
Targeting the Ubiquinol-Reduction (Qi) Site of the Mitochondrial Cytochrome bc1 Complex for the Development of Next Generation Quinolone Antimalarials

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
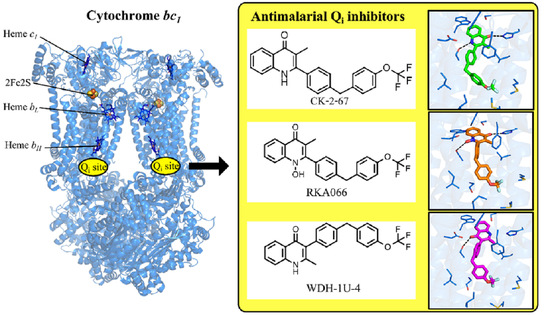
1. Introduction
2. Materials and Methods
2.1. Cytochrome bc1 Purification
2.2. Crystallization, Data Collection, and Structure Refinement
2.3. Parasite Culture and Drug Sensitivity Measurements
2.4. Characterisation of Compounds and Their Purity
2.5. Plasmodium Falciparum Homology Model Generation
2.6. Molecular Docking
2.7. Bovine Cytochrome bc1 Activity Assay
3. Results
3.1. Inhibitory Effect of 4(1H)-Quinolones Binding to the Qi Site of Bovine Cytochrome bc1
3.2. Binding of 4(1H)-Quinolones to the Qi Site of Bovine Cytochrome bc1
3.3. Predicted Binding Mode of 4(1H)-Quinolones within the Plasmodium Falciparum Qi Site
3.4. Suggestions for Future Development of 4(1H)-Quinolones
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- World Health Organization. World Malaria Report 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.A.; Burrows, J.N.; Manyando, C.; Van Huijsduijnen, R.H.; Van Voorhis, W.C.; Wells, T.N.C. Malaria. Nat. Rev. Dis. Prim. 2017, 3, nrdp201750. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.L.; Pidathala, C.; Shone, A.E.; Antoine, T.; Fisher, N.; O’Neill, P.M.; Ward, S.; Biagini, G. Targeting the mitochondrial electron transport chain of Plasmodium falciparum: New strategies towards the development of improved antimalarials for the elimination era. Future Med. Chem. 2013, 5, 1573–1591. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; Link, T.; Engel, W.; von Jagow, G. Isolation of the eleven protein subunits of the bc1 complex from beef heart. Methods Enzymol. 1986, 126, 224–237. [Google Scholar] [CrossRef]
- Mitchell, P. Possible molecular mechanisms of the protonmotive function of cytochrome systems. J. Theor. Biol. 1976, 62, 327–367. [Google Scholar] [CrossRef]
- Erecińska, M.; Chance, B.; Wilson, D.F.; Dutton, P.L. Aerobic Reduction of Cytochrome b566 in Pigeon-Heart Mitochondria (succinate-cytochrome C1 reductase-stopped-flow kinetics). Proc. Natl. Acad. Sci. USA 1972, 69, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Hammond, D.; Burchell, J.; Pudney, M. Inhibition of pyrimidine biosynthesis de novo in Plasmodium falciparum by 2-(4-t-butylcyclohexyl)-3-hydroxy-1,4-naphthoquinone in vitro. Mol. Biochem. Parasitol. 1985, 14, 97–109. [Google Scholar] [CrossRef]
- Painter, H.J.; Morrisey, J.M.; Mather, M.; Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. [Google Scholar] [CrossRef]
- Biagini, G.A.; Fisher, N.; Shone, A.E.; Mubaraki, M.A.; Srivastava, A.; Hill, A.; Antoine, T.; Warman, A.J.; Davies, J.; Pidathala, C.; et al. Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc. Natl. Acad. Sci. USA 2012, 109, 8298–8303. [Google Scholar] [CrossRef] [Green Version]
