
The Role of MTBP as a Replication Origin Firing Factor


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
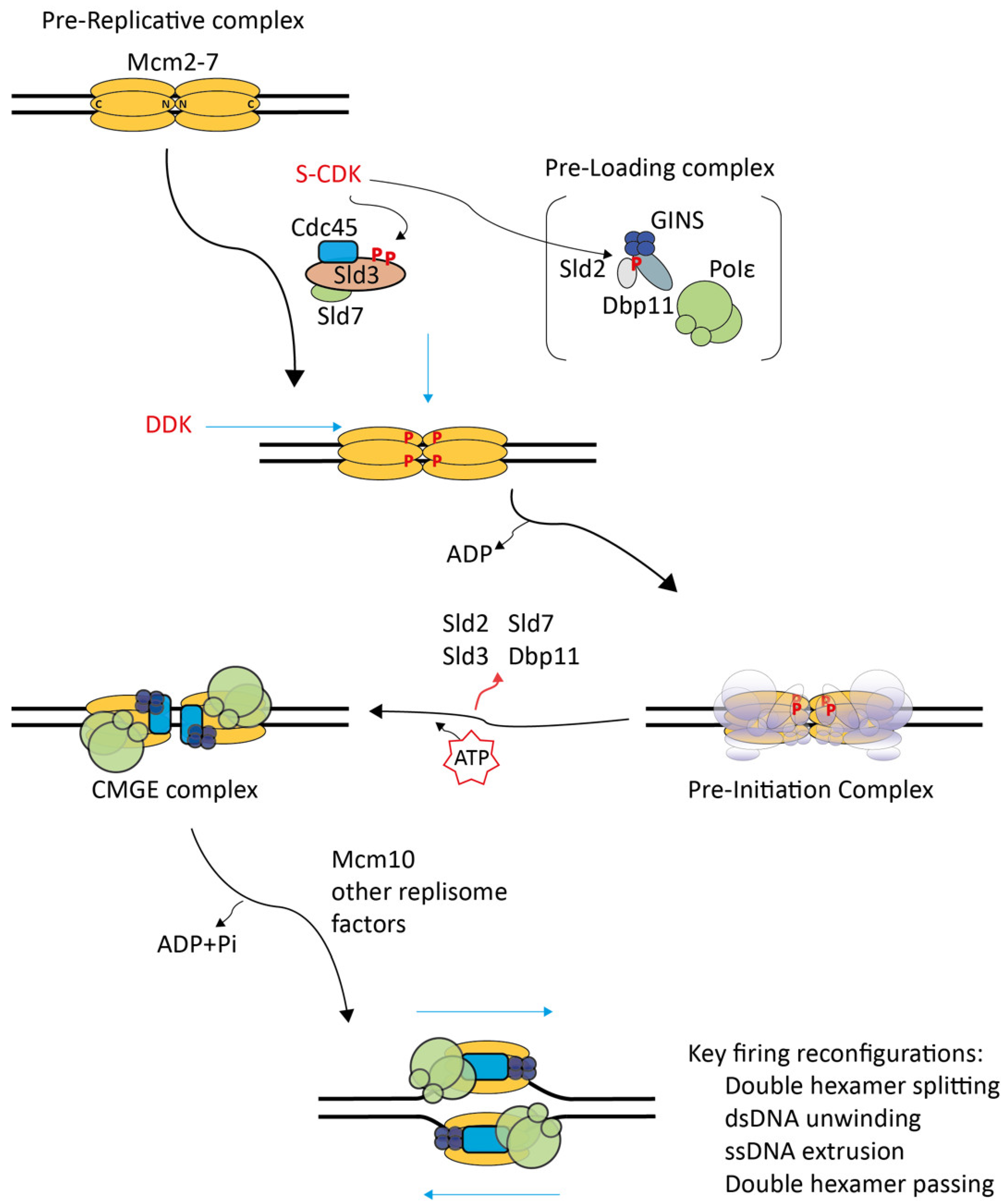
2. Molecular Processes of CMG Formation
2.1. Structural Studies on Pre-RCs and CMGs Outlined the Main Processes of the CMG Formation Reaction
2.2. In Vitro Re-Constituted Eukaryotic Replication Initiation Allowed Initial In-Detail Studies
2.3. Pre-IC Formation Is a Main Regulation Step of Origin Firing
3. The MTBP Origin Firing Factor
3.1. The Yeast MTBP Orthologue Sld7 Has a Little-Defined Important Role in Origin Firing
3.2. The Pre-IC Factor Complex MTBP–Treslin/TICRR–TopBP1 Is Required for S Phase-Specific Origin Firing in Metazoa
3.3. Roles and Characteristics of the MTBP Origin Firing Protein
3.4. Phosphorylation of MTBP by DNA-Damage-Signaling Kinases
3.5. Phosphorylation of MTBP by Cell Cycle and Other Kinases
4. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, S.C.; Bailey, K.J.; Freeland, A. Reduced Mcm2 Expression Results in Severe Stem/Progenitor Cell Deficiency and Cancer. Stem Cells 2007, 25, 3121–3132. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelsen, K.J.; Zanini, I.M.; Mijic, S.; Herrador, R.; Zellweger, R.; Ray Chaudhuri, A.; Creavin, K.D.; Blow, J.J.; Lopes, M. Deregulated origin licensing leads to chromosomal breaks by rereplication of a gapped DNA template. Genes Dev. 2013, 27, 2537–2542. [Google Scholar] [CrossRef] [Green Version]
- Davidson, I.F.; Li, A.; Blow, J.J. Deregulated Replication Licensing Causes DNA Fragmentation Consistent with Head-to-Tail Fork Collision. Mol. Cell 2006, 24, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef]
- Bell, S.P.; Kaguni, J.M. Helicase loading at chromosomal origins of replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010124. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Araki, H. Helicase Activation and Establishment of Replication Forks at Chromosomal Origins of Replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010371. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, K.; On, K.F.; Diffley, J.F. Regulating DNA replication in eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, a012930. [Google Scholar] [CrossRef] [Green Version]
- Leonard, A.C.; Mechali, M. DNA replication origins. Cold Spring Harb. Perspect. Biol. 2013, 5, a010116. [Google Scholar] [CrossRef]
- Bizard, A.H.; Hickson, I.D. Anaphase: A fortune-teller of genomic instability. Curr. Opin. Cell Biol. 2018, 52, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4, a012955. [Google Scholar] [CrossRef]
- Zou, L.; Stillman, B. Formation of a Preinitiation Complex by S-phase Cyclin CDK-Dependent Loading of Cdc45p onto Chromatin. Science 1998, 280, 593–596. [Google Scholar] [CrossRef]
- Kanemaki, M.; Labib, K. Distinct roles for Sld3 and GINS during establishment and progression of eukaryotic DNA replication forks. EMBO J. 2006, 25, 1753–1763. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa-Onami, M.; Araki, H.; Tanaka, S. Pre-initiation complex assembly functions as a molecular switch that splits the Mcm2-7 double hexamer. EMBO Rep. 2017, 18, 1752–1761. [Google Scholar] [CrossRef]
- Boyd, M.T.; Vlatkovic, N.; Haines, D.S. A novel cellular protein (MTBP) binds to MDM2 and induces a G1 arrest that is suppressed by MDM2. J. Biol. Chem. 2000, 275, 31883–31890. [Google Scholar] [CrossRef] [Green Version]
- Boos, D.; Yekezare, M.; Diffley, J.F.X. Identification of a Heteromeric Complex That Promotes DNA Replication Origin Firing in Human Cells. Science 2013, 340, 981–984. [Google Scholar] [CrossRef]
