
NRSF/REST-Mediated Epigenomic Regulation in the Heart: Transcriptional Control of Natriuretic Peptides and Beyond


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
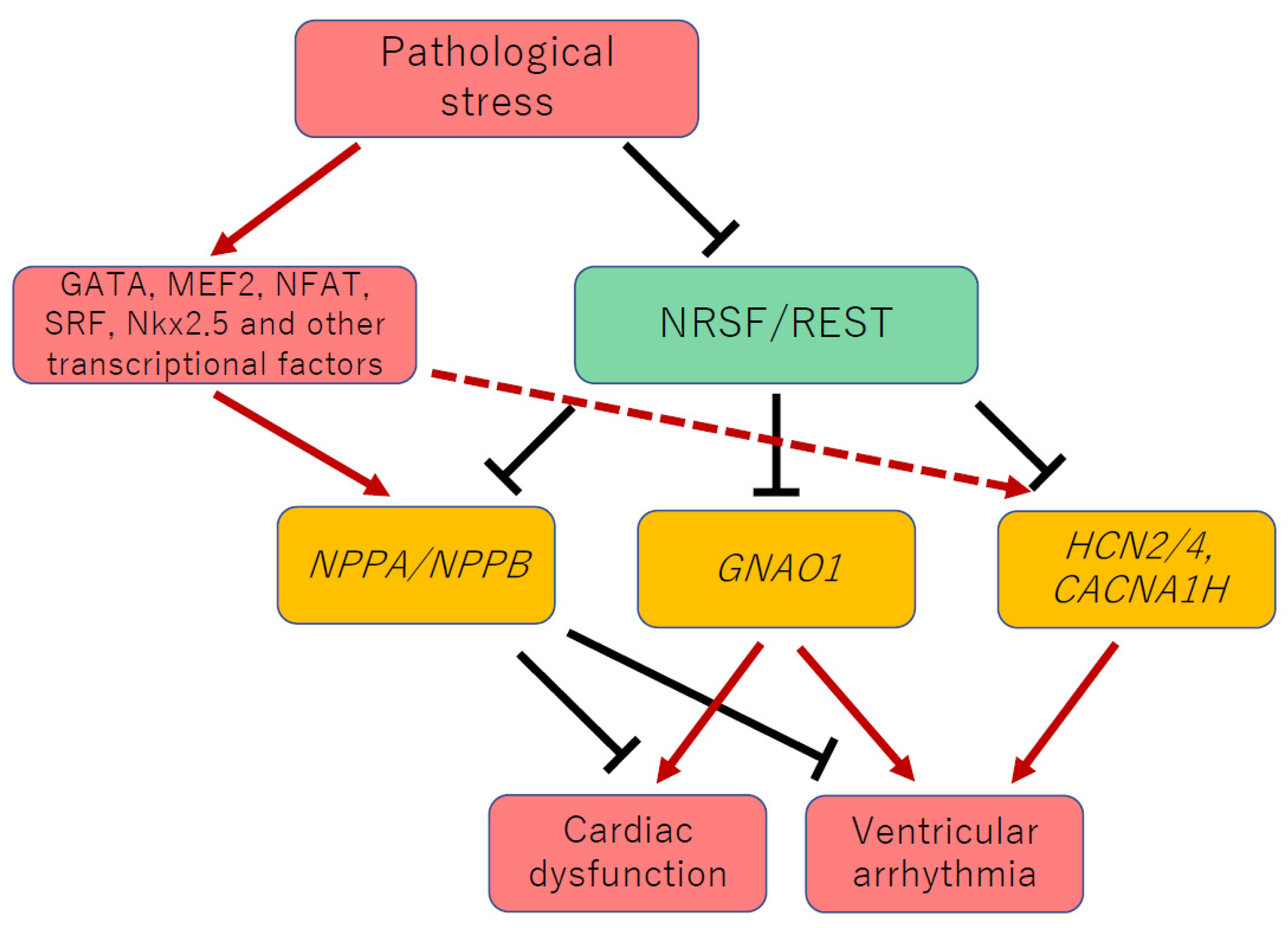
1. Introduction
2. Transcriptional Regulation of ANP and BNP in the Heart
3. NRSF Is a Transcriptional Repressor of Fetal Cardiac Genes, including NPPA and NPPB
4. NRSF Regulates Fetal Cardiac Ion Channels and Maintains Electrical Stability in the Heart
5. NRSF Maintains Ca2+ Homeostasis and Systolic Function in the Heart
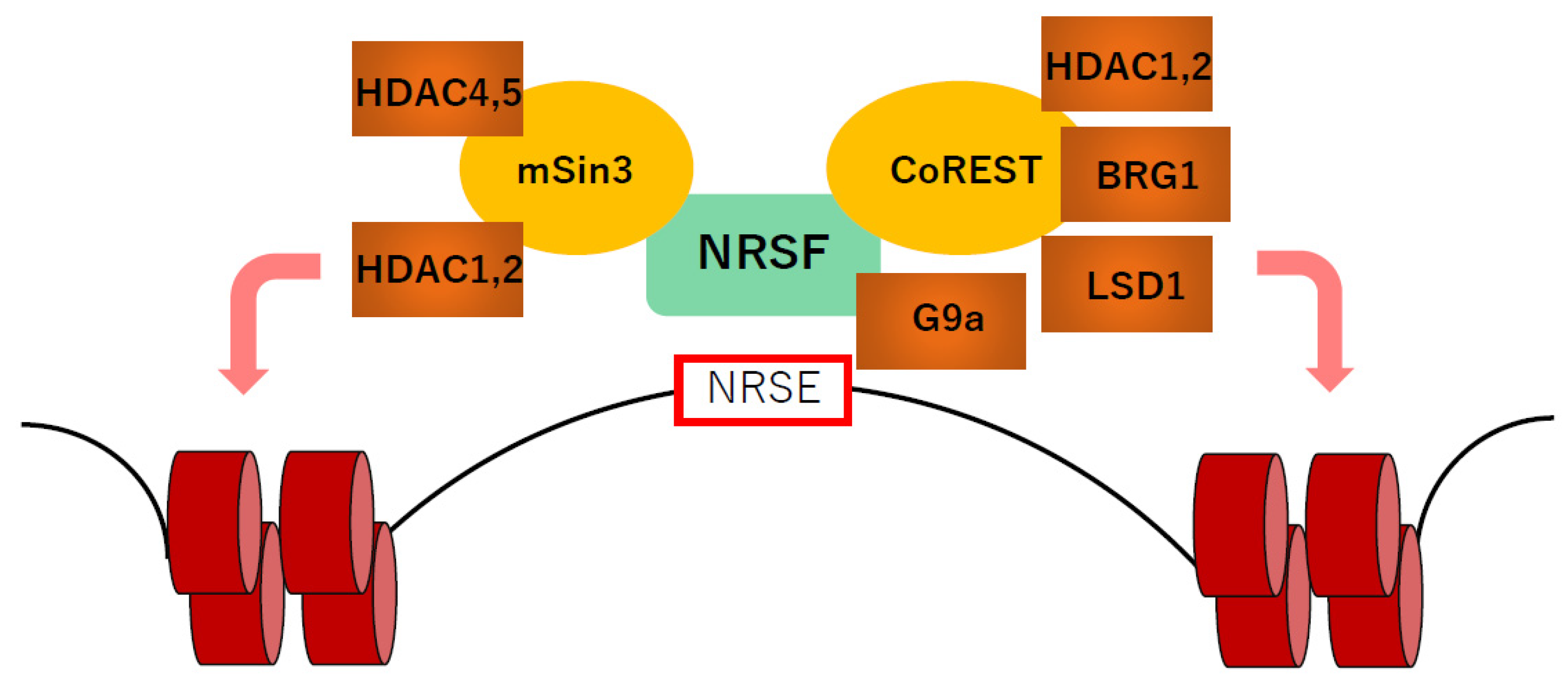
6. Epigenetic Regulators Associated with NRSF
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ahmad, F.B.; Anderson, R.N. The Leading Causes of Death in the US for 2020. JAMA 2021, 325, 1829–1830. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Olson, E.N. Toward transcriptional therapies for the failing heart: Chemical screens to modulate genes. J. Clin. Investig. 2005, 115, 538–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukoyama, M.; Nakao, K.; Hosoda, K.; Suga, S.; Saito, Y.; Ogawa, Y.; Shirakami, G.; Jougasaki, M.; Obata, K.; Yasue, H.; et al. Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J. Clin. Investig. 1991, 87, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, A.; Hesse, B. Heart failure and neuroendocrine activation: Diagnostic, prognostic and therapeutic perspectives. Clin. Physiol. 2001, 21, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Xu, J.; Molkentin, J.D. Re-employment of developmental transcription factors in adult heart disease. Semin. Cell Dev. Biol. 2007, 18, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K.; Saito, Y.; Ogawa, E.; Takahashi, N.; Nakagawa, Y.; Naruse, Y.; Harada, M.; Hamanaka, I.; Izumi, T.; Miyamoto, Y.; et al. The neuron-restrictive silencer element-neuron-restrictive silencer factor system regulates basal and endothelin 1-inducible atrial natriuretic peptide gene expression in ventricular myocytes. Mol. Cell. Biol. 2001, 21, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, E.; Saito, Y.; Kuwahara, K.; Harada, M.; Miyamoto, Y.; Hamanaka, I.; Kajiyama, N.; Takahashi, N.; Izumi, T.; Kawakami, R.; et al. Fibronectin signaling stimulates BNP gene transcription by inhibiting neuron-restrictive silencer element-dependent repression. Cardiovasc. Res. 2002, 53, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K.; Saito, Y.; Takano, M.; Arai, Y.; Yasuno, S.; Nakagawa, Y.; Takahashi, N.; Adachi, Y.; Takemura, G.; Horie, M.; et al. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 2003, 22, 6310–6321. [Google Scholar] [CrossRef]
- Inazumi, H.; Kuwahara, K.; Nakagawa, Y.; Kuwabara, Y.; Numaga-Tomita, T.; Kashihara, T.; Nakada, T.; Kurebayashi, N.; Oya, M.; Nonaka, M.; et al. NRSF-GNAO1 Pathway Contributes to the Regulation of Cardiac Ca2+ Homeostasis. Circ. Res. 2022, 130, 234–248. [Google Scholar] [CrossRef]
- Kuwahara, K.; Nakao, K. Regulation and significance of atrial and brain natriuretic peptides as cardiac hormones. Endocr. J. 2010, 57, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef]
