
Detecting Local Adaptation between North and South European Atlantic Salmon Populations



Abstract
:Simple Summary
Abstract
1. Introduction
- (1)
- Detection of atypically high values of differentiation (FST outliers) associated with particular markers/loci.
- (2)
- Detection of unusual haplotypic patterns associated with a selective carryover effect, such as increased homozygosity in certain regions.
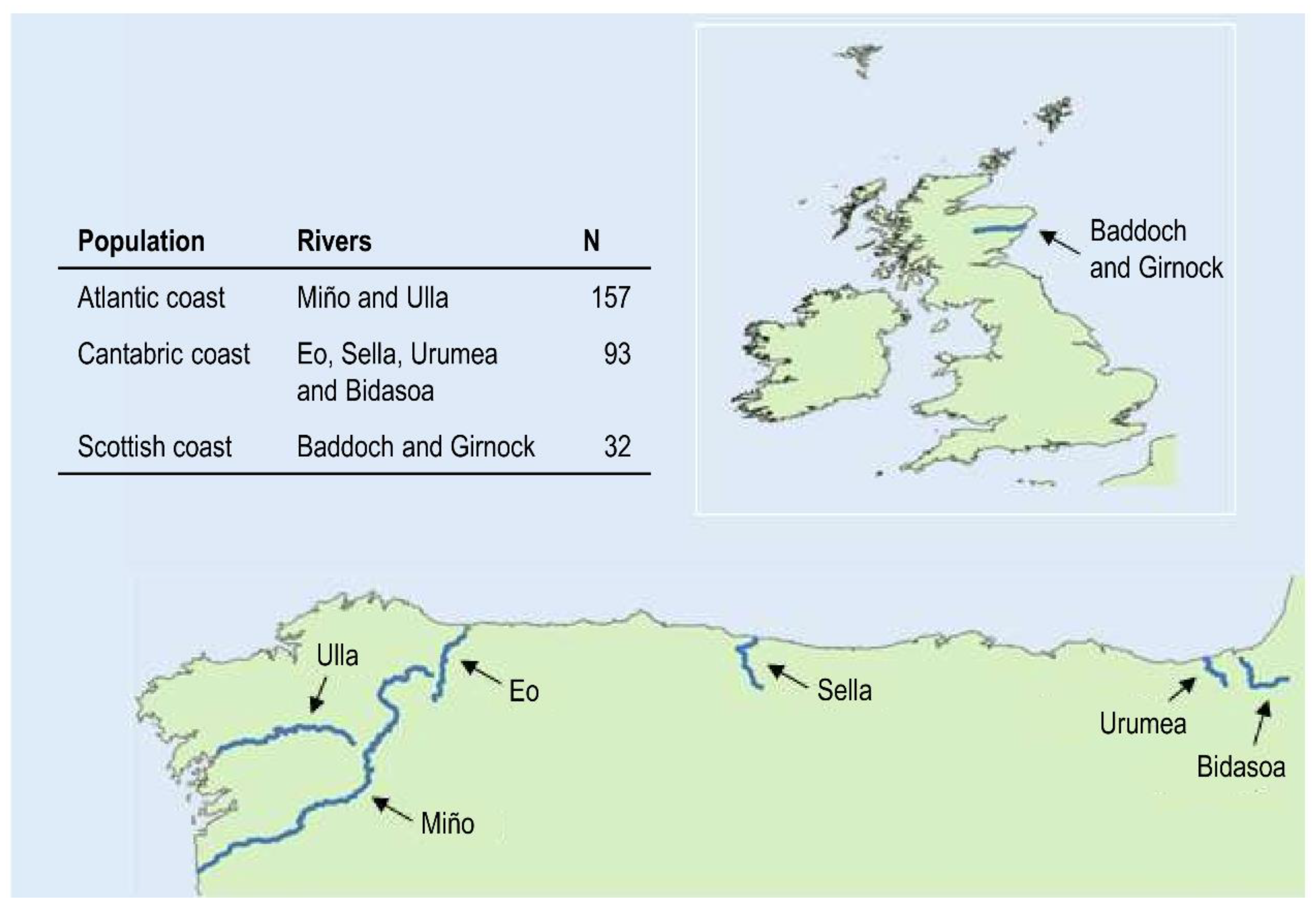
2. Materials and Methods
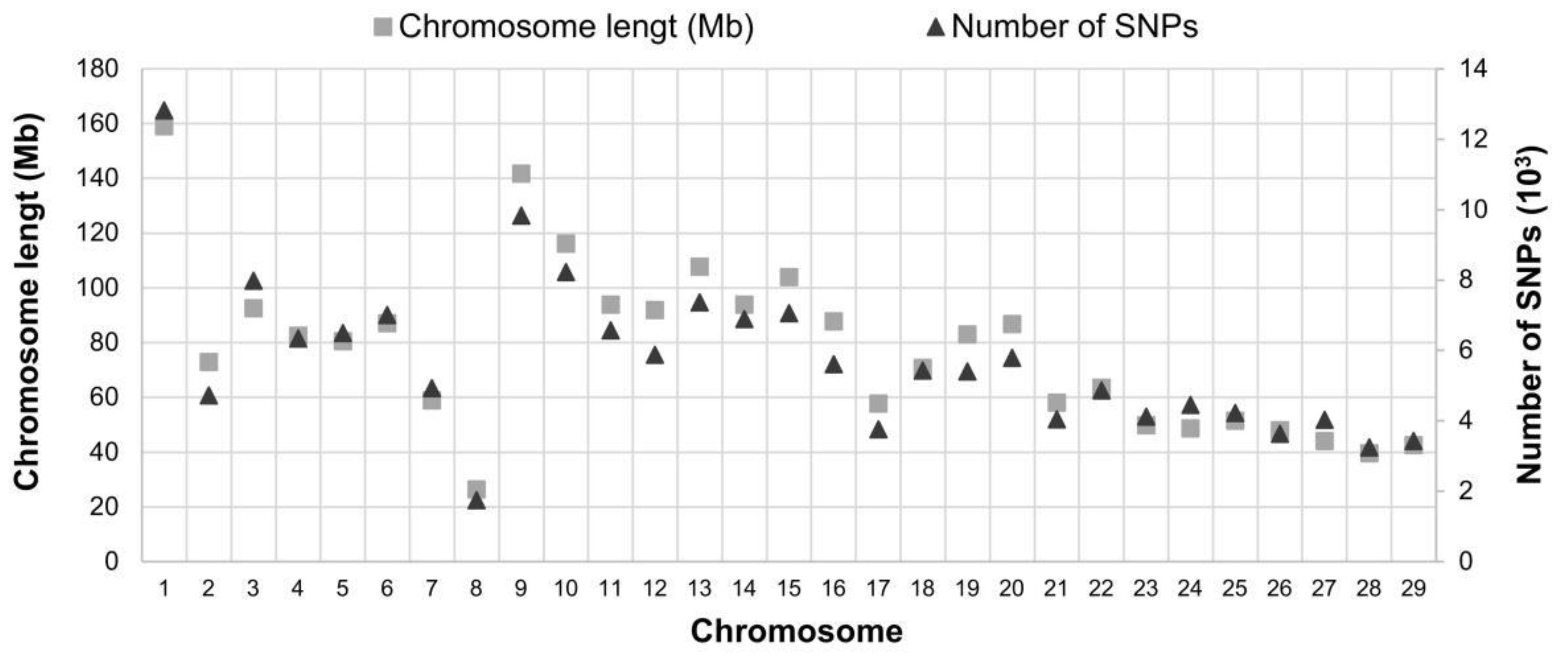
2.1. Genotyping Quality Analyses
2.2. Selection Signatures and Gene Functional Annotation
- (1)
- XP-EHH. The cross-population extended haplotype homozygosity model is based on the inspection of patterns of linkage disequilibrium decay around selected loci and detects selection based on an excess of specific haplotypes in one of the populations. This method requires a linkage map. Since no map was available for our 220 K array, we used the physical distance.
- (2)
- nvdFST. The nvdFST statistic combines two measures: a normalized variance difference (nvd) and an FST index. The nvd measure divides the haplotypes into two sets for each candidate SNP: one set carrying the major allele for the SNP and the other set carrying the minor allele. Only SNPs shared by both populations were considered. A variance of