- Remus, D.; Beuron, F.; Tolun, G.; Griffith, J.D.; Morris, E.P.; Diffley, J.F. Concerted Loading of Mcm2–7 Double Hexamers around DNA during DNA Replication Origin Licensing. Cell 2009, 139, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Evrin, C.; Clarke, P.; Zech, J.; Lurz, R.; Sun, J.; Uhle, S.; Li, H.; Stillman, B.; Speck, C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 2009, 106, 20240–20245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeeles, J.T.P.; Deegan, T.D.; Janska, A.; Early, A.; Diffley, J.F.X. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 2015, 519, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, M.E.; Ali, F.A.; Costa, A.; Diffley, J.F.X. The mechanism of eukaryotic CMG helicase activation. Nature 2018, 555, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhai, Y.; Zhang, Y.; Li, W.; Yang, M.; Lei, J.; Tye, B.-K.; Gao, N. Structure of the eukaryotic MCM complex at 3.8 Å. Nature 2015, 524, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Ilves, I.; Tamberg, N.; Petojevic, T.; Nogales, E.; Botchan, M.R.; Berger, J.M. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 2011, 18, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langston, L.D.; Zhang, D.; Yurieva, O.; Georgescu, R.E.; Finkelstein, J.; Yao, N.Y.; Indiani, C.; O’Donnell, M.E. CMG helicase and DNA polymerase epsilon form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proc. Natl. Acad. Sci. USA 2014, 111, 15390–15395. [Google Scholar] [CrossRef] [Green Version]
- Erzberger, J.P.; Berger, J.M. Evolutionary relationships and structural mechanisms of AAA+proteins. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 93–114. [Google Scholar] [CrossRef]
- Abid Ali, F.; Douglas, M.E.; Locke, J.; Pye, V.E.; Nans, A.; Diffley, J.F.X. Cryo-EM structure of a licensed DNA replication origin. Nat. Commun. 2017, 8, 2241. [Google Scholar] [CrossRef]
- Samel, S.A.; Fernández-Cid, A.; Sun, J.; Riera, A.; Tognetti, S.; Herrera, M.C.; Li, H.; Speck, C. A unique DNA entry gate serves for regulated loading of the eukaryotic replicative helicase MCM2–7 onto DNA. Genes Dev. 2014, 28, 1653–1666. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Renault, L.; Swuec, P.; Petojevic, T.; Pesavento, J.J.; Ilves, I.; MacLellan-Gibson, K.; Fleck, R.A.; Botchan, M.R.; Berger, J.M. DNA binding polarity, dimerization, and ATPase ring remodeling in the CMG helicase of the eukaryotic replisome. eLife 2014, 3, e03273. [Google Scholar] [CrossRef]
- Yuan, Z.; Bai, L.; Sun, J.; Georgescu, R.; Liu, J.; O’Donnell, M.; Li, H. Structure of the eukaryotic replicative CMG helicase suggests a pumpjack motion for translocation. Nat. Struct. Mol. Biol. 2016, 23, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochman, M.L.; Schwacha, A. The Mcm2-7 Complex Has In Vitro Helicase Activity. Mol. Cell 2008, 31, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Frigola, J.; He, J.; Kinkelin, K.; Pye, V.E.; Renault, L.; Douglas, M.E.; Remus, D.; Cherepanov, P.; Costa, A.; Diffley, J.F.X. Cdt1 stabilizes an open MCM ring for helicase loading. Nat. Commun. 2017, 8, 15720. [Google Scholar] [CrossRef] [PubMed]
