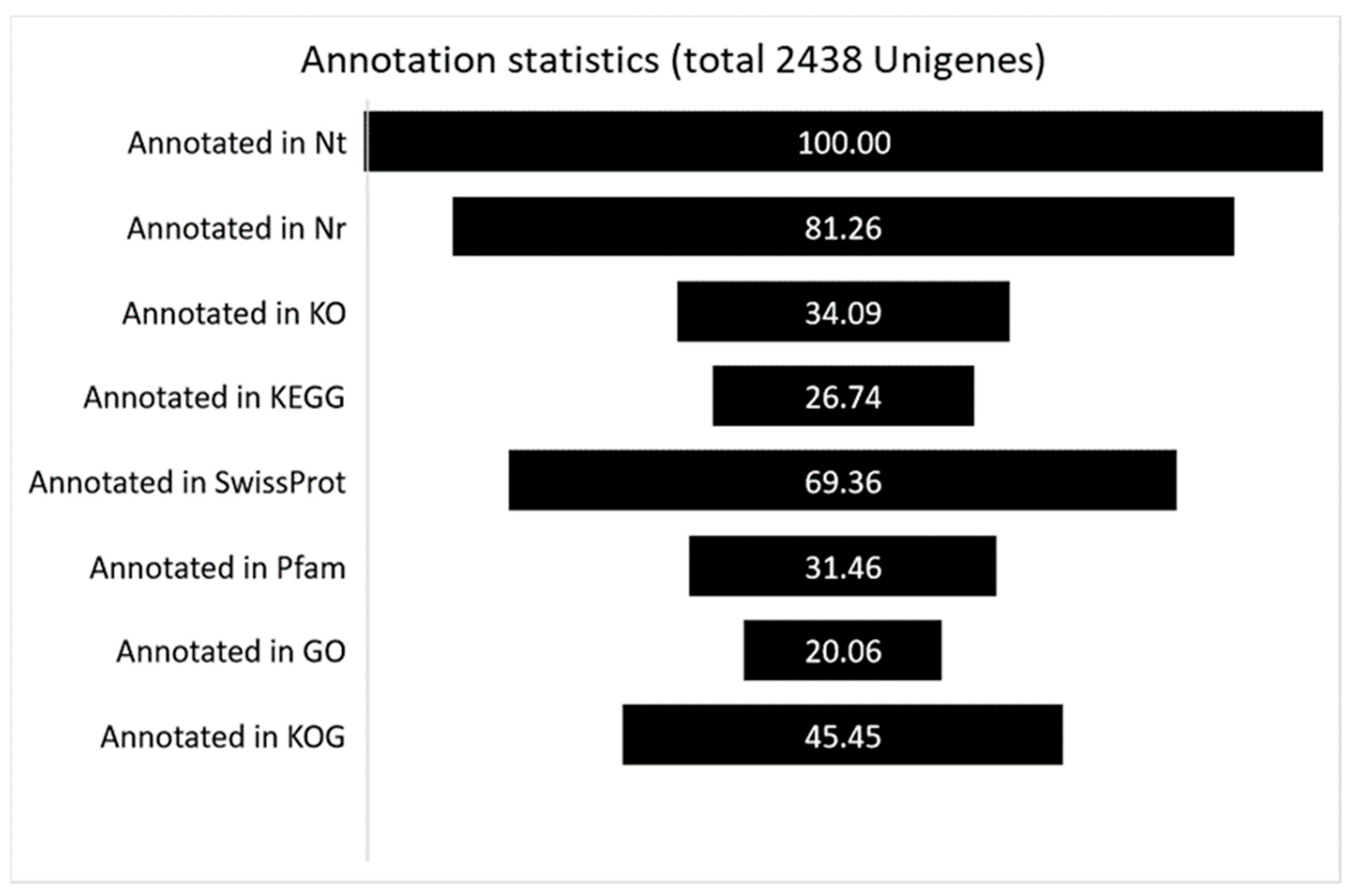
3.2. Fungus Transcriptome Recovering and Transcript Functional Annotation
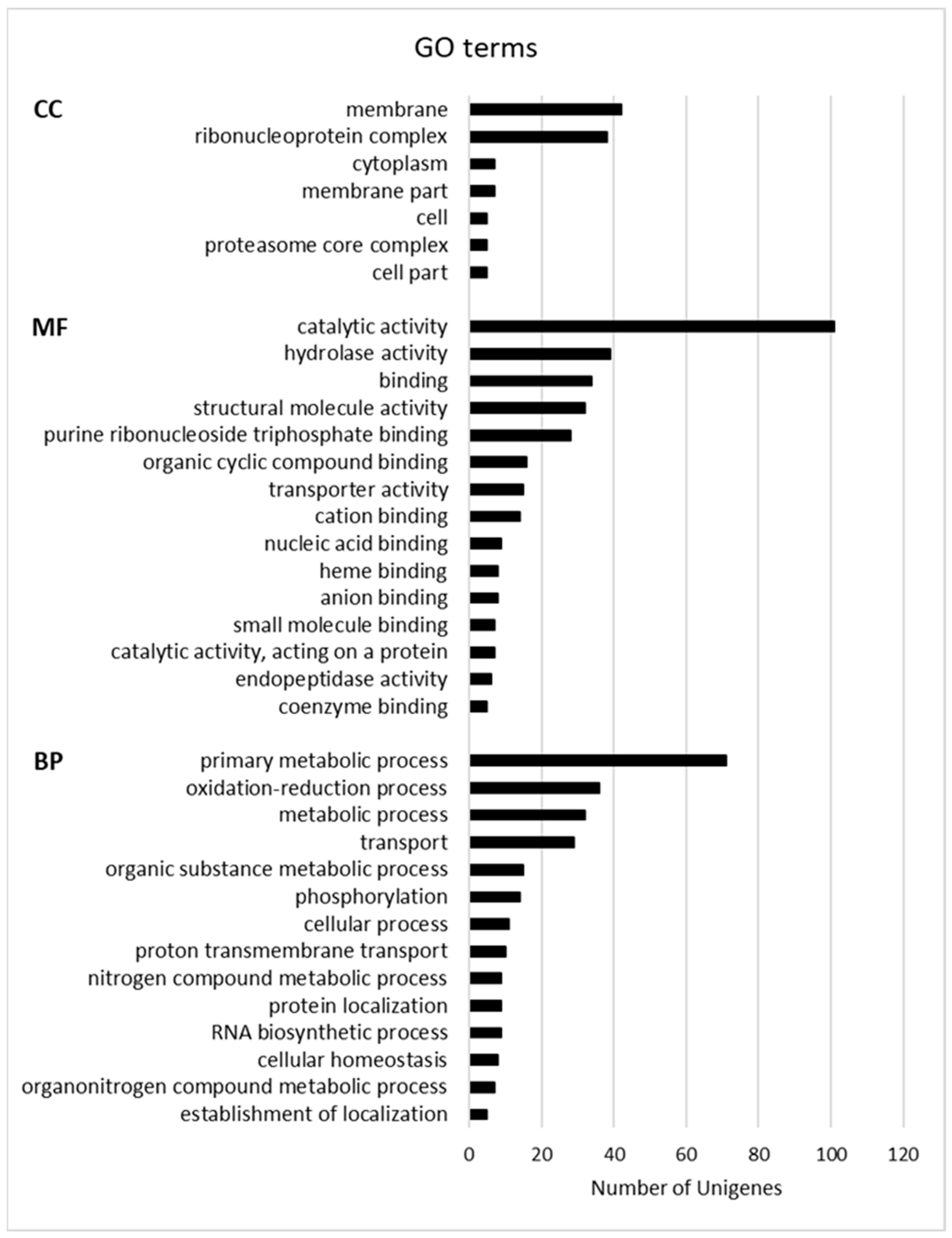
3.3. Functional Classification of Fungus Transcripts
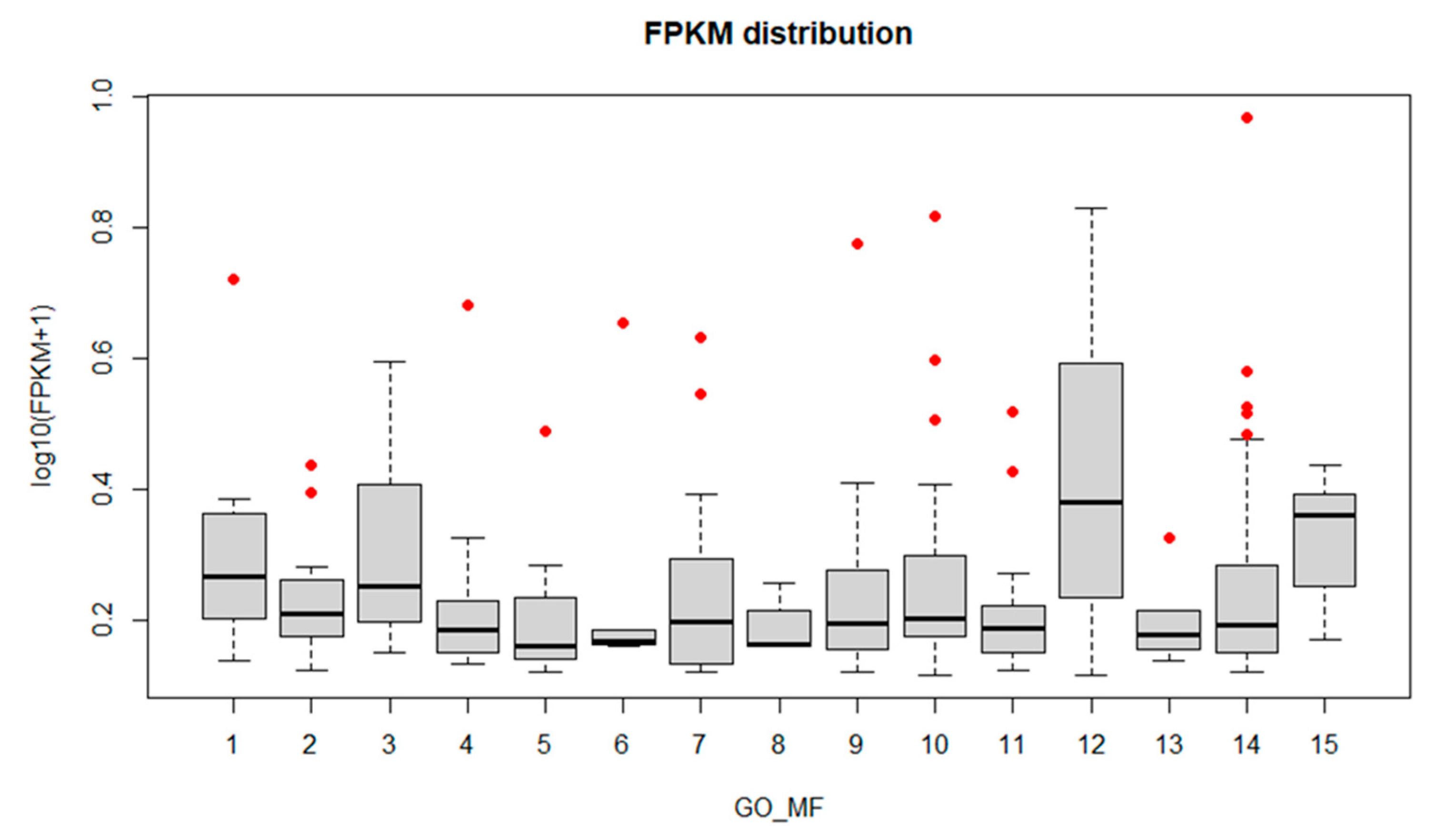
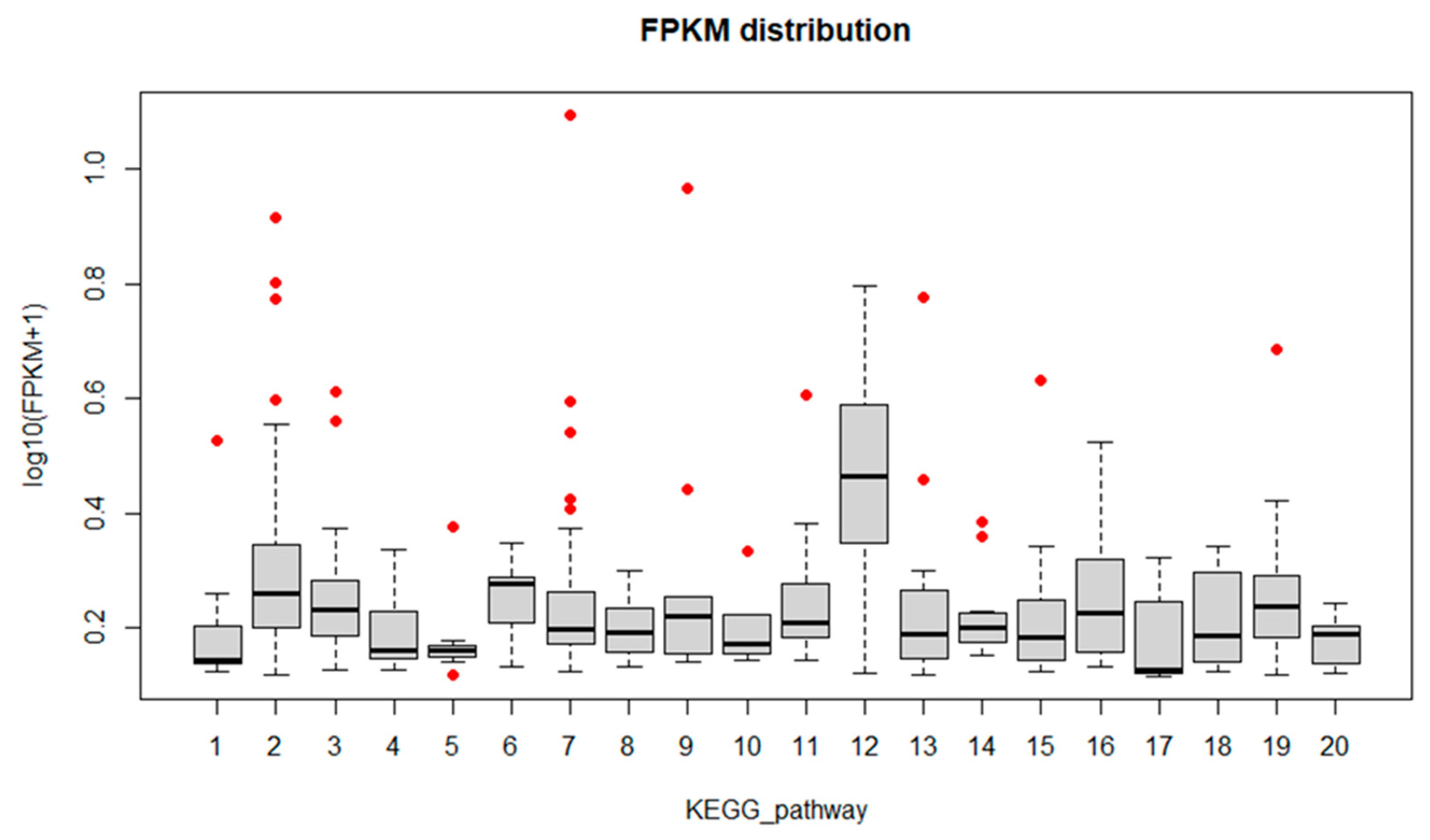
3.4. Classification of Highly Expressed Fungus Genes
3.5. Fungus-Plant Interaction: Identification of Crucial Genes Involved in Fungal Development and Pathogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Gruyter, H.; Woudenberg, J.H.C.; Aveskamp, M.M.; Verkley, G.J.M.; Groenewald, J.Z.; Crous, P.W. Redisposition of phoma-like anamorphs in pleosporales. Stud. Mycol. 2013, 75, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Hamo, M.; Ezra, D.; Krasnov, H.; Blank, L. Spatial and temporal dynamics of Mal Secco disease spread in lemon orchards in Israel. Phytopathology 2020, 110, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Catalano, C.; Di Guardo, M.; Distefano, G.; Caruso, M.; Nicolosi, E.; Deng, Z.; Gentile, A.; La Malfa, S.G. Biotechnological Approaches for Genetic Improvement of Lemon (Citrus limon (L.) Burm. f.) against Mal Secco Disease. Plants 2021, 10, 1002. [Google Scholar] [CrossRef] [PubMed]
- Catara, A.; Catara, V. Il “mal secco” degli agrumi, da un secolo in Sicilia. In Memorie e Rendiconti; Università degli Studi di Catania: Catania, Italy, 2019; pp. 35–58. [Google Scholar]
- Solel, Z. Epidemiology of Mai Secco Disease of Lemons. J. Phytopathol. 1976, 85, 90–92. [Google Scholar] [CrossRef]
- Cutuli, G.; Salerno, M. On the epidemiological meaning of phialospores in Phoma tracheiphila (Petri) Kanc. et Ghik. In Proceedings of the 5th Congress of the Mediterranean Phytopathological Union, Patras, Greece, 21–27 September 1980; pp. 72–73. [Google Scholar]
- Nigro, F.; Ippolito, A.; Salerno, M.G. Mal secco disease of citrus: A journey through a century of research. J. Plant Pathol. 2011, 93, 523–560. [Google Scholar]
- Migheli, Q.; Cacciola, S.O.; Balmas, V.; Pane, A.; Ezra, D.; Di San Lio, G.M. Mal Secco Disease Caused by Phoma tracheiphila: A Potential Threat to Lemon Production Worldwide. Plant Dis. 2009, 93, 852–867. [Google Scholar] [CrossRef] [Green Version]
- Deb, D.; Khan, A.; Dey, N. Phoma diseases: Epidemiology and control. Plant Pathol. 2020, 69, 1203–1217. [Google Scholar] [CrossRef]
- Magnano Di San Lio, G.; Lo Giudice, L. Role of cell wall in gum production in “Citrus”. Caryologia 1982, 35, 388–389. [Google Scholar]