- Alday, P.H.; McConnell, E.V.; Zarella, J.M.B.; Dodean, R.A.; Kancharla, P.; Kelly, J.X.; Doggett, J.S. Acridones Are Highly Potent Inhibitors of Toxoplasma gondii Tachyzoites. ACS Infect. Dis. 2021, 7, 1877–1884. [Google Scholar] [CrossRef]
- Wall, R.J.; Carvalho, S.; Milne, R.; Bueren-Calabuig, J.A.; Moniz, S.; Cantizani-Perez, J.; MacLean, L.; Kessler, A.; Cotillo, I.; Sastry, L.; et al. The Qi Site of Cytochrome b is a Promiscuous Drug Target in Trypanosoma cruzi and Leishmania donovani. ACS Infect. Dis. 2020, 6, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Shanks, G.D.; Gordon, D.M.; Klotz, F.W.; Aleman, G.M.; Oloo, A.J.; Sadie, D.; Scott, T.R. Efficacy and Safety of Atovaquone/Proguanil as Suppressive Prophylaxis for Plasmodium falciparum Malaria. Clin. Infect. Dis. 1998, 27, 494–499. [Google Scholar] [CrossRef] [Green Version]
- Nixon, G.L.; Moss, D.M.; Shone, A.E.; Lalloo, D.G.; Fisher, N.; O‘Neill, P.M.; Ward, S.A.; Biagini, G.A. Antimalarial pharmacology and therapeutics of atovaquone. J. Antimicrob. Chemother. 2013, 68, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Lalloo, D.G.; Shingadia, D.; Bell, D.J.; Beeching, N.J.; Whitty, C.J.; Chiodini, P.L. UK malaria treatment guidelines 2016. J. Infect. 2016, 72, 635–649. [Google Scholar] [CrossRef]
- Araujo, F.G.; Huskinson, J.; Remington, J.S. Remarkable in vitro and in vivo activities of the hydroxynaphthoquinone 566C80 against tachyzoites and tissue cysts of Toxoplasma gondii. Antimicrob. Agents Chemother. 1991, 35, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, I.K.; Vaidya, A.B. A Mechanism for the Synergistic Antimalarial Action of Atovaquone and Proguanil. Antimicrob. Agents Chemother. 1999, 43, 1334–1339. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, I.K.; Morrisey, J.M.; Darrouzet, E.; Daldal, F.; Vaidya, A.B. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 1999, 33, 704–711. [Google Scholar] [CrossRef]
- Fisher, N.; Majid, R.A.; Antoine, T.; Al-Helal, M.; Warman, A.J.; Johnson, D.J.; Lawrenson, A.S.; Ranson, H.; O’Neill, P.M.; Ward, S.A.; et al. Cytochrome b Mutation Y268S Conferring Atovaquone Resistance Phenotype in Malaria Parasite Results in Reduced Parasite bc1 Catalytic Turnover and Protein Expression. J. Biol. Chem. 2012, 287, 9731–9741. [Google Scholar] [CrossRef] [Green Version]
- Birth, D.; Kao, W.-C.; Hunte, C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat. Commun. 2014, 5, 4029. [Google Scholar] [CrossRef] [Green Version]
- Xia, D.; Yu, C.-A.; Kim, H.; Xia, J.-Z.; Kachurin, A.M.; Zhang, L.; Yu, L.; Deisenhofer, J. Crystal Structure of the Cytochrome bc 1 Complex from Bovine Heart Mitochondria. Science 1997, 277, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Huang, L.-S.; Shulmeister, V.M.; Chi, Y.-I.; Kim, K.K.; Hung, L.-W.; Crofts, A.R.; Berry, E.A.; Kim, S.-H. Electron transfer by domain movement in cytochrome bc1. Nature 1998, 392, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Hunte, C.; Koepke, J.; Lange, C.; Roßmanith, T.; Michel, H. Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure 2000, 8, 669–684. [Google Scholar] [CrossRef] [Green Version]