- Ilves, I.; Petojevic, T.; Pesavento, J.; Botchan, M.R. Activation of the MCM2-7 Helicase by Association with Cdc45 and GINS Proteins. Mol. Cell 2010, 37, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.C.; Janska, A.; Goswami, P.; Renault, L.; Ali, F.A.; Kotecha, A.; Diffley, J.F.X.; Costa, A. CMG–Pol epsilon dynamics suggests a mechanism for the establishment of leading-strand synthesis in the eukaryotic replisome. Proc. Natl. Acad. Sci. USA 2017, 114, 4141–4146. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, R.; Yuan, Z.; Bai, L.; Santos, R.D.L.A.; Sun, J.; Zhang, D.; Yurieva, O.; Li, H.; O’Donnell, M.E. Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc. Natl. Acad. Sci. USA 2017, 114, E697–E706. [Google Scholar] [CrossRef] [Green Version]
- Goswami, P.; Ali, F.A.; Douglas, M.E.; Locke, J.; Purkiss, A.; Janska, A.; Eickhoff, P.; Early, A.; Nans, A.; Cheung, A.M.C.; et al. Structure of DNA-CMG-Pol epsilon elucidates the roles of the non-catalytic polymerase modules in the eukaryotic replisome. Nat. Commun. 2018, 9, 5061. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Shi, Y.; Georgescu, R.E.; Yuan, Z.; Chait, B.T.; Li, H.; O’Donnell, M.E. The architecture of a eukaryotic replisome. Nat. Struct. Mol. Biol. 2015, 22, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Georgescu, R.; Schauer, G.D.; O’Donnell, M.E.; Li, H. Structure of the polymerase epsilon holoenzyme and atomic model of the leading strand replisome. Nat. Commun. 2020, 11, 31565. [Google Scholar] [CrossRef]
- Dua, R.; Levy, D.L.; Campbell, J.L. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae pol epsilon and its unexpected ability to support growth in the absence of the DNA polymerase domain. J. Biol. Chem. 1999, 274, 22283–22288. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; D’Urso, G. Schizosaccharomyces pombe Cells Lacking the Amino-Terminal Catalytic Domains of DNA Polymerase Epsilon Are Viable but Require the DNA Damage Checkpoint Control. Mol. Cell. Biol. 2001, 21, 4495–4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; van Deursen, F.; de Piccoli, G.; Labib, K. Dpb2 integrates the leading-strand DNA polymerase into the eukaryotic replisome. Curr. Biol. 2013, 23, 543–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, S.; Hirai, K.; Tak, Y.-S.; Kamimura, Y.; Araki, H. CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ɛ, and GINS in budding yeast. Genes Dev. 2010, 24, 602–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Yardimci, H.; Long, D.; Guainazzi, A.; Bermudez, V.P.; Hurwitz, J.; van Oijen, A.; Schärer, O.D.; Walter, J.C. Selective Bypass of a Lagging Strand Roadblock by the Eukaryotic Replicative DNA Helicase. Cell 2011, 146, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Langston, L.D.; O’Donnell, M. Action of CMG with strand-specific DNA blocks supports an internal unwinding mode for the eukaryotic replicative helicase. eLife 2017, 6, e23449. [Google Scholar] [CrossRef] [Green Version]
- Langston, L.D.; Mayle, R.; Schauer, G.D.; Yurieva, O.; Zhang, D.; Yao, N.Y.; Georgescu, R.E.; E O’Donnell, M. Mcm10 promotes rapid isomerization of CMG-DNA for replisome bypass of lagging strand DNA blocks. eLife 2017, 6, e29118. [Google Scholar] [CrossRef]
- Yeeles, J.; Janska, A.; Early, A.; Diffley, J.F. How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication. Mol. Cell 2016, 65, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Kanke, M.; Kodama, Y.; Takahashi, T.S.; Nakagawa, T.; Masukata, H. Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO J. 2012, 31, 2182–2194. [Google Scholar] [CrossRef] [Green Version]
- van Deursen, F.; Sengupta, S.; De Piccoli, G.; Sanchez-Diaz, A.; Labib, K. Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO J. 2012, 31, 2195–2206. [Google Scholar] [CrossRef] [Green Version]