- Balmas, V.; Scherm, B.; Ghignone, S.; Salem, A.O.M.; Cacciola, S.O.; Migheli, Q. Characterisation of Phoma tracheiphila by RAPD-PCR, microsatellite-primed PCR and ITS rDNA sequencing and development of specific primers for in planta PCR detection. Eur. J. Plant Pathol. 2005, 111, 235–247. [Google Scholar] [CrossRef]
- Grasso, F.M.; Catara, V. Preliminary characterization of Phoma tracheiphila isolates from Italy and Greece by DNA-based typing methods. J. Plant Pathol. 2006, 88, 45. [Google Scholar]
- Licciardello, G.; Grasso, F.M.; Bella, P.; Cirvilleri, G.; Grimaldi, V.; Catara, V. Identification and detection of Phoma tracheiphila, causal agent of citrus mal secco disease, by real-time polymerase chain reaction. Plant Dis. 2006, 90, 1523–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezra, D.; Kroitor, T.; Sadowsky, A. Molecular characterization of Phoma tracheiphila, causal agent of Mal secco disease of citrus, in Israel. Eur. J. Plant Pathol. 2007, 118, 183–191. [Google Scholar] [CrossRef]
- Russo, M.; Grasso, F.M.; Bella, P.; Licciardello, G.; Catara, A.; Catara, V. Molecular diagnostic tools for the detection and characterization of Phoma tracheiphila. Acta Hortic. 2011, 892, 207–214. [Google Scholar] [CrossRef]
- Magnano Di San Lio, G.; Perrotta, G. Variabilità in Phoma tracheiphila. In Integrated Pest Control in Citrus Groves; Cavalloro, R., Di Martino, E., Eds.; A A Balkema Publishers: Rotterdam, Holland, 1986; pp. 267–270. [Google Scholar]
- Cacciola, S.O.; Natoli, M.; Pane, A.; Perrotta, G.; Petrone, G. Characterization of polygalacturonase activities from Phoma tracheiphila. Ital. J. Biochem. 1990, 39, 3. [Google Scholar]
- Shao, D.; Smith, D.L.; Kabbage, M.; Roth, M.G. Effectors of Plant Necrotrophic Fungi. Front. Plant Sci. 2021, 12, 687713. [Google Scholar] [CrossRef] [PubMed]
- Nachmias, A.; Barash, I.; Solel, Z.; Strobel, G.A. Purification and characterization of a phytotoxin produced by Phoma tracheiphila, the causal agent of mal secco disease of citrus. Physiol. Plant Pathol. 1977, 10, 147–152. [Google Scholar] [CrossRef]
- Gentile, A.; Tribulato, E.; Continella, G.; Vardi, A. Differential responses of citrus calli and protoplasts to culture filtrate and toxin of Phoma tracheiphila. Theor. Appl. Genet. 1992, 83, 759–764. [Google Scholar] [CrossRef]
- Fogliano, V.; Marchese, A.; Scaloni, A.; Ritieni, A.; Visconti, A.; Randazzo, G.; Graniti, A. Characterization of a 60 kDa phytotoxic glycoprotein produced by Phoma tracheiphila and its relation to malseccin. Physiol. Mol. Plant Pathol. 1998, 53, 149–161. [Google Scholar] [CrossRef]