- Zeller, R.; Bloch, K.D.; Williams, B.S.; Arceci, R.J.; Seidman, C.E. Localized expression of the atrial natriuretic factor gene during cardiac embryogenesis. Genes Dev. 1987, 1, 693–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, L.J. Atrial natriuretic factor-SV40 T antigen transgenes produce tumors and cardiac arrhythmias in mice. Science 1988, 239, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, K.U.; Rockman, H.A.; Itani, M.; Vovan, A.; Seidman, C.E.; Chien, K.R. Divergent pathways mediate the induction of ANF transgenes in neonatal and hypertrophic ventricular myocardium. J. Clin. Investig. 1995, 96, 1311–1318. [Google Scholar] [CrossRef]
- Argentin, S.; Ardati, A.; Tremblay, S.; Lihrmann, I.; Robitaille, L.; Drouin, J.; Nemer, M. Developmental stage-specific regulation of atrial natriuretic factor gene transcription in cardiac cells. Mol. Cell. Biol. 1994, 14, 777–790. [Google Scholar] [CrossRef]
- LaPointe, M.C.; Wu, J.P.; Greenberg, B.; Gardner, D.G. Upstream sequences confer atrial-specific expression on the human atrial natriuretic factor gene. J. Biol. Chem. 1988, 263, 9075–9078. [Google Scholar] [CrossRef]
- Houweling, A.C.; van Borren, M.M.; Moorman, A.F.; Christoffels, V.M. Expression and regulation of the atrial natriuretic factor encoding gene Nppa during development and disease. Cardiovasc. Res. 2005, 67, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Debrus, S.; Rahbani, L.; Marttila, M.; Delorme, B.; Paradis, P.; Nemer, M. The zinc finger-only protein Zfp260 is a novel cardiac regulator and a nuclear effector of α1-adrenergic signaling. Mol. Cell. Biol. 2005, 25, 8669–8682. [Google Scholar] [CrossRef] [Green Version]
- Sprenkle, A.B.; Murray, S.F.; Glembotski, C.C. Involvement of multiple cis elements in basal- and α-adrenergic agonist-inducible atrial natriuretic factor transcription. Roles for serum response elements and an SP-1-like element. Circ. Res. 1995, 77, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Shioi, T.; Kasahara, H.; Jobe, S.M.; Wiese, R.J.; Markham, B.E.; Izumo, S. The cardiac tissue-restricted homeobox protein Csx/Nkx2.5 physically associates with the zinc finger protein GATA4 and cooperatively activates atrial natriuretic factor gene expression. Mol. Cell. Biol. 1998, 18, 3120–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durocher, D.; Chen, C.Y.; Ardati, A.; Schwartz, R.J.; Nemer, M. The atrial natriuretic factor promoter is a downstream target for Nkx-2.5 in the myocardium. Mol. Cell. Biol. 1996, 16, 4648–4655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiojima, I.; Komuro, I.; Oka, T.; Hiroi, Y.; Mizuno, T.; Takimoto, E.; Monzen, K.; Aikawa, R.; Akazawa, H.; Yamazaki, T.; et al. Context-dependent transcriptional cooperation mediated by cardiac transcription factors Csx/Nkx-2.5 and GATA-4. J. Biol. Chem. 1999, 274, 8231–8239. [Google Scholar] [CrossRef] [Green Version]
- Morin, S.; Charron, F.; Robitaille, L.; Nemer, M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000, 19, 2046–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, A.N.; Ruiz-Lozano, P.; Chen, Y.F.; Sionit, P.; Yu, Y.T.; Lilly, B.; Olson, E.N.; Chien, K.R. A novel A/T-rich element mediates ANF gene expression during cardiac myocyte hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 515–525. [Google Scholar] [CrossRef]