mutational distances is computed within each set and a normalized difference between variances defines the statistic that will increase under selection. The FST measure takes advantage of the fact that if selection acts on a SNP pointed by a high nvd value, then the FST at that site will be higher when compared to the overall FST assuming equilibrium in the presence of migration. A resampling method is used to compute the p-value under the hypothesis of panmixia and the final candidate SNPs are those with highest nvd values that additionally reject panmixia [21]. We used different windows sizes (1000, 500, 250, 125 and 62) for computing nvd and considered as potential candidates the 1% of the SNPs with highest nvd, which were also significant for the FST test under any window size.
- (3)
- SmileFinder. This method uses a resampling-based strategy to infer the significance of multiloci FST variance using sliding windows of haplotypes of increasing size. In this case, the highest values of variance indicate the presence of selection.
3. Results
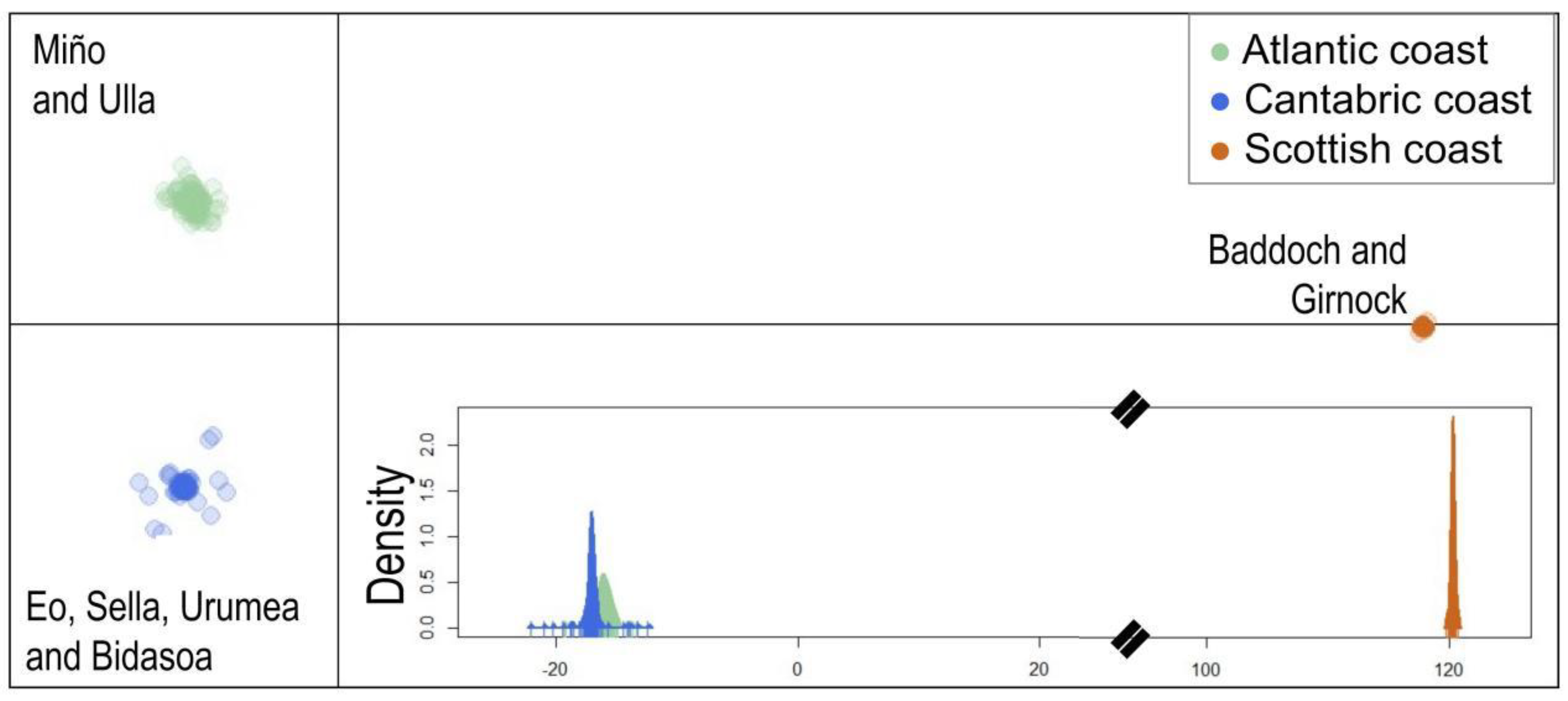
3.1. Population Structure
3.2. Selection Signatures: Outlier Methods
3.3. Selection Signatures: Haplotype Methods
3.4. Gene Functional Annotation
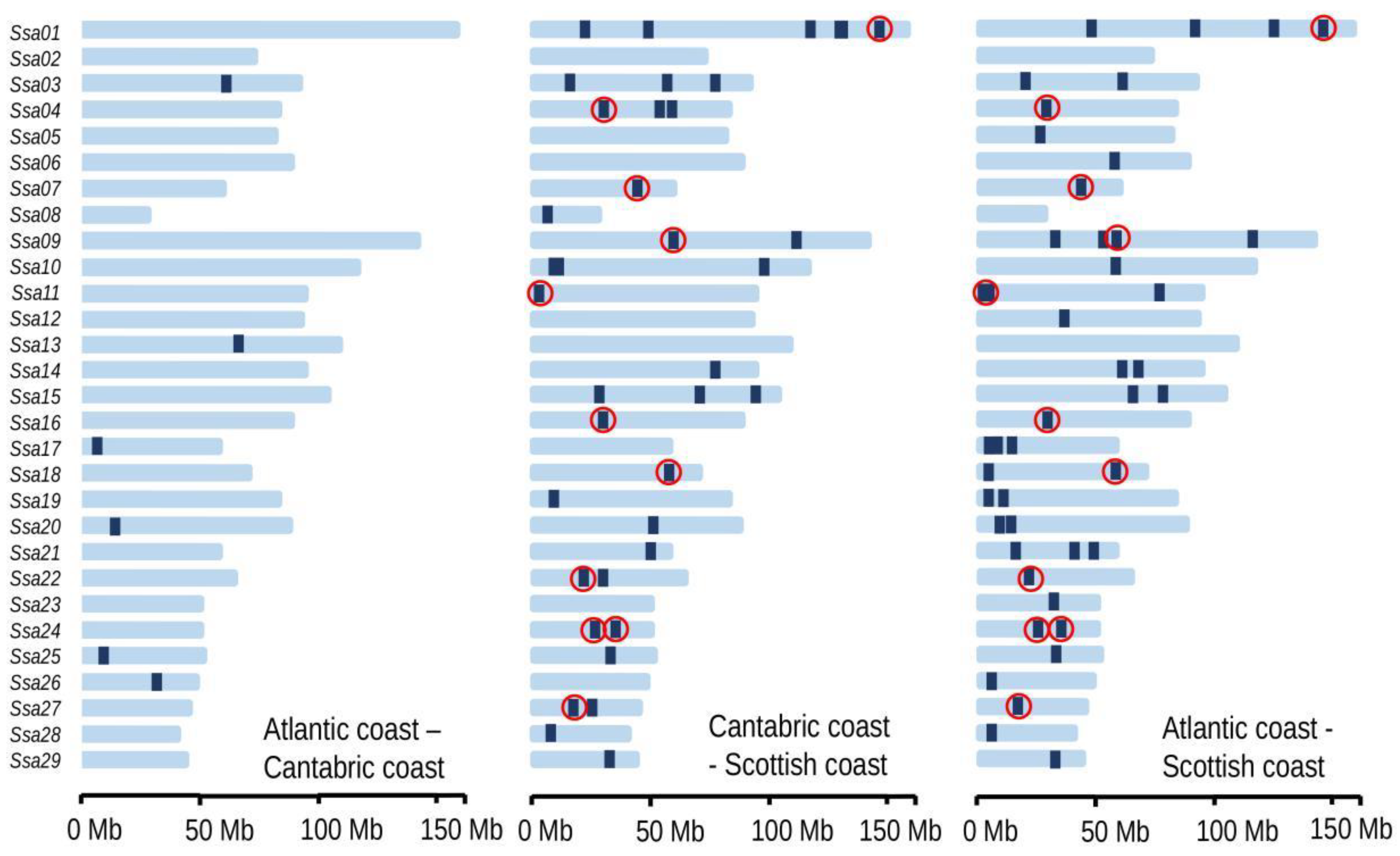
3.5. Atlantic–Cantabric Comparison