- Watase, G.; Takisawa, H.; Kanemaki, M.T. Mcm10 Plays a Role in Functioning of the Eukaryotic Replicative DNA Helicase, Cdc45-Mcm-GINS. Curr. Biol. 2012, 22, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Francis, L.I.; Randell, J.C.; Takara, T.J.; Uchima, L.; Bell, S.P. Incorporation into the prereplicative complex activates the Mcm2–7 helicase for Cdc7–Dbf4 phosphorylation. Genes Dev. 2009, 23, 643–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, Y.-J.; Stillman, B. Cdc7-Dbf4 Phosphorylates MCM Proteins via a Docking Site-Mediated Mechanism to Promote S Phase Progression. Mol. Cell 2006, 24, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Sheu, Y.J.; Stillman, B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 2010, 463, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, R.C.; Kang, S.; Lam, W.M.; Chen, S.; Chan, C.S.; Bell, S.P. Eukaryotic Origin-Dependent DNA Replication In Vitro Reveals Sequential Action of DDK and S-CDK Kinases. Cell 2011, 146, 80–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deegan, T.D.; Yeeles, J.; Diffley, J.F. Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to MCM replicative helicase activation. EMBO J. 2016, 35, 961–973. [Google Scholar] [CrossRef]
- De Jesus-Kim, L.; Friedman, L.J.; Looke, M.; Ramsoomair, C.K.; Gelles, J.; Bell, S.P. DDK regulates replication initiation by controlling the multiplicity of Cdc45-GINS binding to Mcm2-7. eLife 2021, 10, e65471. [Google Scholar] [CrossRef]
- Zegerman, P.; Diffley, J. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 2006, 445, 281–285. [Google Scholar] [CrossRef]
- Tanaka, S.; Umemori, T.; Hirai, K.; Muramatsu, S.; Kamimura, Y.; Araki, H. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 2006, 445, 328–332. [Google Scholar] [CrossRef]
- Rappas, M.; Oliver, A.W.; Pearl, L.H. Structure and function of the Rad9-binding region of the DNA-damage checkpoint adaptor TopBP1. Nucleic Acids Res. 2011, 39, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, H.; Muramatsu, S.; Kamimura, Y.; Araki, H. S-Cdk-dependent phosphorylation of Sld2 essential for chromosomal DNA replication in budding yeast. Nature 2002, 415, 651–655. [Google Scholar] [CrossRef]
- Tak, Y.S.; Tanaka, Y.; Endo, S.; Kamimura, Y.; Araki, H. A CDK-catalysed regulatory phosphorylation for formation of the DNA replication complex Sld2-Dpb11. EMBO J. 2006, 25, 1987–1996. [Google Scholar] [CrossRef] [Green Version]
- Kamimura, Y.; Tak, Y.; Sugino, A.; Araki, H. Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J. 2001, 20, 2097–2107. [Google Scholar] [CrossRef] [Green Version]
- Gambus, A.; Jones, R.C.; Sanchez-Diaz, A.; Kanemaki, M.; Van Deursen, F.; Edmondson, R.D.; Labib, K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 2006, 8, 358–366. [Google Scholar] [CrossRef]
- Mantiero, D.; Mackenzie, A.; Donaldson, A.; Zegerman, P. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 2011, 30, 4805–4814. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Araki, H. Multiple Regulatory Mechanisms to Inhibit Untimely Initiation of DNA Replication Are Important for Stable Genome Maintenance. PLoS Genet. 2011, 7, e1002136. [Google Scholar] [CrossRef]
- Tanaka, S.; Nakato, R.; Katou, Y.; Shirahige, K.; Araki, H. Origin Association of Sld3, Sld7, and Cdc45 Proteins Is a Key Step for Determination of Origin-Firing Timing. Curr. Biol. 2011, 21, 2055–2063. [Google Scholar] [CrossRef] [Green Version]
