
Adipokines in Non-Alcoholic Fatty Liver Disease: Are We on the Road toward New Biomarkers and Therapeutic Targets?

 ,
,  , ,
, , 
Abstract
:Simple Summary
Abstract
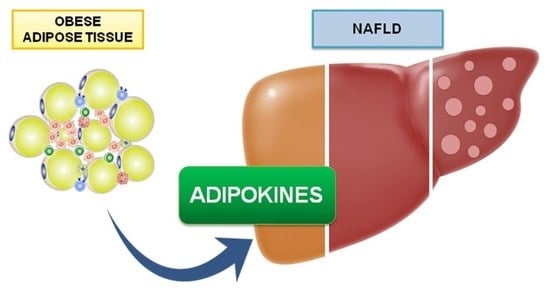
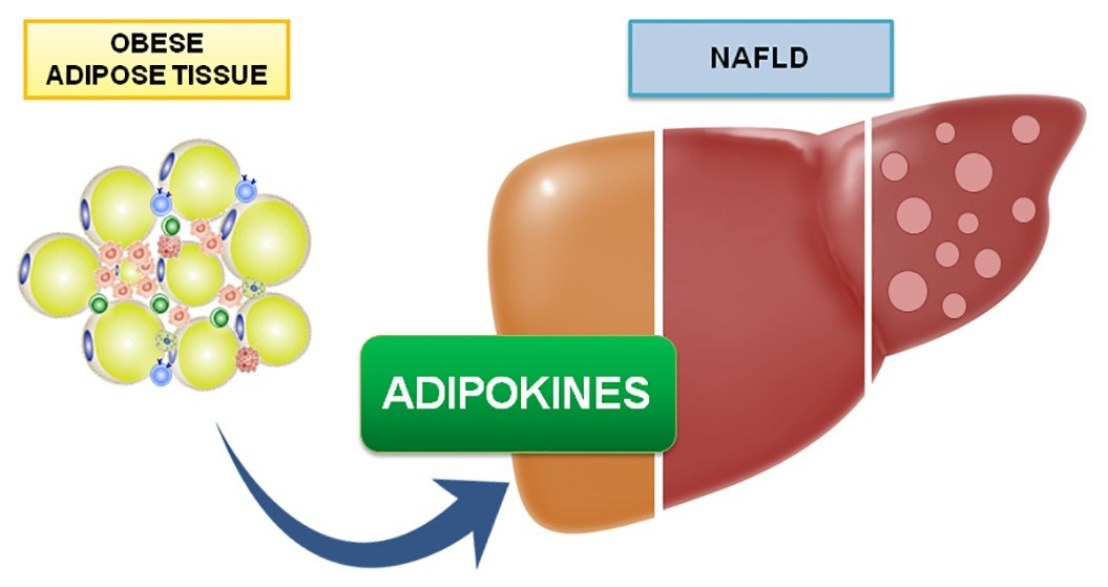
1. Introduction
2. NAFLD, an Unmet Medical Need
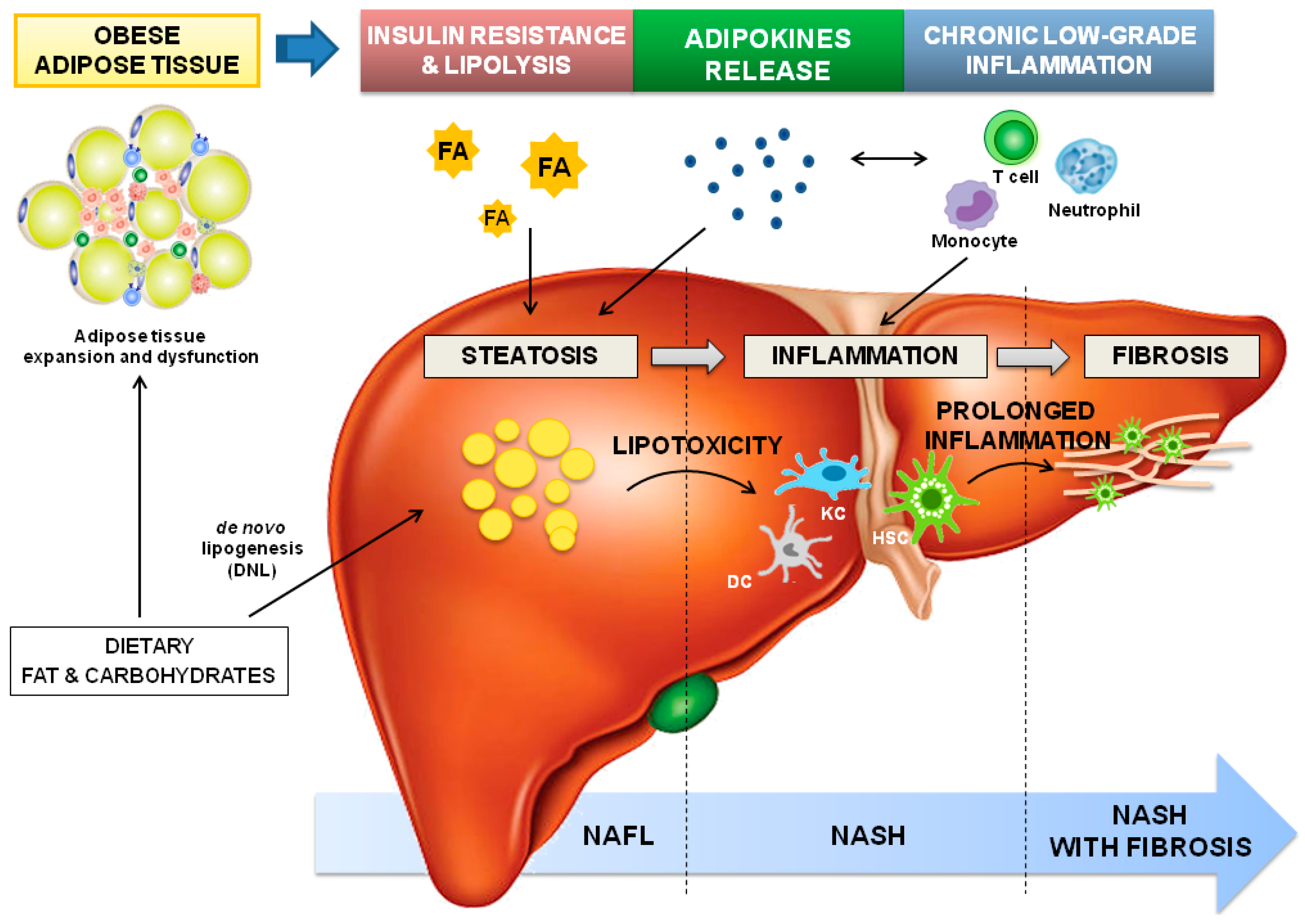
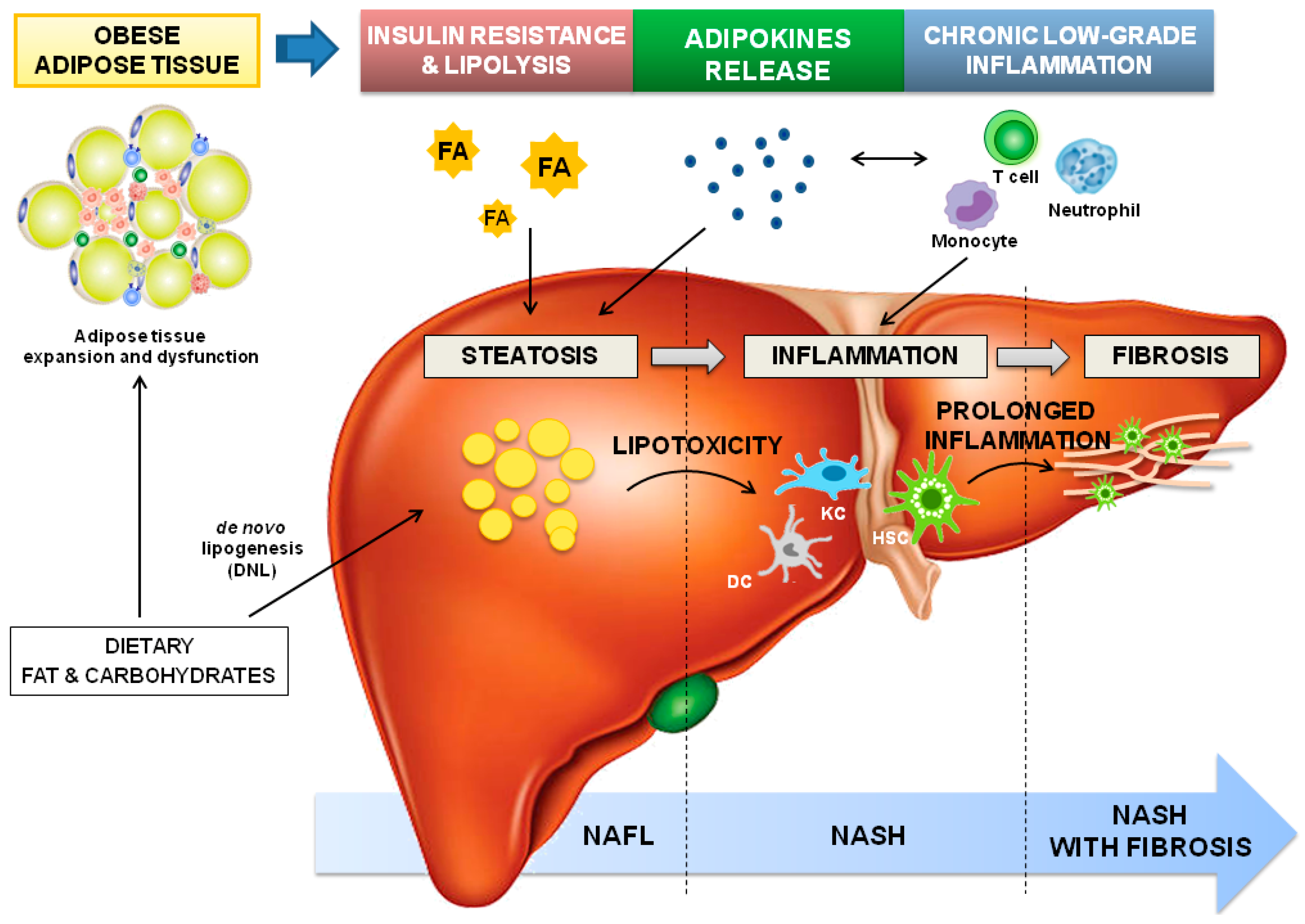
3. Adipose Tissue-Liver Axis in NAFLD Pathophysiology
4. Adipokines as Potential Therapeutic Targets for NAFLD
4.1. Leptin and Adiponectin
4.2. Ghrelin
4.3. Resistin
4.4. Retinol Binding Protein 4 (RBP4)
4.5. Visfatin
4.6. Chemerin
4.7. Adipocyte Fatty Acid-Binding Protein (AFABP)