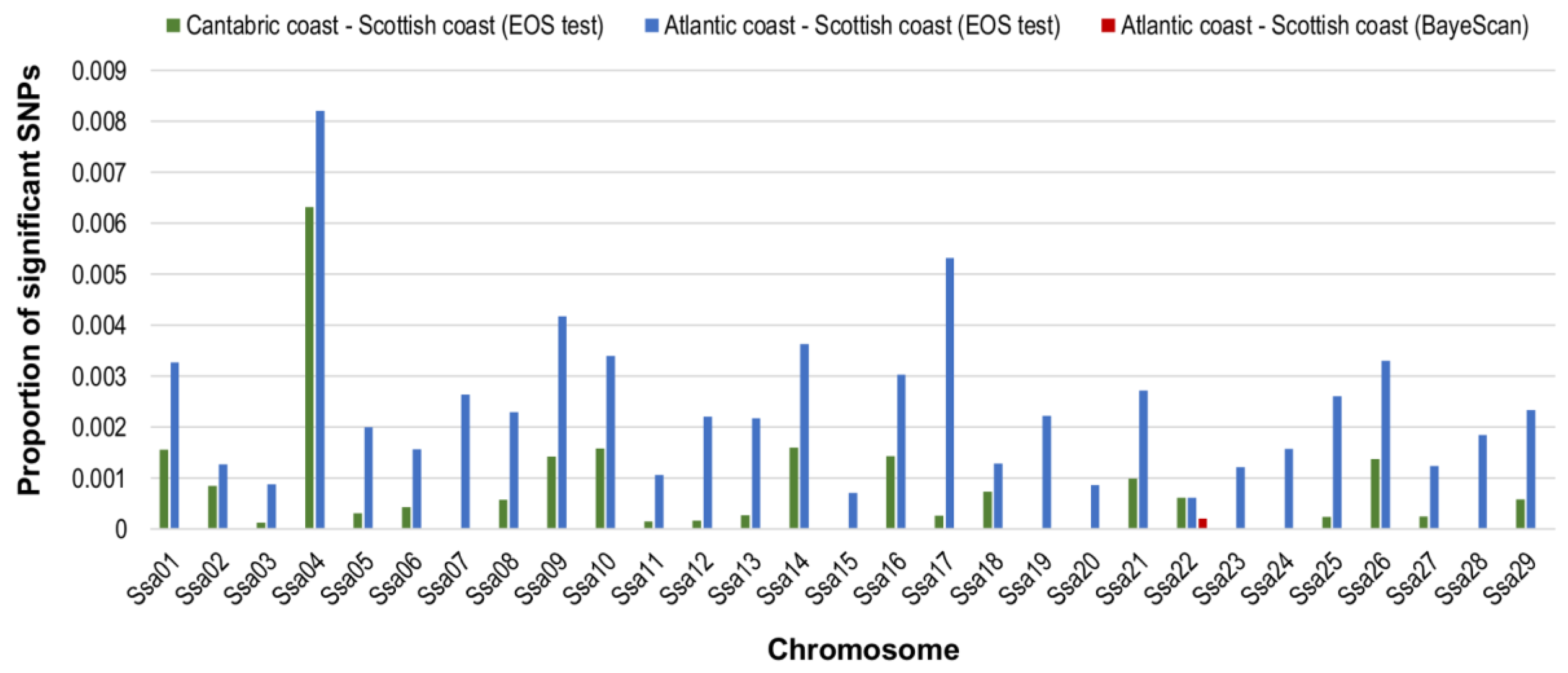
3.6. Comparison with Scotland: SNPs Significant for All Three Haplotype-Based Methods
3.7. Malic Enzyme
4. Discussion
Malic Enzyme
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Malic Enzyme
Appendix A.2. HAC Variance Method
Appendix A.3. Divergent Selection Detection Variance Test
Appendix A.3.1. Major Allele Reference Haplotype and Sample Partitions P1 and P2
Appendix A.3.2. Haplotype Allelic Class (HAC)
Appendix A.3.3. HAC Variance
Appendix A.3.4. Skewness and Kurtosis
Appendix A.3.5. Homogeneity Variance Tests
Appendix A.4. Algorithm for Divergent Selection Variance Test
- Decide a candidate SNP x and the number of markers L (window size).
- For each population, i define its major-allele-reference haplotype Ri, and perform sample partitions Pi1 and Pi2 with the haplotypes carrying the major or minor allele of x, respectively.
- Compute sample variances vi1 for the Pi1 partition of population i and compute v2 as the sample variance of the accumulated HAC values of the P2 partitions for all populations. The reason for accumulating HAC values over the different populations for the second partition is that the sample size is smaller in these partitions and the allele distribution is expected to be random (no sweep) around the candidate site.
- If vi1 ≥ v2 we conclude that there is no selection at the candidate site and if there are more populations, return to the second step to analyze the next population. Otherwise, if vi1 < v2, we continue with step 5.
- For each partition, test skewness and kurtosis, i.e., test the null of no skewness (g1 = 0) and if kurtosis is not platykurtic (g2 ≥ 0, D’Agostino test).
- If data for both partitions are normally distributed, an F test is performed. If data is marginally normal (one partition is normal and the other has a p value above α/2) then a Levene test is performed. Otherwise, we use the composite test BF-OB that applies OB if any of the partitions is platykurtic and BF otherwise. If homogeneity of variances is rejected, we conclude that candidate site x is selective in population i. For the computation of skewness and kurtosis using the D’Agostino test, a sample size of at least 8 is required. If the sample size is below this value, the homogeneity test will be the OB, which is more robust under a low sample size.
- Repeat the above steps 1–6 for the next population. Divergent selection between populations i and j at site x can be concluded if x was selective in at least one population and the FST index that compares the FST for site x with the overall FST is significant. The significance of the FST index was computed by resampling, as in [21].