- Zegerman, P.; Diffley, J.F.X. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef]
- Duch, A.; Palou, G.; Jonsson, Z.O.; Palou, R.; Calvo, E.; Wohlschlegel, J.; Quintana, D.G. A Dbf4 Mutant Contributes to Bypassing the Rad53-mediated Block of Origins of Replication in Response to Genotoxic Stress. J. Biol. Chem. 2011, 286, 2486–2491. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Mosqueda, J.; Maas, N.L.; Jonsson, Z.O.; DeFazio-Eli, L.G.; Wohlschlegel, J.; Toczyski, D.P. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature 2010, 467, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Randell, J.C.; Fan, A.; Chan, C.; Francis, L.I.; Heller, R.C.; Galani, K.; Bell, S.P. Mec1 Is One of Multiple Kinases that Prime the Mcm2-7 Helicase for Phosphorylation by Cdc7. Mol. Cell 2010, 40, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Davé, A.; Cooley, C.; Garg, M.; Bianchi, A. Protein Phosphatase 1 Recruitment by Rif1 Regulates DNA Replication Origin Firing by Counteracting DDK Activity. Cell Rep. 2014, 7, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraga, S.-I.; Alvino, G.M.; Chang, F.; Lian, H.-Y.; Sridhar, A.; Kubota, T.; Brewer, B.J.; Weinreich, M.; Raghuraman, M.; Donaldson, A.D. Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev. 2014, 28, 372–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattarocci, S.; Shyian, M.; Lemmens, L.; Damay, P.; Altintas, D.M.; Shi, T.; Bartholomew, C.R.; Thomä, N.H.; Hardy, C.F.; Shore, D. Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep. 2014, 7, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsume, T.; Müller, C.A.; Katou, Y.; Retkute, R.; Gierlinski, M.; Araki, H.; Blow, J.J.; Shirahige, K.; Nieduszynski, C.; Tanaka, T. Kinetochores Coordinate Pericentromeric Cohesion and Early DNA Replication by Cdc7-Dbf4 Kinase Recruitment. Mol. Cell 2013, 50, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S. Interaction of replication factor Sld3 and histone acetyl transferase Esa1 alleviates gene silencing and promotes the activation of late and dormant replication origins. Genetics 2020, 217, iyaa001. [Google Scholar] [CrossRef]
- Kamimura, Y.; Masumoto, H.; Sugino, A.; Araki, H. Sld2, which interacts with Dpb11 in Saccharomyces cerevisiae, is required for chromosomal DNA replication. Mol. Cell. Biol. 1998, 18, 6102–6109. [Google Scholar] [CrossRef] [Green Version]
- Reusswig, K.U.; Zimmermann, F.; Galanti, L.; Pfander, B. Robust Replication Control Is Generated by Temporal Gaps between Licensing and Firing Phases and Depends on Degradation of Firing Factor Sld2. Cell Rep. 2016, 17, 556–569. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.C.; Can, G.; Santos, M.M.; Alexander, D.; Zegerman, P. Checkpoint inhibition of origin firing prevents inappropriate replication outside of S-phase. eLife 2021, 10, e63589. [Google Scholar] [CrossRef]
- Boos, D.; Ferreira, P. Origin Firing Regulations to Control Genome Replication Timing. Genes 2019, 10, 199. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Umemori, T.; Endo, S.; Muramatsu, S.; Kanemaki, M.; Kamimura, Y.; Obuse, C.; Araki, H. Sld7, an Sld3-associated protein required for efficient chromosomal DNA replication in budding yeast. EMBO J. 2011, 30, 2019–2030. [Google Scholar] [CrossRef]