- Russo, R.; Caruso, M.; Arlotta, C.; Piero, A.R.L.; Nicolosi, E.; Di Silvestro, S. Identification of field tolerance and resistance to mal secco disease in a citrus germplasm collection in sicily. Agronomy 2020, 10, 1806. [Google Scholar] [CrossRef]
- Russo, R.; Sicilia, A.; Caruso, M.; Arlotta, C.; Di Silvestro, S.; Gmitter, F.G.; Nicolosi, E.; Lo Piero, A.R. De novo transcriptome sequencing of rough lemon leaves (Citrus jambhiri Lush.) in response to Plenodomus tracheiphilus infection. Int. J. Mol. Sci. 2021, 22, 882. [Google Scholar] [CrossRef]
- Sicilia, A.; Testa, G.; Santoro, D.F.; Cosentino, S.L.; Lo Piero, A.R. RNASeq analysis of giant cane reveals the leaf transcriptome dynamics under long-term salt stress. BMC Plant Biol. 2019, 19, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicilia, A.; Santoro, D.F.; Testa, G.; Cosentino, S.L.; Lo Piero, A.R. Transcriptional response of giant reed (Arundo donax L.) low ecotype to long-term salt stress by unigene-based RNAseq. Phytochemistry 2020, 177, 112436. [Google Scholar] [CrossRef] [PubMed]
- Grasso, F.M. Caratterizzazione Fenotipica di Isolati del Fungo Phoma Tracheiphila e Sviluppo di un Metodo di Rilevamento Quantitativo Mediante Real-Time PCR; University of Catania: Catania, Italy, 2008. [Google Scholar]
- Salerno, M.; Catara, V. Ricerche sul “mal secco” degli Agrumi (Deutero-phoma tracheiphila Petri). IV. Comportamento parassitario del fungo in ospiti diversi dagli Agrumi. Tec. Agric. 1967, 19, 290–297. [Google Scholar]
- Hart, T.; Komori, H.K.; LaMere, S.; Podshivalova, K.; Salomon, D.R. Finding the active genes in deep RNA-seq gene expression studies. BMC Genom. 2013, 14, 778. [Google Scholar] [CrossRef] [Green Version]
- Hebenstreit, D.; Fang, M.; Gu, M.; Charoensawan, V.; Van Oudenaarden, A.; Teichmann, S.A. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 2011, 7, 497. [Google Scholar] [CrossRef] [Green Version]
- Baccelli, I. Cerato-platanin family proteins: One function for multiple biological roles? Front. Plant Sci. 2015, 5, 769. [Google Scholar] [CrossRef]
- Frischmann, A.; Neudl, S.; Gaderer, R.; Bonazza, K.; Zach, S.; Gruber, S.; Spadiut, O.; Friedbacher, G.; Grothe, H.; Seidl-Seiboth, V. Self-assembly at air/water interfaces and carbohydrate binding properties of the small secreted protein EPL1 from the fungus Trichoderma atroviride. J. Biol. Chem. 2013, 288, 4278–4287. [Google Scholar] [CrossRef] [Green Version]
- Gaderer, R.; Bonazza, K.; Seidl-Seiboth, V. Cerato-platanins: A fungal protein family with intriguing properties and application potential. Appl. Microbiol. Biotechnol. 2014, 98, 4795–4803. [Google Scholar] [CrossRef] [Green Version]