Appendix A.5. Simulations
Analysis of the Simulated Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| File Name | θ | ρ | Mean Window Size | FPR |
|---|---|---|---|---|
| C4 | 12 | 0 | 25 | 1% |
| C4 | 12 | 0 | 87 | 1% |
| C5 | 12 | 4 | 25 | 2% |
| C5 | 12 | 4 | 87 | 1% |
| C6 | 12 | 12 | 25 | 2% |
| C6 | 12 | 12 | 86 | 1% |
| C16 | 60 | 0 | 25 | 1% |
| C16 | 60 | 0 | 51 | 1% |
| C16 | 60 | 0 | 125 | 2% |
| C16 | 60 | 0 | 436 | 1% |
| C17 | 60 | 4 | 25 | 2% |
| C17 | 60 | 4 | 51 | 2% |
| C17 | 60 | 4 | 125 | 2% |
| C17 | 60 | 4 | 429 | 2% |
| C18 | 60 | 60 | 25 | 2% |
| C18 | 60 | 60 | 51 | 2% |
| C18 | 60 | 60 | 125 | 2% |
| C18 | 60 | 60 | 431 | 1% |
References
- Morán, P.; Cal, L.; Cobelo-García, A.; Almécija, C.; Caballero, P.; García de Leániz, C. Historical legacies of river pollution reconstructed from fish scales. Environ. Pollut. 2018, 234, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, M.; Caballero, A.; Caballero, P.; Morán, P. Impact of precocious male parr on the effective size of a wild population of Atlantic salmon. Freshw. Biol. 2008, 53, 2375–2384. [Google Scholar] [CrossRef]
- Álvarez, J.J.; Antón, A.; Azpíroz, I.; Caballero, P.; Hervella, F.; De la Hoz, J.; Urrizalki, I. Atlas de los Ríos Salmoneros de la Península Ibérica; Ekolur SLL: España, Spain, 2010. [Google Scholar]
- Campos, J.L.; Posada, D.; Morán, P. Introgression and genetic structure in northern Spanish Atlantic salmon (Salmo salar L.) populations according to mtDNA data. Conserv. Genet. 2008, 9, 157–169. [Google Scholar] [CrossRef]
- Morán, P.; Pendás, A.M.; Garcia-Vázquez, E.; Izquierdo, J.T.; Rutherford, D.T. Electrophoretic Assessment of the Contribution of Transplanted Scottish Atlantic Salmon (Salmo salar) to the Esva River (Northern Spain). Can. J. Fish. Aquat. Sci. 1994, 51, 248–252. [Google Scholar] [CrossRef]
- Morán, P.; Perez, J.; Dumas, J.; Beall, E.; García-Vázquez, E. Stocking-related patterns of genetic variation at enzymatic loci in south European Atlantic salmon populations. J. Fish Biol. 2005, 67, 185–199. [Google Scholar] [CrossRef]
- Horreo, J.L.; Machado-Schiaffino, G.; Ayllon, F.; Griffiths, A.M.; Bright, D.; Stevens, J.R.; Garcia-Vazquez, E. Impact of climate change and human-mediated introgression on southern European Atlantic salmon populations. Glob. Change Biol. 2011, 17, 1778–1787. [Google Scholar] [CrossRef]
- García de Leániz, C.; Fleming, I.A.; Einum, S.; Verspoor, E.; Jordan, W.C.; Consuegra, S.; Aubin-Horth, N.; Lajus, D.; Letcher, B.H.; Youngson, A.F.; et al. A critical review of adaptive genetic variation in Atlantic salmon: Implications for conservation. Biol. Rev. Camb. Philos. Soc. 2007, 82, 173–211. [Google Scholar] [CrossRef]
- Fraser, D.J.; Weir, L.K.; Bernatchez, L.; Hansen, M.M.; Taylor, E.B. Extent and scale of local adaptation in salmonid fishes: Review and meta-analysis. Heredity 2011, 106, 404–420. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, V.L.; Mäkinen, H.; Vähä, J.-P.; Erkinaro, J.; Orell, P.; Primmer, C.R. Genomic signatures of fine-scale local selection in Atlantic salmon suggest involvement of sexual maturation, energy homeostasis and immune defence-related genes. Mol. Ecol. 2018, 27, 2560–2575. [Google Scholar] [CrossRef]
- Mobley, K.B.; Granroth-Wilding, H.; Ellmen, M.; Vähä, J.-P.; Aykanat, T.; Johnston, S.E.; Orell, P.; Erkinaro, J.; Primmer, C.R. Home ground advantage: Local Atlantic salmon have higher reproductive fitness than dispersers in the wild. Sci. Adv. 2019, 5, eaav1112. [Google Scholar] [CrossRef] [Green Version]
- García de Leániz, C.; Morán, P.; Pendás, A.M.; Caballero, C.; López-Beiras, P.; Valero, E.; García-Vázquez, E. Freshwater performance of native and foreign Atlantic salmon populations stocked in two Galician rivers (Spain). In Fifth International Symposium on Genetics in Aquaculture, Halifax; Halifax University: Halifax, NS, Canada, 1994. [Google Scholar]
- Morán, P.; Perez, J.; Garcia-Vazquez, E. The malic enzyme MEP-2* locus in Spanish populations of Atlantic salmon: Sea age and foreign stocking. Aquat. Sci. 1998, 60, 359–366. [Google Scholar] [CrossRef]
- Saavedra-Nieves, P.; Crujeiras, R.M.; Caballero, P.; Cobo, F. Assessing the effect of recovery programs for salmon (Salmo salar Linnaeus, 1758) at its Southern limit in Europe: Application of segmented regression models to long-term data from the Ulla River. Limnetica 2021, 40, 189–203. [Google Scholar] [CrossRef]
- Altukhov, Y.P.; Salmenkova, E.A.; Omelchenko, V.T. Salmonid Fishes: Population Biology, Genetics and Management; John Wiley & Sons: Hoboken, NJ, USA, 2000. [Google Scholar]
- Jeffery, N.W.; Wringe, B.F.; McBride, M.C.; Hamilton, L.C.; Stanley, R.R.E.; Bernatchez, L.; Kent, M.; Clément, M.; Gilbey, J.; Sheehan, T.F.; et al. Range-wide regional assignment of Atlantic salmon (Salmo salar) using genome wide single-nucleotide polymorphisms. Fish. Res. 2018, 206, 163–175. [Google Scholar] [CrossRef]
- Blanquart, F.; Kaltz, O.; Nuismer, S.L.; Gandon, S. A practical guide to measuring local adaptation. Ecol. Lett. 2013, 16, 1195–1205. [Google Scholar] [CrossRef]
- Savolainen, O.; Lascoux, M.; Merila, J. Ecological genomics of local adaptation. Nat. Rev. Genet. 2013, 14, 807. [Google Scholar] [CrossRef]
- Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef] [Green Version]