- Berry, E.A.; Huang, L.-S.; Saechao, L.K.; Pon, N.G.; Valkova-Valchanova, M.; Daldal, F. X-ray Structure of Rhodobacter Capsulatus Cytochrome bc1: Comparison with its Mitochondrial and Chloroplast Counterparts. Photosynth. Res. 2004, 81, 251–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, L.; Gong, X.; Yang, S.; Yu, L.; Yu, C.-A.; Xia, D. Surface-modulated motion switch: Capture and release of iron–sulfur protein in the cytochrome bc1 complex. Proc. Natl. Acad. Sci. USA 2006, 103, 13045–13050. [Google Scholar] [CrossRef] [Green Version]
- Kleinschroth, T.; Castellani, M.; Trinh, C.H.; Morgner, N.; Brutschy, B.; Ludwig, B.; Hunte, C. X-ray structure of the dimeric cytochrome bc1 complex from the soil bacterium Paracoccus denitrificans at 2.7-Å resolution. Biochim. Biophys. Acta 2011, 1807, 1606–1615. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell 2016, 167, 1598–1609. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Amporndanai, K.; Johnson, R.M.; O’Neill, P.M.; Fishwick, C.W.G.; Jamson, A.H.; Rawson, S.; Muench, S.P.; Hasnain, S.S.; Antonyuk, S.V. X-ray and cryo-EM structures of inhibitor-bound cytochromebc1complexes for structure-based drug discovery. IUCrJ 2018, 5, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Wen, X.; Esser, L.; Quinn, B.; Yu, L.; Yu, C.-A.; Xia, D. Structural Basis for the Quinone Reduction in the bc1 Complex: A Comparative Analysis of Crystal Structures of Mitochondrial Cytochrome bc1 with Bound Substrate and Inhibitors at the Qi Site. Biochemistry 2003, 42, 9067–9080. [Google Scholar] [CrossRef]
- Esser, L.; Quinn, B.; Li, Y.-F.; Zhang, M.; Elberry, M.; Yu, L.; Yu, C.-A.; Xia, D. Crystallographic Studies of Quinol Oxidation Site Inhibitors: A Modified Classification of Inhibitors for the Cytochrome bc1 Complex. J. Mol. Biol. 2004, 341, 281–302. [Google Scholar] [CrossRef]
- Charoensutthivarakul, S.; Hong, W.D.; Leung, S.C.; Gibbons, P.D.; Bedingfield, P.T.P.; Nixon, G.L.; Lawrenson, A.S.; Berry, N.G.; Ward, S.A.; Biagini, G.A.; et al. 2-Pyridylquinolone antimalarials with improved antimalarial activity and physicochemical properties. MedChemComm 2015, 6, 1252–1259. [Google Scholar] [CrossRef]
- Capper, M.J.; O’Neill, P.M.; Fisher, N.; Strange, R.W.; Moss, D.; Ward, S.A.; Berry, N.G.; Lawrenson, A.S.; Hasnain, S.S.; Biagini, G.A.; et al. Antimalarial 4(1H)-pyridones bind to the Qi site of cytochrome bc1. Proc. Natl. Acad. Sci. USA 2015, 112, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Bueno, J.M.; Herreros, E.; Angulo-Barturen, I.; Ferrer, S.; Fiandor, J.M.; Gamo, F.J.; Gargallo-Viola, D.; Derimanov, G. Exploration of 4(1H)-pyridones as a novel family of potent antimalarial inhibitors of the plasmodial cytochrome bc1. Future Med. Chem. 2012, 4, 2311–2323. [Google Scholar] [CrossRef]
- Schlitzer, M. Antimalarial drugs–What is in use and what is in the pipeline. Arch. Pharm. 2008, 341, 149–163. [Google Scholar] [CrossRef]
- Stephen, J.M.L.; Tonkin, I.M.; Walker, J. 192. Tetrahydroacridones and related compounds as antimalarials. J. Chem. Soc. 1947, 1034–1039. [Google Scholar] [CrossRef]