- Small, E.M.; Krieg, P.A. Transgenic analysis of the atrialnatriuretic factor (ANF) promoter: Nkx2-5 and GATA-4 binding sites are required for atrial specific expression of ANF. Dev. Biol. 2003, 261, 116–131. [Google Scholar] [CrossRef] [Green Version]
- Horsthuis, T.; Houweling, A.C.; Habets, P.E.; de Lange, F.J.; el Azzouzi, H.; Clout, D.E.; Moorman, A.F.; Christoffels, V.M. Distinct regulation of developmental and heart disease-induced atrial natriuretic factor expression by two separate distal sequences. Circ. Res. 2008, 102, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Argentin, S.; Sun, Y.L.; Lihrmann, I.; Schmidt, T.J.; Drouin, J.; Nemer, M. Distal cis-acting promoter sequences mediate glucocorticoid stimulation of cardiac atrial natriuretic factor gene transcription. J. Biol. Chem. 1991, 266, 23315–23322. [Google Scholar] [CrossRef]
- Chun, Y.S.; Hyun, J.Y.; Kwak, Y.G.; Kim, I.S.; Kim, C.H.; Choi, E.; Kim, M.S.; Park, J.W. Hypoxic activation of the atrial natriuretic peptide gene promoter through direct and indirect actions of hypoxia-inducible factor-1. Biochem. J. 2003, 370, 149–157. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Wang, D.; Yang, X.P.; Carretero, O.A.; LaPointe, M.C. Inducible regulation of human brain natriuretic peptide promoter in transgenic mice. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H368–H376. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, M.C.; Wu, G.; Garami, M.; Yang, X.P.; Gardner, D.G. Tissue-specific expression of the human brain natriuretic peptide gene in cardiac myocytes. Hypertension 1996, 27, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, O.; Ogawa, Y.; Itoh, H.; Suga, S.; Komatsu, Y.; Kishimoto, I.; Nishino, K.; Yoshimasa, T.; Nakao, K. Rapid transcriptional activation and early mRNA turnover of brain natriuretic peptide in cardiocyte hypertrophy. Evidence for brain natriuretic peptide as an “emergency” cardiac hormone against ventricular overload. J. Clin. Investig. 1995, 96, 1280–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhelper, M.E. Structure, expression, and genomic mapping of the mouse natriuretic peptide type-B gene. Circ. Res. 1993, 72, 984–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thuerauf, D.J.; Hanford, D.S.; Glembotski, C.C. Regulation of rat brain natriuretic peptide transcription. A potential role for GATA-related transcription factors in myocardial cell gene expression. J. Biol. Chem. 1994, 269, 17772–17775. [Google Scholar] [CrossRef]
- Kuwahara, K.; Kinoshita, H.; Kuwabara, Y.; Nakagawa, Y.; Usami, S.; Minami, T.; Yamada, Y.; Fujiwara, M.; Nakao, K. Myocardin-related transcription factor A is a common mediator of mechanical stress-and neurohumoral stimulation-induced cardiac hypertrophic signaling leading to activation of brain natriuretic peptide gene expression. Mol. Cell. Biol. 2010, 30, 4134–4148. [Google Scholar] [CrossRef] [Green Version]
- Akazawa, H.; Komuro, I. Roles of cardiac transcription factors in cardiac hypertrophy. Circ. Res. 2003, 92, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, N.; Smith, G.; Izumo, S. Both a ubiquitous factor mTEF-1 and a distinct muscle-specific factor bind to the M-CAT motif of the myosin heavy chain β gene. Nucleic Acids Res. 1993, 21, 4103–4110. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Olson, E.N. GATA4: A novel transcriptional regulator of cardiac hypertrophy? Circulation 1997, 96, 3833–3835. [Google Scholar]