- Barreiro, L.B.; Laval, G.; Quach, H.; Patin, E.; Quintana-Murci, L. Natural selection has driven population differentiation in modern humans. Nat. Genet. 2008, 40, 340–345. [Google Scholar] [CrossRef]
- Carvajal-Rodríguez, A. HacDivSel: Two new methods (haplotype-based and outlier-based) for the detection of divergent selection in pairs of populations. PLoS ONE 2017, 12, e0175944. [Google Scholar] [CrossRef] [Green Version]
- Pavlidis, P.; Alachiotis, N. A survey of methods and tools to detect recent and strong positive selection. J. Biol. Res.-Thessalon. 2017, 24, 7. [Google Scholar] [CrossRef] [Green Version]
- Weigand, H.; Leese, F. Detecting signatures of positive selection in non-model species using genomic data. Zool. J. Linn. Soc. 2018, 187, 528–583. [Google Scholar] [CrossRef]
- Granka, J.M.; Henn, B.M.; Gignoux, C.R.; Kidd, J.M.; Bustamante, C.D.; Feldman, M.W. Limited evidence for classic selective sweeps in African populations. Genetics 2012, 192, 1049–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horscroft, C.; Ennis, S.; Pengelly, R.J.; Sluckin, T.J.; Collins, A. Sequencing era methods for identifying signatures of selection in the genome. Brief. Bioinform. 2018, 20, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Jordan, W.C.; Youngson, A.F. Genetic protein variation and natural selection in Atlantic salmon (Salmo salav, L.) parr. J. Fish Biol. 1991, 39, 185–192. [Google Scholar] [CrossRef]
- Verspoor, E.; Jordan, W.C. Genetic variation at the Me-2 locus in the Atlantic salmon within and between rivers: Evidence for its selective maintenance. J. Fish Biol. 1989, 35, 205–213. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Schaffner, S.F.; Fry, B.; Lohmueller, J.; Varilly, P.; Shamovsky, O.; Palma, A.; Mikkelsen, T.S.; Altshuler, D.; Lander, E.S. Positive natural selection in the human lineage. Science 2006, 312, 1614–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, S.H.; Hubisz, M.J.; Clark, A.G.; Payseur, B.A.; Bustamante, C.D.; Nielsen, R. Localizing Recent Adaptive Evolution in the Human Genome. PLoS Genet. 2007, 3, e90. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Yano, A.; Nicol, B.; Jouanno, E.; Quillet, E.; Fostier, A.; Guyomard, R.; Guiguen, Y. The sexually dimorphic on the Y-chromosome gene (sdY) is a conserved male-specific Y-chromosome sequence in many salmonids. Evol. Appl. 2013, 6, 486–496. [Google Scholar] [CrossRef]
- Quéméré, E.; Perrier, C.; Besnard, A.-L.; Evanno, G.; Baglinière, J.-L.; Guiguen, Y.; Launey, S. An improved PCR-based method for faster sex determination in brown trout (Salmo trutta) and Atlantic salmon (Salmo salar). Conserv. Genet. Resour. 2014, 6, 825–827. [Google Scholar] [CrossRef]
- Barson, N.J.; Aykanat, T.; Hindar, K.; Baranski, M.; Bolstad, G.H.; Fiske, P.; Jacq, C.; Jensen, A.J.; Johnston, S.E.; Karlsson, S.; et al. Sex-dependent dominance at a single locus maintains variation in age at maturity in salmon. Nature 2015, 528, 405–408. [Google Scholar] [CrossRef]
- Lewontin, R.C.; Krakauer, J. Testing Heterogeneity of F-Values. Genetics 1975, 80, 397–398. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.A. Adaptation and speciation: What can F(st) tell us? Trends Ecol. Evol. 2005, 20, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, M.C.; Lotterhos, K.E. Reliable Detection of Loci Responsible for Local Adaptation: Inference of a Null Model through Trimming the Distribution of FST. Am. Nat. 2015, 186, S24–S36. [Google Scholar] [CrossRef] [PubMed]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinform. Oxf. Engl. 2012, 28, 298–299. [Google Scholar] [CrossRef] [Green Version]
- Lotterhos, K.E.; Whitlock, M.C. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol. Ecol. 2014, 23, 2178. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Figueroa, A.; Garcia-Pereira, M.J.; Saura, M.; Rolan-Alvarez, E.; Caballero, A. Comparing three different methods to detect selective loci using dominant markers. J. Evol. Biol. 2010, 23, 2267–2276. [Google Scholar] [CrossRef]
- Bierne, N.; Roze, D.; Welch, J.J. Pervasive selection or is it…? why are FST outliers sometimes so frequent? Mol. Ecol. 2013, 22, 2061–2064. [Google Scholar] [CrossRef]
- De Mita, S.; Thuillet, A.-C.; Gay, L.; Ahmadi, N.; Manel, S.; Ronfort, J.; Vigouroux, Y. Detecting selection along environmental gradients: Analysis of eight methods and their effectiveness for outbreeding and selfing populations. Mol. Ecol. 2013, 22, 1383. [Google Scholar] [CrossRef]