- Winter, R.W.; Kelly, J.X.; Smilkstein, M.J.; Dodean, R.; Hinrichs, D.; Riscoe, M.K. Antimalarial quinolones: Synthesis, potency, and mechanistic studies. Exp. Parasitol. 2008, 118, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, A.; LaCrue, A.N.; White, K.L.; Forquer, I.P.; Cross, R.M.; Marfurt, J.; Mather, M.W.; Delves, M.J.; Shackleford, D.M.; Saenz, F.E.; et al. Quinolone-3-Diarylethers: A New Class of Antimalarial Drug. Sci. Transl. Med. 2013, 5, 177ra37-177ra37. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, A.; Miley, G.P.; Forquer, I.P.; Mather, M.W.; Katneni, K.; Li, Y.; Pou, S.; Pershing, A.M.; Stickles, A.M.; Ryan, E.; et al. Discovery, Synthesis, and Optimization of Antimalarial 4(1H)-Quinolone-3-Diarylethers. J. Med. Chem. 2014, 57, 3818–3834. [Google Scholar] [CrossRef]
- Smilkstein, M.J.; Pou, S.; Krollenbrock, A.; Bleyle, L.A.; Dodean, R.A.; Frueh, L.; Hinrichs, D.J.; Li, Y.; Martinson, T.; Munar, M.Y.; et al. ELQ-331 as a prototype for extremely durable chemoprotection against malaria. Malar. J. 2019, 18, 1–17. [Google Scholar] [CrossRef] [Green Version]
- De Souza, J.O.; Almeida, S.M.; Souza, G.E.; Zanini, C.L.; da Silva, E.M.; Calit, J.; Bargieri, D.Y.; Amporndanai, K.; Antonyuk, S.; Hasnain, S.S.; et al. Parasitological profiling shows 4(1H)-quinolone derivatives as new lead candidates for malaria. Eur. J. Med. Chem. Rep. 2021, 3, 100012. [Google Scholar] [CrossRef]
- Doggett, J.S.; Nilsen, A.; Forquer, I.; Wegmann, K.W.; Jones-Brando, L.; Yolken, R.H.; Bordón, C.; Charman, S.A.; Katneni, K.; Schultz, T.; et al. Endochin-like quinolones are highly efficacious against acute and latent experimental toxoplasmosis. Proc. Natl. Acad. Sci. USA 2012, 109, 15936–15941. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Iorga, B.I.; Mounkoro, P.; Fisher, N.; Meunier, B. The antimalarial compound ELQ-400 is an unusual inhibitor of the bc1 complex, targeting both Qo and Qi sites. FEBS Lett. 2018, 592, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Stickles, A.M.; de Almeida, M.J.; Morrisey, J.M.; Sheridan, K.A.; Forquer, I.P.; Nilsen, A.; Winter, R.W.; Burrows, J.N.; Fidock, D.A.; Vaidya, A.B.; et al. Subtle Changes in Endochin-Like Quinolone Structure Alter the Site of Inhibition within the Cytochrome bc1 Complex of Plasmodium falciparum. Antimicrob. Agents Chemother. 2015, 59, 1977–1982. [Google Scholar] [CrossRef] [Green Version]
- Stickles, A.M.; Nilsen, A.; Forquer, I.P.; Riscoe, M.K.; Morrisey, J.M.; Miley, G.P.; Winter, R.W.; Mather, M.W.; Ting, L.-M.; Kelly, J.X.; et al. Inhibition of Cytochrome bc1 as a Strategy for Single-Dose, Multi-Stage Antimalarial Therapy. Am. J. Trop. Med. Hyg. 2015, 92, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Pidathala, C.; Amewu, R.; Pacorel, B.; Nixon, G.L.; Gibbons, P.; Hong, W.D.; Leung, S.C.; Berry, N.G.; Sharma, R.; Stocks, P.A.; et al. Identification, Design and Biological Evaluation of Bisaryl Quinolones Targeting Plasmodium falciparum Type II NADH:Quinone Oxidoreductase (PfNDH2). J. Med. Chem. 2012, 55, 1831–1843. [Google Scholar] [CrossRef]