- Grépin, C.; Dagnino, L.; Robitaille, L.; Haberstroh, L.; Antakly, T.; Nemer, M. A hormone-encoding gene identifies a pathway for cardiac but not skeletal muscle gene transcription. Mol. Cell. Biol. 1994, 14, 3115–3129. [Google Scholar] [CrossRef]
- LaPointe, M.C. Molecular regulation of the brain natriuretic peptide gene. Peptides 2005, 26, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Gardner, D.G. Mechanical strain activates BNP gene transcription through a p38/NF-kappaB-dependent mechanism. J. Clin. Investig. 1999, 104, 1603–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, E.; Saito, Y.; Harada, M.; Kamitani, S.; Kuwahara, K.; Miyamoto, Y.; Ishikawa, M.; Hamanaka, I.; Kajiyama, N.; Takahashi, N.; et al. Outside-in signalling of fibronectin stimulates cardiomyocyte hypertrophy in cultured neonatal rat ventricular myocytes. J. Mol. Cell. Cardiol. 2000, 32, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Webb, P.; Marimuthu, A.; Zhang, S.; Gardner, D.G. Triiodothyronine increases brain natriuretic peptide (BNP) gene transcription and amplifies endothelin-dependent BNP gene transcription and hypertrophy in neonatal rat ventricular myocytes. J. Biol. Chem. 2003, 278, 15073–15083. [Google Scholar] [CrossRef] [Green Version]
- Sergeeva, I.A.; Hooijkaas, I.B.; Ruijter, J.M.; van der Made, I.; de Groot, N.E.; van de Werken, H.J.; Creemers, E.E.; Christoffels, V.M. Identification of a regulatory domain controlling the Nppa-Nppb gene cluster during heart development and stress. Development 2016, 143, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- de Bold, A.J.; Bruneau, B.G.; Kuroski de Bold, M.L. Mechanical and neuroendocrine regulation of the endocrine heart. Cardiovasc. Res. 1996, 31, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Saito, Y.; Kuwahara, K.; Ogawa, E.; Ishikawa, M.; Nakagawa, O.; Miyamoto, Y.; Kamitani, S.; Hamanaka, I.; Kajiyama, N.; et al. Interaction of myocytes and nonmyocytes is necessary for mechanical stretch to induce ANP/BNP production in cardiocyte culture. J. Cardiovasc. Pharmacol. 1998, 31 (Suppl. 1), S357–S359. [Google Scholar] [CrossRef]
- Kinnunen, P.; Vuolteenaho, O.; Uusimaa, P.; Ruskoaho, H. Passive mechanical stretch releases atrial natriuretic peptide from rat ventricular myocardium. Circ. Res. 1992, 70, 1244–1253. [Google Scholar] [CrossRef] [Green Version]
- Sergeeva, I.A.; Christoffels, V.M. Regulation of expression of atrial and brain natriuretic peptide, biomarkers for heart development and disease. Biochim. Biophys. Acta 2013, 1832, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Mori, N.; Schoenherr, C.; Vandenbergh, D.J.; Anderson, D.J. A common silencer element in the SCG10 and type II Na+ channel genes binds a factor present in nonneuronal cells but not in neuronal cells. Neuron 1992, 9, 45–54. [Google Scholar] [CrossRef]