- Hoban, S.; Kelley, J.L.; Lotterhos, K.E.; Antolin, M.F.; Bradburd, G.; Lowry, D.B.; Poss, M.L.; Reed, L.K.; Storfer, A.; Whitlock, M.C. Finding the Genomic Basis of Local Adaptation: Pitfalls, Practical Solutions, and Future Directions. Am. Nat. 2016, 188, 379–397. [Google Scholar] [CrossRef] [Green Version]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Szpiech, Z.A.; Hernandez, R.D. selscan: An Efficient Multithreaded Program to Perform EHH-Based Scans for Positive Selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiblet, W.M.; Zhao, K.; O’Brien, S.J.; Massey, S.E.; Roca, A.L.; Oleksyk, T.K. SmileFinder: A resampling-based approach to evaluate signatures of selection from genome-wide sets of matching allele frequency data in two or more diploid populations. GigaScience 2015, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Scheet, P.; Stephens, M. A Fast and Flexible Statistical Model for Large-Scale Population Genotype Data: Applications to Inferring Missing Genotypes and Haplotypic Phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samy, J.K.A.; Mulugeta, T.D.; Nome, T.; Sandve, S.R.; Grammes, F.; Kent, M.P.; Lien, S.; Våge, D.I. SalmoBase: An integrated molecular data resource for Salmonid species. BMC Genom. 2017, 18, 482. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourret, V.; Kent, M.P.; Primmer, C.R.; Vasemägi, A.; Karlsson, S.; Hindar, K.; McGinnity, P.; Verspoor, E.; Bernatchez, L.; Lien, S. SNP-array reveals genome-wide patterns of geographical and potential adaptive divergence across the natural range of Atlantic salmon (Salmo salar). Mol. Ecol. 2013, 22, 532–551. [Google Scholar] [CrossRef]
- Moore, J.-S.; Bourret, V.; Dionne, M.; Bradbury, I.; O’Reilly, P.; Kent, M.; Chaput, G.; Bernatchez, L. Conservation genomics of anadromous Atlantic salmon across its North American range: Outlier loci identify the same patterns of population structure as neutral loci. Mol. Ecol. 2014, 23, 5680–5697. [Google Scholar] [CrossRef]
- De Villemereuil, P.; Frichot, É.; Bazin, É.; François, O.; Gaggiotti, O.E. Genome scan methods against more complex models: When and how much should we trust them? Mol. Ecol. 2014, 23, 2006–2019. [Google Scholar] [CrossRef] [Green Version]
- Reis Neto, R.V.; Yoshida, G.M.; Lhorente, J.P.; Yáñez, J.M. Genome-wide association analysis for body weight identifies candidate genes related to development and metabolism in rainbow trout (Oncorhynchus mykiss). Mol. Genet. Genom. MGG 2019, 294, 563–571. [Google Scholar] [CrossRef]
- Boison, S.A.; Gjerde, B.; Hillestad, B.; Makvandi-Nejad, S.; Moghadam, H.K. Genomic and Transcriptomic Analysis of Amoebic Gill Disease Resistance in Atlantic Salmon (Salmo salar L.). Front. Genet. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.; Xu, G.; Li, Y.; Nie, Z.; Xu, P. Glyoxalase 1 gene of Coilia nasus: Molecular characterization and differential expression during transport stress. Fish. Sci. 2016, 82, 719–728. [Google Scholar] [CrossRef]
- Sano, S.; Takashima, S.; Niwa, H.; Yokoi, H.; Shimada, A.; Arenz, A.; Wittbrodt, J.; Takeda, H. Characterization of teleost Mdga1 using a gene-trap approach in medaka (Oryzias latipes). genesis 2009, 47, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Kjærner-Semb, E.; Ayllon, F.; Furmanek, T.; Wennevik, V.; Dahle, G.; Niemelä, E.; Ozerov, M.; Vähä, J.-P.; Glover, K.A.; Rubin, C.J.; et al. Atlantic salmon populations reveal adaptive divergence of immune related genes—A duplicated genome under selection. BMC Genom. 2016, 17, 610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, L.-J.; He, A.-Y.; Li, J.-M.; Lu, D.-L.; Jiao, J.-G.; Li, L.-Y.; Li, D.-L.; Zhang, M.-L.; Chen, L.-Q.; Du, Z.-Y. Mechanisms and metabolic regulation of PPARα activation in Nile tilapia (Oreochromis niloticus). Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2016, 1861, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Hasenbein, M.; Werner, I.; Deanovic, L.A.; Geist, J.; Fritsch, E.B.; Javidmehr, A.; Foe, C.; Fangue, N.A.; Connon, R.E. Transcriptomic profiling permits the identification of pollutant sources and effects in ambient water samples. Sci. Total Environ. 2014, 468–469, 688–698. [Google Scholar] [CrossRef]
- Ma, K.; Liao, M.; Liu, F.; Ye, B.; Sun, F.; Yue, G.H. Charactering the ZFAND3 gene mapped in the sex-determining locus in hybrid tilapia (Oreochromis spp.). Sci. Rep. 2016, 6, 25471. [Google Scholar] [CrossRef] [Green Version]
- Skaggs, H.S.; Xing, H.; Wilkerson, D.C.; Murphy, L.A.; Hong, Y.; Mayhew, C.N.; Sarge, K.D. HSF1-TPR interaction facilitates export of stress-induced HSP70 mRNA. J. Biol. Chem. 2007, 282, 33902–33907. [Google Scholar] [CrossRef] [Green Version]
- Robledo, D.; Rubiolo, J.A.; Cabaleiro, S.; Martínez, P.; Bouza, C. Differential gene expression and SNP association between fast- and slow-growing turbot (Scophthalmus maximus). Sci. Rep. 2017, 7, 12105. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.-Y.; Hamilton, A.; Tinch, A.E.; Guy, D.R.; Gharbi, K.; Stear, M.J.; Matika, O.; Bishop, S.C.; Houston, R.D. Genome wide association and genomic prediction for growth traits in juvenile farmed Atlantic salmon using a high density SNP array. BMC Genom. 2015, 16, 969. [Google Scholar]
- Gutierrez, A.P.; Yáñez, J.M.; Fukui, S.; Swift, B.; Davidson, W.S. Genome-wide association study (GWAS) for growth rate and age at sexual maturation in Atlantic salmon (Salmo salar). PLoS ONE 2015, 10, e0119730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, W.S.; Koop, B.F.; Jones, S.J.; Iturra, P.; Vidal, R.; Maass, A.; Jonassen, I.; Lien, S.; Omholt, S.W. Sequencing the genome of the Atlantic salmon (Salmo salar). Genome Biol. 2010, 11, 403. [Google Scholar] [CrossRef] [PubMed]
- Gabián, M.; Morán, P.; Fernández, A.I.; Villanueva, B.; Chtioui, A.; Kent, M.P.; Covelo-Soto, L.; Fernández, A.; Saura, M. Identification of genomic regions regulating sex determination in Atlantic salmon using high density SNP data. BMC Genom. 2019, 20, 764. [Google Scholar] [CrossRef] [PubMed]