- Itou, H.; Shirakihara, Y.; Araki, H. The quaternary structure of the eukaryotic DNA replication proteins Sld7 and Sld3. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Mäkiniemi, M.; Hillukkala, T.; Tuusa, J.; Reini, K.; Vaara, M.; Huang, D.; Pospiech, H.; Majuri, I.; Westerling, T.; Mäkelä, T.P.; et al. BRCT Domain-containing Protein TopBP1 Functions in DNA Replication and Damage Response. J. Biol. Chem. 2001, 276, 30399–30406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hatten, R.A.; Tutter, A.V.; Holway, A.H.; Khederian, A.M.; Walter, J.C.; Michael, W.M. The Xenopus Xmus101 protein is required for the recruitment of Cdc45 to origins of DNA replication. J. Cell Biol. 2002, 159, 541–547. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Takisawa, H. Xenopus Cut5 is essential for a CDK-dependent process in the initiation of DNA replication. EMBO J. 2003, 22, 2526–2535. [Google Scholar] [CrossRef] [Green Version]
- Sansam, C.L.; Cruz, N.M.; Danielian, P.S.; Amsterdam, A.; Lau, M.L.; Hopkins, N.; Lees, J.A. A vertebrate gene, ticrr, is an essential checkpoint and replication regulator. Genes Dev. 2010, 24, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Treslin Collaborates with TopBP1 in Triggering the Initiation of DNA Replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J. Cell Biol. 2011, 193, 995–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boos, D.; Sanchez-Pulido, L.; Rappas, M.; Pearl, L.H.; Oliver, A.W.; Ponting, C.P.; Diffley, J.F. Regulation of DNA Replication through Sld3-Dpb11 Interaction Is Conserved from Yeast to Humans. Curr. Biol. 2011, 21, 1152–1157. [Google Scholar] [CrossRef] [Green Version]
- Sansam, C.G.; Goins, D.; Siefert, J.C.; Clowdus, E.A.; Sansam, C.L. Cyclin-dependent kinase regulates the length of S phase through TICRR/TRESLIN phosphorylation. Genes Dev. 2015, 29, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Volpi, I.; Gillespie, P.J.; Chadha, G.S.; Blow, J.J. The role of DDK and Treslin–MTBP in coordinating replication licensing and pre-initiation complex formation. Open Biol. 2021, 11, 210121. [Google Scholar] [CrossRef]
- Matsuno, K.; Kumano, M.; Kubota, Y.; Hashimoto, Y.; Takisawa, H. The N-Terminal Noncatalytic Region of Xenopus RecQ4 Is Required for Chromatin Binding of DNA Polymerase α in the Initiation of DNA Replication. Mol. Cell. Biol. 2006, 26, 4843–4852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangrithi, M.N.; Bernal, J.A.; Madine, M.; Philpott, A.; Lee, J.; Dunphy, W.G.; Venkitaraman, A.R. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 2005, 121, 887–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zegerman, P. Evolutionary conservation of the CDK targets in eukaryotic DNA replication initiation. Chromosoma 2015, 124, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Yoshimura, A.; Hosono, Y.; Tada, S.; Seki, M.; Enomoto, T. The N-terminal region of RECQL4 lacking the helicase domain is both essential and sufficient for the viability of vertebrate cells: Role of the N-terminal region of RECQL4 in cells. Biochim. Biophys. Acta 2011, 1813, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Gaggioli, V.; Zeiser, E.; Rivers, D.; Bradshaw, C.R.; Ahringer, J.; Zegerman, P. CDK phosphorylation of SLD-2 is required for replication initiation and germline development in C. elegans. J. Cell Biol. 2014, 204, 507–522. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.W.; Bell, M.; Mir, M.; Kao, J.A.; Darzacq, X.; Botchan, M.R.; Berger, J.M. A new class of disordered elements controls DNA replication through initiator self-assembly. eLife 2019, 8, e48562. [Google Scholar] [CrossRef]