- Schoenherr, C.J.; Anderson, D.J. The neuron-restrictive silencer factor (NRSF): A coordinate repressor of multiple neuron-specific genes. Science 1995, 267, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.A.; Tapia-Ramírez, J.; Kim, S.; Toledo-Aral, J.J.; Zheng, Y.; Boutros, M.C.; Altshuller, Y.M.; Frohman, M.A.; Kraner, S.D.; Mandel, G. REST: A mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 1995, 80, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Ooi, L.; Wood, I.C. Chromatin crosstalk in development and disease: Lessons from REST. Nat. Rev. Genet. 2007, 8, 544–554. [Google Scholar] [CrossRef]
- Schoenherr, C.J.; Paquette, A.J.; Anderson, D.J. Identification of potential target genes for the neuron-restrictive silencer factor. Proc. Natl. Acad. Sci. USA 1996, 93, 9881–9886. [Google Scholar] [CrossRef] [Green Version]
- Yasuno, S.; Usami, S.; Kuwahara, K.; Nakanishi, M.; Arai, Y.; Kinoshita, H.; Nakagawa, Y.; Fujiwara, M.; Murakami, M.; Ueshima, K.; et al. Endogenous cardiac natriuretic peptides protect the heart in a mouse model of dilated cardiomyopathy and sudden death. Am. J. Physiol.-Heart Circ. Physiol. 2009, 296, H1804–H1810. [Google Scholar] [CrossRef] [Green Version]
- Takano, M.; Kinoshita, H.; Shioya, T.; Itoh, M.; Nakao, K.; Kuwahara, K. Pathophysiological remodeling of mouse cardiac myocytes expressing dominant negative mutant of neuron restrictive silencing factor. Circ. J. 2010, 74, 2712–2719. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K.; Takano, M.; Nakao, K. Pathophysiological significance of T-type Ca2+ channels: Transcriptional regulation of T-type Ca2+ channel—Regulation of CACNA1H by neuron-restrictive silencer factor. J. Pharmacol. Sci. 2005, 99, 211–213. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Iijima, T. Cardiac T-type Ca2+ channels in the heart. J. Mol. Cell. Cardiol. 2010, 48, 65–70. [Google Scholar] [CrossRef]
- Yasui, K.; Niwa, N.; Takemura, H.; Opthof, T.; Muto, T.; Horiba, M.; Shimizu, A.; Lee, J.K.; Honjo, H.; Kamiya, K.; et al. Pathophysiological significance of T-type Ca2+ channels: Expression of T-type Ca2+ channels in fetal and diseased heart. J. Pharmacol. Sci. 2005, 99, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Niwa, N.; Yasui, K.; Opthof, T.; Takemura, H.; Shimizu, A.; Horiba, M.; Lee, J.K.; Honjo, H.; Kamiya, K.; Kodama, I. Cav3.2 subunit underlies the functional T-type Ca2+ channel in murine hearts during the embryonic period. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H2257–H2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clozel, J.P.; Ertel, E.A.; Ertel, S.I. Voltage-gated T-type Ca2+ channels and heart failure. Proc. Assoc. Am. Physicians 1999, 111, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Kuwahara, K.; Takano, M.; Arai, Y.; Kuwabara, Y.; Yasuno, S.; Nakagawa, Y.; Nakanishi, M.; Harada, M.; Fujiwara, M.; et al. T-type Ca2+ channel blockade prevents sudden death in mice with heart failure. Circulation 2009, 120, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, R.B.; Siegelbaum, S.A. Hyperpolarization-activated cation currents: From molecules to physiological function. Annu. Rev. Physiol. 2003, 65, 453–480. [Google Scholar] [CrossRef] [Green Version]
- Wahl-Schott, C.; Biel, M. HCN channels: Structure, cellular regulation and physiological function. Cell. Mol. Life Sci. 2009, 66, 470–494. [Google Scholar] [CrossRef]