- Talibi, D.; Raymond, M. Isolation of a Putative Candida albicans Transcriptional Regulator Involved in Pleiotropic Drug Resistance by Functional Complementation of a pdr1 pdr3 Mutation in Saccharomyces cerevisiae. J. Bacteriol. 1999, 181, 231. [Google Scholar] [CrossRef] [Green Version]
- Bari, V.K.; Sharma, S.; Alfatah, M.; Mondal, A.K.; Ganesan, K. Plasma Membrane Proteolipid 3 Protein Modulates Amphotericin B Resistance throughSphingolipid Biosynthetic Pathway. Sci. Rep. 2015, 5, 9685. [Google Scholar] [CrossRef] [Green Version]
- de Block, J.; Szopinska, A.; Guerriat, B.; Dodzian, J.; Villers, J.; Hochstenbach, J.F.; Morsomme, P. Yeast Pmp3p has an important role in plasma membrane organization. J. Cell Sci. 2015, 128, 3646–3659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soal, N.C.; Coetzee, M.P.A.; van der Nest, M.A.; Hammerbacher, A.; Wingfield, B.D. Phenolic degradation by catechol dioxygenases is associated with pathogenic fungi with a necrotrophic lifestyle in the Ceratocystidaceae. G3 Genes|Genome|Genet. 2022, 12, jkac008. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.E.; Fox, E.M.; Jarvis, R.S.; Howlett, B.J. The cross-pathway control system regulates production of the secondary metabolite toxin, sirodesmin PL, in the ascomycete, Leptosphaeria maculans. BMC Microbiol. 2011, 11, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langner, T.; Göhre, V. Fungal chitinases: Function, regulation, and potential roles in plant/pathogen interactions. Curr. Genet. 2016, 62, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Ibe, C.; Munro, C.A. Fungal Cell Wall Proteins and Signaling Pathways Form a Cytoprotective Network to Combat Stresses. J. Fungi 2021, 7, 739. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Walsh, C.T. Cyclopiazonic acid biosynthesis in Aspergillus sp.: Characterization of a reductase-like R* domain in cyclopiazonate synthetase that forms and releases cyclo-acetoacetyl-L-tryptophan. Biochemistry 2009, 48, 8746–8757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalivendra, S.C.; De Robertis, C.; Chang, P.K.; Damann, K.E. Cyclopiazonic acid is a pathogenicity factor for Aspergillus flavus and a promising target for screening germplasm for ear rot resistance. Mol. Plant-Microbe Interact. 2017, 30, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xie, X.; Liu, J.; Wang, G.L.; Qiu, D. Nascent Polypeptide-Associated Complex Involved in the Development and Pathogenesis of Fusarium graminearum on Wheat. Engineering 2020, 6, 546–552. [Google Scholar] [CrossRef]