- López, M.E.; Benestan, L.; Moore, J.-S.; Perrier, C.; Gilbey, J.; Genova, A.D.; Maass, A.; Diaz, D.; Lhorente, J.-P.; Correa, K.; et al. Comparing genomic signatures of domestication in two Atlantic salmon (Salmo salar L.) populations with different geographical origins. Evol. Appl. 2019, 12, 137–156. [Google Scholar] [CrossRef] [Green Version]
- Mäkinen, H.; Vasemägi, A.; McGinnity, P.; Cross, T.F.; Primmer, C.R. Population genomic analyses of early-phase Atlantic Salmon (Salmo salar) domestication/captive breeding. Evol. Appl. 2015, 8, 93–107. [Google Scholar] [CrossRef]
- Oleksyk, T.K.; Smith, M.W.; O’Brien, S.J. Genome-wide scans for footprints of natural selection. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 185–205. [Google Scholar] [CrossRef] [Green Version]
- Lehnert, S.J.; Kess, T.; Bentzen, P.; Clément, M.; Bradbury, I.R. Divergent and linked selection shape patterns of genomic differentiation between European and North American Atlantic salmon (Salmo salar). Mol. Ecol. 2020, 29, 2160–2175. [Google Scholar] [CrossRef]
- Verspoor, E.; Beardmore, J.A.; Consuegra, S.; García de Leániz, C.; Hindar, K.; Jordan, W.C.; Koljonen, M.-L.; Mahkrov, A.A.; Paaver, T.; Sánchez, J.A.; et al. Population structure in the Atlantic salmon: Insights from 40 years of research into genetic protein variation. J. Fish Biol. 2005, 67, 3–54. [Google Scholar] [CrossRef]
- Morán, P.; Pérez, J.; Garcia-Vazquez, E. Genetic variation at enzyme loci in the southernmost European populations of Atlantic salmon. J. Fish Biol. 2005, 67, 206–212. [Google Scholar] [CrossRef]
- Hussin, J.; Nadeau, P.; Lefebvre, J.-F.; Labuda, D. Haplotype allelic classes for detecting ongoing positive selection. BMC Bioinform. 2010, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Joanes, D.N.; Gill, C.A. Comparing measures of sample skewness and kurtosis. J. R. Stat. Soc. Ser. Stat. 1998, 47, 183–189. [Google Scholar] [CrossRef]
- D’Agostino, R.B.; Belanger, A. A Suggestion for Using Powerful and Informative Tests of Normality. Am. Stat. 1990, 44, 316–321. [Google Scholar]
- Ramsey, P.H.; Ramsey, P.P. Testing Variability in the Two-Sample Case. Commun. Stat.—Simul. Comput. 2007, 36, 233–248. [Google Scholar] [CrossRef]
- Brown, M.B.; Forsythe, A.B. Robust Tests for the Equality of Variances. J. Am. Stat. Assoc. 1974, 69, 364. [Google Scholar] [CrossRef]
- O’Brien, R.G. A simple test for variance effects in experimental designs. Psychol. Bull. 1981, 89, 570–574. [Google Scholar] [CrossRef]





| Quality Filter | N° Removed SNP |
|---|---|
| Not mapped | 1112 |
| Low quality | 47,432 |
| MAF < 0.01 | 5679 |
| Erratic genotypes | 3 |
| Total analysed SNPs | 165,774 |
| Statistic | Software | Number of Significant SNPs | ||
|---|---|---|---|---|
| Atl-Can | Can-Scot | Atl-Scot | ||
| FST outliers detection | HacDivSel (EOS test) | 0 | 142 | 412 |
| BayeScan (logBF = 1.5) | 0 | 0 | 2 | |
| Haplotype based methods | HacDivSel (nvdFST) | 748 | 1504 | 2607 |
| SmileFinder | 631 | 1346 | 2786 | |
| selscan (XP-EHH) | 201 | 1863 | 2880 | |
| Comparison | Methods | ||||
|---|---|---|---|---|---|
| X-N | X-S | N-S | X-N-S | Total | |
| Atl-Can | 0 | 4 | 14 | 0 | 16 |
| Atl-Scot | 210 | 275 | 147 | 64 | 506 |
| Can-Scot | 59 | 31 | 82 | 19 | 170 |
| Chromosome | # SNPs | SNP IDs | Gene (Mb) |
|---|---|---|---|
| Ssa09 | 23 | H181–186, H188 | fat4 (59) |
| Ssa11 | 37 | H256 *, H259, H261–H264, H269, H275 *, H281 *, (H297–300) *, H304–310 *, H313, H324–325 | wdr43 (1), trmt61b (1), atts-glupro (1), glo1 (1), zfand3 (1), mdga1 (1) |
| Ssa24 | 2 | H479 *, H488 * | ppp6c (25), golga1 (25), rpl35 (25), ofml2a (25) |
| Ssa27 | 2 | H615–616 | - |
| Comparison | WINDOW SIZE | Var Test p-Value | FST p-Value | Divergence Significance Test |
|---|---|---|---|---|
| Atl-Can | 25 | 4 × 10−8 | 0.014 | * |
| 51 | 0.003 | 0.004 | * | |
| 125 | 0.164 | 0.002 | ns | |
| Can-Scot | 25 | 0.011 | 0.038 | * |
| 51 | 0.005 | 0.030 | * | |
| 125 | 0.653 | 0.024 | ns | |
| Atl-Scot | 25 | 8 × 10−10 | 0 | * |
| 51 | 0.021 | 0 | * | |
| 125 | 1 | 0 | ns |
| ID | Window Size | Var Test p-Value | FST p-Value | Divergence Significance Test |
|---|---|---|---|---|
| 3454 | 25 | 1 | 1 | ns |
| 3529 | 25 | 1 | 1 | ns |
| 3578 | 25 | 0.026 | 3 × 10−9 | * |
| 3579 | 25 | 4 × 10−8 | 0.002 | * |
| 3580 | 25 | 0.002 | 0.013 | * |
| 3629 | 25 | 1 | 1 | ns |
| 3704 | 25 | 2 × 10−8 | 0.008 | * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabián, M.; Morán, P.; Saura, M.; Carvajal-Rodríguez, A. Detecting Local Adaptation between North and South European Atlantic Salmon Populations. Biology 2022, 11, 933. https://doi.org/10.3390/biology11060933
Gabián M, Morán P, Saura M, Carvajal-Rodríguez A. Detecting Local Adaptation between North and South European Atlantic Salmon Populations. Biology. 2022; 11(6):933. https://doi.org/10.3390/biology11060933
Chicago/Turabian StyleGabián, María, Paloma Morán, María Saura, and Antonio Carvajal-Rodríguez. 2022. "Detecting Local Adaptation between North and South European Atlantic Salmon Populations" Biology 11, no. 6: 933. https://doi.org/10.3390/biology11060933
APA StyleGabián, M., Morán, P., Saura, M., & Carvajal-Rodríguez, A. (2022). Detecting Local Adaptation between North and South European Atlantic Salmon Populations. Biology, 11(6), 933. https://doi.org/10.3390/biology11060933