- Cerbai, E.; Mugelli, A. If in non-pacemaker cells: Role and pharmacological implications. Pharmacol. Res. 2006, 53, 416–423. [Google Scholar] [CrossRef]
- Yasui, K.; Liu, W.; Opthof, T.; Kada, K.; Lee, J.K.; Kamiya, K.; Kodama, I. If current and spontaneous activity in mouse embryonic ventricular myocytes. Circ. Res. 2001, 88, 536–542. [Google Scholar] [CrossRef] [Green Version]
- Stillitano, F.; Lonardo, G.; Zicha, S.; Varro, A.; Cerbai, E.; Mugelli, A.; Nattel, S. Molecular basis of funny current (If) in normal and failing human heart. J. Mol. Cell. Cardiol. 2008, 45, 289–299. [Google Scholar] [CrossRef]
- Kuratomi, S.; Ohmori, Y.; Ito, M.; Shimazaki, K.; Muramatsu, S.; Mizukami, H.; Uosaki, H.; Yamashita, J.K.; Arai, Y.; Kuwahara, K.; et al. The cardiac pacemaker-specific channel Hcn4 is a direct transcriptional target of MEF2. Cardiovasc. Res. 2009, 83, 682–687. [Google Scholar] [CrossRef] [Green Version]
- Kuratomi, S.; Kuratomi, A.; Kuwahara, K.; Ishii, T.M.; Nakao, K.; Saito, Y.; Takano, M. NRSF regulates the developmental and hypertrophic changes of HCN4 transcription in rat cardiac myocytes. Biochem. Biophys. Res. Commun. 2007, 353, 67–73. [Google Scholar] [CrossRef]
- Kuwabara, Y.; Kuwahara, K.; Takano, M.; Kinoshita, H.; Arai, Y.; Yasuno, S.; Nakagawa, Y.; Igata, S.; Usami, S.; Minami, T.; et al. Increased expression of HCN channels in the ventricular myocardium contributes to enhanced arrhythmicity in mouse failing hearts. J. Am. Heart Assoc. 2013, 2, e000150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusch, G. Heart rate and heart failure. Not a simple relationship. Circ. J. 2011, 75, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- Hurowitz, E.H.; Melnyk, J.M.; Chen, Y.J.; Kouros-Mehr, H.; Simon, M.I.; Shizuya, H. Genomic characterization of the human heterotrimeric G protein α, β, and γ subunit genes. DNA Res. 2000, 7, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strathmann, M.P.; Simon, M.I. G α12 and G α13 subunits define a fourth class of G protein α subunits. Proc. Natl. Acad. Sci. USA 1991, 88, 5582–5586. [Google Scholar] [CrossRef] [Green Version]
- He, J.C.; Neves, S.R.; Jordan, J.D.; Iyengar, R. Role of the Go/i signaling network in the regulation of neurite outgrowth. Can. J. Physiol. Pharmacol. 2006, 84, 687–694. [Google Scholar] [CrossRef]
- Neumann, J.; Schmitz, W.; Scholz, H.; von Meyerinck, L.; Doring, V.; Kalmar, P. Increase in myocardial Gi-proteins in heart failure. Lancet 1988, 2, 936–937. [Google Scholar] [CrossRef]
- Feldman, A.M.; Cates, A.E.; Veazey, W.B.; Hershberger, R.E.; Bristow, M.R.; Baughman, K.L.; Baumgartner, W.A.; Van Dop, C. Increase of the 40,000-mol wt pertussis toxin substrate (G protein) in the failing human heart. J. Clin. Investig. 1988, 82, 189–197. [Google Scholar] [CrossRef] [Green Version]
- DeGeorge, B.R., Jr.; Gao, E.; Boucher, M.; Vinge, L.E.; Martini, J.S.; Raake, P.W.; Chuprun, J.K.; Harris, D.M.; Kim, G.W.; Soltys, S.; et al. Targeted inhibition of cardiomyocyte Gi signaling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation 2008, 117, 1378–1387. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Mende, U.; Lewis, C.; Neer, E.J. Maintenance of cellular levels of G-proteins: Different efficiencies of αs and αo synthesis in GH3 cells. Biochem. J. 1996, 318 Pt 3, 1071–1077. [Google Scholar] [CrossRef]