- Yu, J.; Bhatnagar, D.; Cleveland, T.E. Completed sequence of aflatoxin pathway gene cluster in Aspergillus parasiticus. FEBS Lett. 2004, 564, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Shima, Y.; Shiina, M.; Shinozawa, T.; Ito, Y.; Nakajima, H.; Adachi, Y.; Yabe, K. Participation in aflatoxin biosynthesis by a reductase enzyme encoded by vrdA gene outside the aflatoxin gene cluster. Fungal Genet. Biol. 2009, 46, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Artigot, M.P.; Loiseau, N.; Laffitte, J.; Mas-Reguieg, L.; Tadrist, S.; Oswald, I.P.; Puel, O. Molecular cloning and functional characterization of two CYP619 cytochrome P450s involved in biosynthesis of patulin in Aspergillus clavatus. Microbiology 2009, 155, 1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tragni, V.; Cotugno, P.; De Grassi, A.; Cavalluzzi, M.M.; Mincuzzi, A.; Lentini, G.; Sanzani, S.M.; Ippolito, A.; Pierro, C.L. Targeting Penicillium expansum GMC Oxidoreductase with High Affinity Small Molecules for Reducing Patulin Production. Biology 2021, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Fiore, M.C.; Mercati, F.; Spina, A.; Blangiforti, S.; Venora, G.; Dell’Acqua, M.; Sunseri, F. High-Throughput Genotype, Morphology, and Quality Traits Evaluation for the Assessment of Genetic Diversity of Wheat Landraces from Sicily. Plants 2019, 8, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazawa, T.; Kawahigashi, H.; Matsumoto, T.; Mizuno, H. Simultaneous Transcriptome Analysis of Sorghum and Bipolaris sorghicola by Using RNA-seq in Combination with De Novo Transcriptome Assembly. PLoS ONE 2013, 8, e62460. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Minio, A.; Ferrarini, A.; Valente, M.T.; Bagnaresi, P.; Orrù, L.; Tononi, P.; Zamperin, G.; Infantino, A.; Valè, G.; et al. De novo genome assembly of the soil-borne fungus and tomato pathogen Pyrenochaeta lycopersici. BMC Genom. 2014, 15, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, R.G.T.; Cassin, A.; Grandaubert, J.; Clark, B.L.; Van De Wouw, A.P.; Rouxel, T.; Howlett, B.J. Genomes and transcriptomes of partners in plant-fungal- interactions between canola (Brassica napus) and two Leptosphaeria species. PLoS ONE 2014, 9, e103098. [Google Scholar] [CrossRef]
- Ghosh, S.; Kanwar, P.; Jha, G. Identification of candidate pathogenicity determinants of Rhizoctonia solani AG1-IA, which causes sheath blight disease in rice. Curr. Genet. 2018, 64, 729–740. [Google Scholar] [CrossRef]