- Biagini, G.A.; Viriyavejakul, P.; O’Neill, P.M.; Bray, P.G.; Ward, S.A. Functional Characterization and Target Validation of Alternative Complex I of Plasmodium falciparum Mitochondria. Antimicrob. Agents Chemother. 2006, 50, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Fisher, N.; Bray, P.G.; Ward, S.A.; Biagini, G.A. The malaria parasite type II NADH:quinone oxidoreductase: An alternative enzyme for an alternative lifestyle. Trends Parasitol. 2007, 23, 305–310. [Google Scholar] [CrossRef]
- Smith, A.L. Preparation, properties, and conditions for assay of mitochondria: Slaughterhouse material, small-scale. Methods Enzymol. 1967, 10, 81–86. [Google Scholar] [CrossRef]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.R. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Struct. Biol. Crystallogr. 2004, D60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebedev, A.A.; Young, P.; Isupov, M.N.; Moroz, O.V.; Vagin, A.A.; Murshudov, G.N. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trager, W.; Jensen, J.B. Human Malaria Parasites in Continuous Culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum Erythrocytic Stages in Culture. J. Parasitol. 1979, 65, 418. [Google Scholar] [CrossRef]
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and Inexpensive Fluorescence-Based Technique for High-Throughput Antimalarial Drug Screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [Google Scholar] [CrossRef] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Kiefer, F.; Arnold, K.; Künzli, M.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009, 37, D387–D392. [Google Scholar] [CrossRef] [Green Version]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2010, 27, 343–350. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [Green Version]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
- Kuhn, B.; Fuchs, J.E.; Reutlinger, M.; Stahl, M.; Taylor, N.R. Rationalizing Tight Ligand Binding through Cooperative Interaction Networks. J. Chem. Inf. Model. 2011, 51, 3180–3198. [Google Scholar] [CrossRef]
- Zhang, Y.; Clark, J.A.; Connelly, M.C.; Zhu, F.; Min, J.; Guiguemde, W.A.; Pradhan, A.; Iyer, L.; Furimsky, A.; Gow, J.; et al. Lead Optimization of 3-Carboxyl-4(1H)-Quinolones to Deliver Orally Bioavailable Antimalarials. J. Med. Chem. 2012, 55, 4205–4219. [Google Scholar] [CrossRef] [Green Version]
- Winter, R.; Kelly, J.X.; Smilkstein, M.J.; Hinrichs, D.; Koop, D.R.; Riscoe, M.K. Optimization of endochin-like quinolones for antimalarial activity. Exp. Parasitol. 2011, 127, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Huang, L.-S.; Cobessi, D.; Tung, E.Y.; Berry, E.A. Binding of the Respiratory Chain Inhibitor Antimycin to the Mitochondrial bc1 Complex: A New Crystal Structure Reveals an Altered Intramolecular Hydrogen-bonding Pattern. J. Mol. Biol. 2005, 351, 573–597. [Google Scholar] [CrossRef] [Green Version]