- Kohler, K.; Sanchez-Pulido, L.; Hofer, V.; Marko, A.; Ponting, C.P.; Snijders, A.P.; Feederle, R.; Schepers, A.; Boos, D. The Cdk8/19-cyclin C transcription regulator functions in genome replication through metazoan Sld7. PLoS Biol. 2019, 17, e2006767. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, A.; Dunphy, W.G. MTBP, the partner of Treslin, contains a novel DNA-binding domain that is essential for proper initiation of DNA replication. Mol. Biol. Cell 2017, 28, 2998–3012. [Google Scholar] [CrossRef]
- Ferreira, P.; Sanchez-Pulido, L.; Marko, A.; Ponting, C.P.; Boos, D. Refining the domain architecture model of the replication origin firing factor Treslin/TICRR. Life Sci. Alliance 2022, 5, e202101088. [Google Scholar] [CrossRef]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Cayrou, C.; Coulombe, P.; Puy, A.; Rialle, S.; Kaplan, N.; Segal, E.; Méchali, M. New insights into replication origin characteristics in metazoans. Cell Cycle 2012, 11, 658–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guédin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, P.; Höfer, V.; Kronshage, N.; Marko, A.; Reusswig, K.-U.; Tetik, B.; Dießel, C.; Köhler, K.; Tschernoster, N.; Altmüller, J.; et al. MTBP phosphorylation controls DNA replication origin firing. Sci. Rep. 2021, 11, 4242. [Google Scholar] [CrossRef] [PubMed]
- Ciardo, D.; Haccard, O.; Narassimprakash, H.; Cornu, D.; Guerrera, I.C.; Goldar, A.; Marheineke, K. Polo-like kinase 1 (Plk1) regulates DNA replication origin firing and interacts with Rif1 in Xenopus. Nucleic Acids Res. 2021, 49, 9851–9869. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in Tumors Induces Early and Sustained Phosphorylation Changes in Stress Kinase Pathways but Does Not Affect Global Protein Levels. Mol. Cell. Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [Green Version]
- Grimsrud, P.A.; Carson, J.J.; Hebert, A.S.; Hubler, S.L.; Niemi, N.M.; Bailey, D.J.; Jochem, A.; Stapleton, D.S.; Keller, M.P.; Westphall, M.S.; et al. A Quantitative Map of the Liver Mitochondrial Phosphoproteome Reveals Posttranslational Control of Ketogenesis. Cell Metab. 2012, 16, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM–Chk2–Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef]
- Shechter, D.; Costanzo, V.; Gautier, J. ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell Biol. 2004, 6, 648–655. [Google Scholar] [CrossRef]
- Martinez-Val, A.; Lynch, C.J.; Calvo, I.; Ximénez-Embún, P.; Garcia, F.; Zarzuela, E.; Serrano, M.; Munoz, J. Dissection of two routes to naïve pluripotency using different kinase inhibitors. Nat. Commun. 2021, 12, 1863. [Google Scholar] [CrossRef]
- Poss, Z.C.; Ebmeier, C.C.; Odell, A.T.; Tangpeerachaikul, A.; Lee, T.; Pelish, H.E.; Shair, M.D.; Dowell, R.; Old, W.M.; Taatjes, D.J. Identification of Mediator Kinase Substrates in Human Cells using Cortistatin A and Quantitative Phosphoproteomics. Cell Rep. 2016, 15, 436–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poss, Z.C.; Ebmeier, C.C.; Taatjes, D.J. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 575–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenz, K.; Errico, A.; Costanzo, V. Plx1 is required for chromosomal DNA replication under stressful conditions. EMBO J. 2008, 27, 876–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Adaptation of a DNA Replication Checkpoint Response Depends upon Inactivation of Claspin by the Polo-like Kinase. Cell 2004, 117, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Collart, C.; Allen, G.E.; Bradshaw, C.R.; Smith, J.C.; Zegerman, P. Titration of Four Replication Factors Is Essential for the Xenopus laevis Midblastula Transition. Science 2013, 341, 893–896. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Kumagai, A.; Schlacher, K.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Interaction of Chk1 with Treslin Negatively Regulates the Initiation of Chromosomal DNA Replication. Mol. Cell 2014, 57, 492–505. [Google Scholar] [CrossRef] [Green Version]