- Reboledo, G.; Agorio, A.; Vignale, L.; Batista-García, R.A.; Ponce De León, I. Botrytis cinerea transcriptome during the infection process of the bryophyte physcomitrium patens and angiosperms. J. Fungi 2021, 7, 11. [Google Scholar] [CrossRef]
- Strano, C.P.; Bella, P.; Licciardello, G.; Fiore, A.; Lo Piero, A.R.; Fogliano, V.; Venturi, V.; Catara, V. Pseudomonas corrugata crpCDE is part of the cyclic lipopeptide corpeptin biosynthetic gene cluster and is involved in bacterial virulence in tomato and in hypersensitive response in Nicotiana benthamiana. Mol. Plant Pathol. 2015, 16, 495–506. [Google Scholar] [CrossRef]
- Pusztahelyi, T. Chitin and chitin-related compounds in plant-fungal interactions. Mycology 2018, 9, 189–201. [Google Scholar] [CrossRef]
- Skinner, W.; Keon, J.; Hargreaves, J. Gene information for fungal plant pathogens from expressed sequences. Curr. Opin. Microbiol. 2001, 4, 381–386. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Y.; Liang, Y.; Dong, Y.; Yang, X.; Yuan, J.; Qiu, D. The verticillium dahliae snodprot1-like protein VdCP1 contributes to virulence and triggers the plant immune system. Front. Plant Sci. 2017, 8, 1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barash, I.; Pupkin, G.; Koren, L.; Ben-Hayyim, G.; Strobel, G.A. A low molecular weight phytotoxin produced by Phoma tracheiphila, the cause of mal secco disease in citrus. Physiol. Plant Pathol. 1981, 19, 17–29. [Google Scholar] [CrossRef]
- Graniti, A. Host-parasite relations in citrus diseases as exemplified by Phytophthora gummosis and Deuterophoma mal secco. In Proceedings of the 1st International Citrus Symposium, Riverside, CA, USA, 16–26 March 1968; pp. 1187–1200. [Google Scholar]
- Parisi, A.; Tringali, C.; Magnano Di San Lio, G.; Cacciola, S.O. Phytotoxic activity of mellein; a low-molecular weight metabolite of Phoma tracheiphila. In Proceedings of the International Society of Citriculture, Acireale, Italy, 8–13 March 1992; pp. 884–886. [Google Scholar]
- Parisi, A.; Piattelli, M.; Tringali, C.; Di San Lio, G.M. Identification of the phytotoxin mellein in culture fluids of Phoma tracheiphila. Phytochemistry 1993, 32, 865–867. [Google Scholar] [CrossRef]
- Pennisi, A.M.; Di Pasquale, G.; Bonforte, M.; Sesto, F. Phytotoxic metabolites of ipo-virulent Phoma tracheiphila isolates. In Citriculture, Proceedings of the Sixth International Citrus Congress, Tel Aviv, Israel, 6–11 March, 1988; Goren, R., Mendel, K., Eds.; Balaban: Rehovot, Israel, 1988; pp. 817–827. [Google Scholar]