- Hong, W.D.; Leung, S.C.; Amporndanai, K.; Davies, J.; Priestley, R.S.; Nixon, G.L.; Berry, N.G.; Hasnain, S.S.; Antonyuk, S.; Ward, S.A.; et al. Potent Antimalarial 2-Pyrazolyl Quinolone bc1 (Qi) Inhibitors with Improved Drug-like Properties. ACS Med. Chem. Lett. 2018, 9, 1205–1210. [Google Scholar] [CrossRef]
- McPhillie, M.; Zhou, Y.; Hickman, M.R.; Gordon, J.A.; Weber, C.R.; Li, Q.; Lee, P.J.; Amporndanai, K.; Johnson, R.M.; Darby, H.; et al. Potent Tetrahydroquinolone Eliminates Apicomplexan Parasites. Front. Cell. Infect. Microbiol. 2020, 10, 203. [Google Scholar] [CrossRef]
- Berry, E.A.; Huang, L.-S.; Lee, D.-W.; Daldal, F.; Nagai, K.; Minagawa, N. Ascochlorin is a novel, specific inhibitor of the mitochondrial cytochrome bc1 complex. Biochim. Biophys. Acta 2010, 1797, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Barton, V.; Fisher, N.; Biagini, G.; Ward, S.; O’Neill, P.M. Inhibiting Plasmodium cytochrome bc1: A complex issue. Curr. Opin. Chem. Biol. 2010, 14, 440–446. [Google Scholar] [CrossRef]
- Rathod, P.K.; McErlean, T.; Lee, P.-C. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1997, 94, 9389–9393. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.B.R.; Vieira, J.B.; Lobato, C.C.; Hage-Melim, L.I.S.; Souto, R.N.P.; Lima, C.S.; Costa, E.V.M.; Brasil, D.S.B.; Macêdo, W.J.C.; Carvalho, J.C.T. A SAR and QSAR Study of New Artemisinin Compounds with Antimalarial Activity. Molecules 2013, 19, 367–399. [Google Scholar] [CrossRef] [Green Version]
- Vieira, J.B.; Braga, F.S.; Lobato, C.C.; Santos, C.F.; Costa, J.S.; Bittencourt, J.A.H.M.; Brasil, D.S.B.; Silva, J.O.; Hage-Melim, L.I.S.; Macêdo, W.J.C.; et al. A QSAR, Pharmacokinetic and Toxicological Study of New Artemisinin Compounds with Anticancer Activity. Molecules 2014, 19, 10670–10697. [Google Scholar] [CrossRef] [Green Version]
- Flores-Sumoza, M.; Alcázar, J.J.; Márquez, E.; Mora, J.R.; Lezama, J.; Puello, E. Classical QSAR and Docking Simulation of 4-Pyridone Derivatives for Their Antimalarial Activity. Molecules 2018, 23, 3166. [Google Scholar] [CrossRef] [Green Version]
- Lane, K.D.; Mu, J.; Lu, J.; Windle, S.T.; Liu, A.; Sun, P.D.; Wellems, T.E. Selection of Plasmodium falciparum cytochrome B mutants by putative PfNDH2 inhibitors. Proc. Natl. Acad. Sci. USA 2018, 115, 6285–6290. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (nM) (3D7) | IC50 (nM) (TM90C2B) | IC50 (nM) (PfNDH2) | IC50 (nM) (Pfbc1) * | Bovine Cytochrome bc1 Inhibition (%) | |
|---|---|---|---|---|---|---|
| at 0.1 µM | at 1 µM | |||||
| ELQ300 | 2.2 | 1.7 | NA | 0.56 | 22 ± 5 | 51 ± 7 |
| GSK932121 | 6 | 6 † | >10,000 * | 7 | 65 ± 4 | 82 ± 5 |
| GW844520 | 5 | 2 † | >10,000 * | 2 | 59 ± 6 | 79 ± 3 |
| SCR0911 | 12 ± 3 | 7.2 | >1000 * | 80% inhibition at 100 nM * | 9 ± 2 | 72 ± 5 |
| CK-2-67 | 117 ± 27 | 122 ± 26 | 16 | 38 | 90 ± 3 | 95 ± 1 |
| RKA066 | 22 ± 0.4 | 217 ± 18 | 55 | 28 | 81 ± 4 | 93 ± 2 |
| WDH-1U-4 | 36 ± 6 | 5 ± 2 | 492 | <10 | 7 ± 1 | 61 ± 3 |
| bc1-CK-2-67 | bc1-RKA066 | bc1-WDH-1U-4 | |
|---|---|---|---|
| Data collection | |||