- Wittig, K.A.; Sansam, C.G.; Noble, T.D.; Goins, D.; Sansam, C.L. The CRL4DTL E3 ligase induces degradation of the DNA replication initiation factor TICRR/TRESLIN specifically during S phase. Nucleic Acids Res. 2021, 49, 10507–10523. [Google Scholar] [CrossRef]
- Charrasse, S.; Gharbi-Ayachi, A.; Burgess, A.; Vera, J.; Hached, K.; Raynaud, P.; Schwob, E.; Lorca, T.; Castro, A. Ensa controls S-phase length by modulating Treslin levels. Nat. Commun. 2017, 8, 206. [Google Scholar] [CrossRef]
- Agarwal, N.; Tochigi, Y.; Adhikari, A.S.; Cui, S.; Cui, Y.; Iwakuma, T. MTBP plays a crucial role in mitotic progression and chromosome segregation. Cell Death Differ. 2011, 18, 1208–1219. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, N.; Adhikari, A.S.; Iyer, S.V.; Hekmatdoost, K.; Welch, D.R.; Iwakuma, T. MTBP suppresses cell migration and filopodia formation by inhibiting ACTN4. Oncogene 2013, 32, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Odvody, J.; Vincent, T.; Arrate, M.P.; Grieb, B.; Wang, S.; Garriga, J.; Lozano, G.; Iwakuma, T.; Haines, D.S.; Eischen, C.M. A deficiency in Mdm2 binding protein inhibits Myc-induced B-cell proliferation and lymphomagenesis. Oncogene 2010, 29, 3287–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieb, B.; Gramling, M.W.; Arrate, M.P.; Chen, X.; Beauparlant, S.L.; Haines, D.S.; Xiao, H.; Eischen, C.M. Oncogenic Protein MTBP Interacts with MYC to Promote Tumorigenesis. Cancer Res. 2014, 74, 3591–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, M.; Vlatkovic, N.; Boyd, M.T. Regulation of p53 and MDM2 activity by MTBP. Mol. Cell. Biol. 2005, 25, 545–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlatković, N.; El-Fert, A.; Devling, T.; Ray-Sinha, A.; Gore, D.M.; Rubbi, C.P.; Dodson, A.; Jones, A.S.; Helliwell, T.R.; Jones, T.M.; et al. Loss of MTBP expression is associated with reduced survival in a biomarker-defined subset of patients with squamous cell carcinoma of the head and neck. Cancer 2011, 117, 2939–2950. [Google Scholar] [CrossRef]
- Iwakuma, T.; Tochigi, Y.; Van Pelt, C.S.; Caldwell, L.C.; Terzian, T.; Parant, J.M.; Chau, G.P.; Koch, J.G.; Eischen, C.M.; Lozano, G. Mtbp haploinsufficiency in mice increases tumor metastasis. Oncogene 2007, 27, 1813–1820. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaffar, E.; Ferreira, P.; Sanchez-Pulido, L.; Boos, D. The Role of MTBP as a Replication Origin Firing Factor. Biology 2022, 11, 827. https://doi.org/10.3390/biology11060827
Zaffar E, Ferreira P, Sanchez-Pulido L, Boos D. The Role of MTBP as a Replication Origin Firing Factor. Biology. 2022; 11(6):827. https://doi.org/10.3390/biology11060827
Chicago/Turabian StyleZaffar, Eman, Pedro Ferreira, Luis Sanchez-Pulido, and Dominik Boos. 2022. "The Role of MTBP as a Replication Origin Firing Factor" Biology 11, no. 6: 827. https://doi.org/10.3390/biology11060827
APA StyleZaffar, E., Ferreira, P., Sanchez-Pulido, L., & Boos, D. (2022). The Role of MTBP as a Replication Origin Firing Factor. Biology, 11(6), 827. https://doi.org/10.3390/biology11060827