- Tringali, C.; Parisi, A.; Piattelli, M.; Magnano Di San Lio, G. Phomenin A and B, bioactive polypropionate pyrones from culture fluids of Phoma tracheiphila. Nat. Prod. Lett. 1993, 3, 101–106. [Google Scholar] [CrossRef]
- Nachmias, A.; Barash, I.; Buchner, V.; Solel, Z.; Strobel, G.A. A phytotoxic glycopeptide from lemon leaves infected with Phoma fracheiphila. Physiol. Plant Pathol. 1979, 14, 135–140. [Google Scholar] [CrossRef]
- Kato, N.; Tokuoka, M.; Shinohara, Y.; Kawatani, M.; Uramoto, M.; Seshime, Y.; Fujii, I.; Kitamoto, K.; Takahashi, T.; Takahashi, S.; et al. Genetic Safeguard against Mycotoxin Cyclopiazonic Acid Production in Aspergillus oryzae. ChemBioChem 2011, 12, 1376–1382. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster ID | Database Description | Percent Identity | Evalue | FPKM |
|---|---|---|---|---|
| 17189.0 | GPI-anchored cupredoxin ARB SwissProt: P0DN32 (P. tracheiphilus), Sequence ID: KAF2852624.1 | 100% | 4 × 10−89 | 9.374 |
| 16536.0 | Protein SnodProt1 SwissProt: O74238 (Parastagonospora nodorum), Sequence ID: O74238.1 | 79% | 3 × 10−75 | 8.880 |
| 1627.0 | Glucose-repressible gene SwissProt: P22151 (Akanthomyces lecanii RCEF 1005), Sequence ID: OAA78938.1 | 85% | 1 × 10−33 | 5.533 |
| 14701.25619 | Plasma membrane proteolipid 3 SwissProt: Q4HXT6 (Stemphylium lycopersici), Sequence ID: KNG45495.1 | 98% | 3 × 10−30 | 3.663 |
| 15912.0 | Endochitinase B1 SwissProt: Q873X9 (Leptosphaeria biglobosa), SequenceID: KAH9877693.1 | 78% | 0.0 | 2.660 |
| 4528.0 | Cross-pathway control protein 1 SwissProt: P87090 (Alternaria alternata), Sequence ID: KAH8629237.1 | 67% | 5 × 10−148 | 2.079 |
| 18021.0 | Catechol 1,2-dioxygenase SwissProt: P86029 (Alternaria panax), Sequence ID: KAG9186190.1 | 94% | 0.0 | 1.602 |
| 12526.0 | Cell wall mannoprotein PIR3 SwissProt: A6ZZG1 (Plenodomus tracheiphilus), Sequence ID: KAF2851031.1 | 100% | 2 × 10−170 | 1.411 |
| 20096.0 | Beta-cyclopiazonate dehydrogenase SwissProt: Q2UG11 (Colletotrichum truncatum), Sequence ID: XP_036575998.1 | 62% | 0.0 | 1.320 |
| 14701.7227 | Fluconazole resistance protein 1 C6 transcription factor-like protein SwissProt: O93870 (Plenodomus tracheiphilus), Sequence ID: AF057038.1 | 99% | 0.0 | 1.320 |
| 17612.0 | Nascent polypeptide associated complex subunit beta SwissProt: Q0ULD0 (Parastagonospora nodorum), Sequence ID: Q0ULD0.1 | 92% | 2 × 10−91 | 1.127 |
| 17977.0 | Nascent polypeptide associated complex subunit alpha SwissProt: Q0UKB5 (Plenodomus tracheiphilus), Sequence ID: KAF2851025.1 | 100% | 4 × 10−58 | 1.113 |
| Cluster ID | Database Description | Percent Identity | Evalue | FPKM |
|---|---|---|---|---|