- Asano, T.; Morishita, R.; Semba, R.; Itoh, H.; Kaziro, Y.; Kato, K. Identification of lung major GTP-binding protein as Gi2 and its distribution in various rat tissues determined by immunoassay. Biochemistry 1989, 28, 4749–4754. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; McDonald, A.D.; Nasir, K.; Peller, L.; Rade, J.J.; Miller, J.M.; Heldman, A.W.; Donahue, J.K. Inhibitory G protein overexpression provides physiologically relevant heart rate control in persistent atrial fibrillation. Circulation 2004, 110, 3115–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, K.; Maass, M.; Dizayee, S.; Leiss, V.; Annala, S.; Koth, J.; Seemann, W.K.; Muller-Ehmsen, J.; Mohr, K.; Nurnberg, B.; et al. Lack of Gαi2 leads to dilative cardiomyopathy and increased mortality in β1-adrenoceptor overexpressing mice. Cardiovasc. Res. 2015, 108, 348–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, K.; Parra, S.; Chen, R.; Charbeneau, R.A.; Wade, S.M.; Jay, P.Y.; Neubig, R.R. Gα2 signaling: Friend or foe in cardiac injury and heart failure? Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Nishiga, M.; Horie, T.; Kuwabara, Y.; Nagao, K.; Baba, O.; Nakao, T.; Nishino, T.; Hakuno, D.; Nakashima, Y.; Nishi, H.; et al. MicroRNA-33 Controls Adaptive Fibrotic Response in the Remodeling Heart by Preserving Lipid Raft Cholesterol. Circ. Res. 2017, 120, 835–847. [Google Scholar] [CrossRef]
- Du, C.K.; Morimoto, S.; Nishii, K.; Minakami, R.; Ohta, M.; Tadano, N.; Lu, Q.W.; Wang, Y.Y.; Zhan, D.Y.; Mochizuki, M.; et al. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ. Res. 2007, 101, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Nomura, S.; Satoh, M.; Fujita, T.; Higo, T.; Sumida, T.; Ko, T.; Yamaguchi, T.; Tobita, T.; Naito, A.T.; Ito, M.; et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 2018, 9, 4435. [Google Scholar] [CrossRef]
- Bingham, A.J.; Ooi, L.; Wood, I.C. Multiple chromatin modifications important for gene expression changes in cardiac hypertrophy. Biochem. Soc. Trans. 2006, 34, 1138–1140. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Kuwahara, K.; Harada, M.; Takahashi, N.; Yasuno, S.; Adachi, Y.; Kawakami, R.; Nakanishi, M.; Tanimoto, K.; Usami, S.; et al. Class II HDACs mediate CaMK-dependent signaling to NRSF in ventricular myocytes. J. Mol. Cell. Cardiol. 2006, 41, 1010–1022. [Google Scholar] [CrossRef]
- Roopra, A.; Qazi, R.; Schoenike, B.; Daley, T.J.; Morrison, J.F. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol. Cell. 2004, 14, 727–738. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000, 408, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Sohn, I.S.; Nam, K.I.; Park, J.E.; Qian, Y.R.; Yin, Z.; Ahn, Y.; Jeong, M.H.; Bang, Y.J.; Kim, N.; et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 2006, 113, 51–59. [Google Scholar] [CrossRef]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef]