| Space group | P6522 | P6522 | P6522 |
| Cell dimensions | |||
| a, b, c (Å) | 209.59, 209.59, 342.42 | 210.74, 210.74, 343.94 | 209.56, 209.56, 343.36 |
| α, β, γ(°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å) | 49.81–3.20 (3.27–3.20) | 89.85–3.50 (3.60–3.50) | 90.74–3.50 (3.60–3.50) |
| Rmerge | 0.21 (1.63) | 0.27 (1.42) | 0.20 (0.87) |
| Rpim | 0.09 (0.69) | 0.09 (0.49) | 0.09 (0.40) |
| CC1/2 | 0.997(0.357) | 0.987(0.450) | 0.968(0.543) |
| I/σ | 8.5 (1.7) | 5.0 (1.3) | 7.0 (1.8) |
| Completeness (%) | 95.7 (97.2) | 100 (100) | 97.4 (98.7) |
| Redundancy | 9.4 (8.8) | 9.0 (8.8) | 5.4 (5.5) |
| Refinement | |||
| Resolution (Å) | 49.86–3.20 | 89.85–3.50 | 90.74–3.50 |
| No. reflections | 66,560 | 54,431 | 52,188 |
| Rwork/Rfree(%) | 21.4/25.02 | 22.09/23.82 | 21.71/24.57 |
| No. atoms | |||
| Protein | 16,142 | 15,688 | 15,617 |
| Inhibitor | 30 | 31 | 30 |
| Other ligands | 570 | 565 | 558 |
| Water | 41 | 8 | 36 |
| B-factors (Å2) | |||
| Protein | 90.68 | 140.24 | 133.88 |
| Inhibitor | 110.02 | 83.02 | 142.60 |
| Other ligands | 110.02 | 151.01 | 149.60 |
| Water | 54.87 | 45.87 | 40.79 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.007 | 0.009 | 0.008 |
| Bond angles (°) | 1.681 | 1.505 | 1.437 |
| Ramachandran Plot | |||
| Preferred (%) | 96.02 | 95.98 | 96.17 |
| Allowed (%) | 3.59 | 3.66 | 3.63 |
| Outliers (%) | 0.39 | 0.35 | 0.20 |
| PDB code | 7R3V | 6ZFU | 6ZFS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amporndanai, K.; Pinthong, N.; O’Neill, P.M.; Hong, W.D.; Amewu, R.K.; Pidathala, C.; Berry, N.G.; Leung, S.C.; Ward, S.A.; Biagini, G.A.; et al. Targeting the Ubiquinol-Reduction (Qi) Site of the Mitochondrial Cytochrome bc1 Complex for the Development of Next Generation Quinolone Antimalarials. Biology 2022, 11, 1109. https://doi.org/10.3390/biology11081109
Amporndanai K, Pinthong N, O’Neill PM, Hong WD, Amewu RK, Pidathala C, Berry NG, Leung SC, Ward SA, Biagini GA, et al. Targeting the Ubiquinol-Reduction (Qi) Site of the Mitochondrial Cytochrome bc1 Complex for the Development of Next Generation Quinolone Antimalarials. Biology. 2022; 11(8):1109. https://doi.org/10.3390/biology11081109
Chicago/Turabian StyleAmporndanai, Kangsa, Nattapon Pinthong, Paul M. O’Neill, W. David Hong, Richard K. Amewu, Chandrakala Pidathala, Neil G. Berry, Suet C. Leung, Stephen A. Ward, Giancarlo A. Biagini, and et al. 2022. "Targeting the Ubiquinol-Reduction (Qi) Site of the Mitochondrial Cytochrome bc1 Complex for the Development of Next Generation Quinolone Antimalarials" Biology 11, no. 8: 1109. https://doi.org/10.3390/biology11081109
APA StyleAmporndanai, K., Pinthong, N., O’Neill, P. M., Hong, W. D., Amewu, R. K., Pidathala, C., Berry, N. G., Leung, S. C., Ward, S. A., Biagini, G. A., Hasnain, S. S., & Antonyuk, S. V. (2022). Targeting the Ubiquinol-Reduction (Qi) Site of the Mitochondrial Cytochrome bc1 Complex for the Development of Next Generation Quinolone Antimalarials. Biology, 11(8), 1109. https://doi.org/10.3390/biology11081109