| 21106.0 | Pectinesterase [Alternaria arborescens] | 74.03% | 1 × 10−157 | 1.725 |
| 6634.0 | Pectin lyase-like protein [Setomelanomma holmii] | 88.16% | 0.0 | 0.700 |
| 8430.0 | pectate lyase-like protein [Alternaria alternata] | 82.62% | 1 × 10−173 | 0.479 |
| 19444.0 | Putative endo-beta-1,4-glucanase D [Leotiomycetes sp. MPI-SDFR-AT-0126] | 73.08% | 3 × 10−120 | 0.413 |
| 15121.0 | GMC oxidoreductase [Plenodomus tracheiphilus IPT5] | 100.00% | 2 × 10−69 | 0.637 |
| 7879.0 | Norsolorinic acid reductase A [Alternaria arborescens] | 88.00% | 0.0 | 0.706 |
| 22064.0 | Versiconal hemiacetal acetate reductase [Alternaria arborescens] | 87.00% | 0.0 | 0.322 |
| 12949.0 | zinc-binding oxidoreductase-like protein ToxD [Plenodomus tracheiphilus IPT5] Trans-enoyl reductase fsr4 | 100% | 0.0 | 0.469 |
| 11498.0 | Di-copper centre-containing protein [Plenodomus tracheiphilus IPT5] N-acetyl-6-hydroxytryptophan oxidase ivoB | 100% | 0.0 | 0.357 |
| 8092.0 | Developmental regulator flbA [Alternaria gaisen] KAB2108722.1 | 89% | 6 × 10−102 | 0.683 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sicilia, A.; Russo, R.; Caruso, M.; Arlotta, C.; Di Silvestro, S.; Gmitter, F.G., Jr.; Gentile, A.; Nicolosi, E.; Lo Piero, A.R. Transcriptome Analysis of Plenodomus tracheiphilus Infecting Rough Lemon (Citrus jambhiri Lush.) Indicates a Multifaceted Strategy during Host Pathogenesis. Biology 2022, 11, 761. https://doi.org/10.3390/biology11050761
Sicilia A, Russo R, Caruso M, Arlotta C, Di Silvestro S, Gmitter FG Jr., Gentile A, Nicolosi E, Lo Piero AR. Transcriptome Analysis of Plenodomus tracheiphilus Infecting Rough Lemon (Citrus jambhiri Lush.) Indicates a Multifaceted Strategy during Host Pathogenesis. Biology. 2022; 11(5):761. https://doi.org/10.3390/biology11050761
Chicago/Turabian StyleSicilia, Angelo, Riccardo Russo, Marco Caruso, Carmen Arlotta, Silvia Di Silvestro, Frederick G. Gmitter, Jr., Alessandra Gentile, Elisabetta Nicolosi, and Angela Roberta Lo Piero. 2022. "Transcriptome Analysis of Plenodomus tracheiphilus Infecting Rough Lemon (Citrus jambhiri Lush.) Indicates a Multifaceted Strategy during Host Pathogenesis" Biology 11, no. 5: 761. https://doi.org/10.3390/biology11050761
APA StyleSicilia, A., Russo, R., Caruso, M., Arlotta, C., Di Silvestro, S., Gmitter, F. G., Jr., Gentile, A., Nicolosi, E., & Lo Piero, A. R. (2022). Transcriptome Analysis of Plenodomus tracheiphilus Infecting Rough Lemon (Citrus jambhiri Lush.) Indicates a Multifaceted Strategy during Host Pathogenesis. Biology, 11(5), 761. https://doi.org/10.3390/biology11050761