- Gillette, T.G.; Hill, J.A. Readers, writers, and erasers: Chromatin as the whiteboard of heart disease. Circ. Res. 2015, 116, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Morales, C.R.; Li, D.L.; Pedrozo, Z.; May, H.I.; Jiang, N.; Kyrychenko, V.; Cho, G.W.; Kim, S.Y.; Wang, Z.V.; Rotter, D.; et al. Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2-dependent mTOR repression. Sci. Signal. 2016, 9, ra34. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Jeong, M.Y.; Lin, Y.H.; Wennersten, S.A.; Demos-Davies, K.M.; Cavasin, M.A.; Mahaffey, J.H.; Monzani, V.; Saripalli, C.; Mascagni, P.; Reece, T.B.; et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci. Transl. Med. 2018, 10, eaao0144. [Google Scholar] [CrossRef] [Green Version]
- Wallner, M.; Eaton, D.M.; Berretta, R.M.; Liesinger, L.; Schittmayer, M.; Gindlhuber, J.; Wu, J.; Jeong, M.Y.; Lin, Y.H.; Borghetti, G.; et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci. Transl. Med. 2020, 12, eaay7205. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Wennersten, S.A.; Peña, B.; Bagchi, R.A.; Smith, H.E.; Hirsch, R.A.; Vanderlinden, L.A.; Lin, Y.H.; Dobrinskikh, E.; Demos-Davies, K.M.; et al. HDAC Inhibition Reverses Preexisting Diastolic Dysfunction and Blocks Covert Extracellular Matrix Remodeling. Circulation 2021, 143, 1874–1890. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.L.; Jiao, L.; An, Q.; Chen, X.; Qi, Y.; Wei, B.; Zheng, Y.; Shi, X.; Gao, E.; Liu, H.M.; et al. Myofibroblast Deficiency of LSD1 Alleviates TAC-Induced Heart Failure. Circ. Res. 2021, 129, 400–413. [Google Scholar] [CrossRef]
- Foster, C.T.; Dovey, O.M.; Lezina, L.; Luo, J.L.; Gant, T.W.; Barlev, N.; Bradley, A.; Cowley, S.M. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol. Cell. Biol. 2010, 30, 4851–4863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, M.; Matsumura, Y.; Fukuda, M.; Kimura, H.; Shinkai, Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 2008, 27, 2681–2690. [Google Scholar] [CrossRef]
- Thienpont, B.; Aronsen, J.M.; Robinson, E.L.; Okkenhaug, H.; Loche, E.; Ferrini, A.; Brien, P.; Alkass, K.; Tomasso, A.; Agrawal, A.; et al. The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J. Clin. Investig. 2017, 127, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Bingham, A.J.; Ooi, L.; Kozera, L.; White, E.; Wood, I.C. The repressor element 1-silencing transcription factor regulates heart-specific gene expression using multiple chromatin-modifying complexes. Mol. Cell. Biol. 2007, 27, 4082–4092. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inazumi, H.; Kuwahara, K. NRSF/REST-Mediated Epigenomic Regulation in the Heart: Transcriptional Control of Natriuretic Peptides and Beyond. Biology 2022, 11, 1197. https://doi.org/10.3390/biology11081197
Inazumi H, Kuwahara K. NRSF/REST-Mediated Epigenomic Regulation in the Heart: Transcriptional Control of Natriuretic Peptides and Beyond. Biology. 2022; 11(8):1197. https://doi.org/10.3390/biology11081197
Chicago/Turabian StyleInazumi, Hideaki, and Koichiro Kuwahara. 2022. "NRSF/REST-Mediated Epigenomic Regulation in the Heart: Transcriptional Control of Natriuretic Peptides and Beyond" Biology 11, no. 8: 1197. https://doi.org/10.3390/biology11081197
APA StyleInazumi, H., & Kuwahara, K. (2022). NRSF/REST-Mediated Epigenomic Regulation in the Heart: Transcriptional Control of Natriuretic Peptides and Beyond. Biology, 11(8), 1197. https://doi.org/10.3390/biology11081197