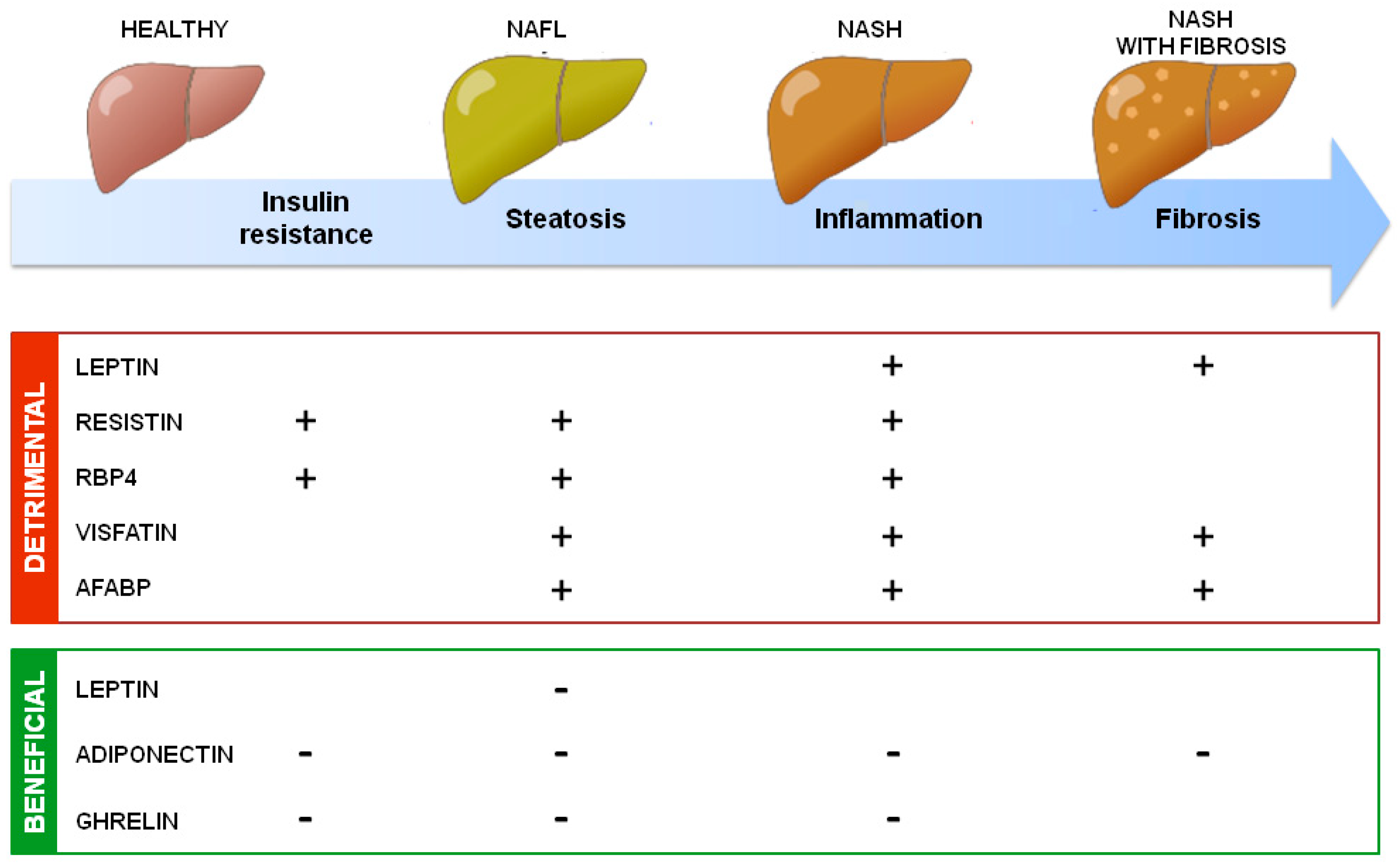
5. Adipokines in NAFLD: Evidence from Clinical Studies
5.1. Ghrelin
5.2. Resistin
5.3. Retinol Binding Protein 4 (RBP4)
5.4. Visfatin
5.5. Chemerin
5.6. Adipocyte Fatty Acid-Binding Protein (AFABP)
6. Therapeutic Interventions and Modulation of Adipokines’ Levels
7. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Leoni, S.; Tovoli, F.; Napoli, L.; Serio, I.; Ferri, S.; Bolondi, L. Current guidelines for the management of non-alcoholic fatty liver disease: A systematic review with comparative analysis. World J. Gastroenterol. 2018, 24, 3361–3373. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Perakakis, N.; Mantzoros, C.S. Fatty liver in lipodystrophy: A review with a focus on therapeutic perspectives of adiponectin and/or leptin replacement. Metabolism 2019, 96, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, fat mass and immune system: Role for leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Francisco, V.; Pino, J.; González-Gay, M.Á.; Lago, F.; Karppinen, J.; Tervonen, O.; Mobasheri, A.; Gualillo, O. A new immunometabolic perspective of intervertebral disc degeneration. Nat. Rev. Rheumatol. 2022, 18, 47–60. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipokines in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1062–1079. [Google Scholar] [CrossRef]
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 Diseases and Injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Papatheodoridi, M.; Cholongitas, E. Diagnosis of Non-alcoholic Fatty Liver Disease (NAFLD): Current Concepts. Curr. Pharm. Des. 2018, 24, 4574–4586. [Google Scholar] [CrossRef]
- Aller, R.; Fernández-Rodríguez, C.; lo Iacono, O.; Bañares, R.; Abad, J.; Carrión, J.A.; García-Monzón, C.; Caballería, J.; Berenguer, M.; Rodríguez-Perálvarez, M.; et al. Consensus document. Management of non-alcoholic fatty liver disease (NAFLD). Clinical practice guideline. Gastroenterol. Hepatol. 2018, 41, 328–349. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Sinha, R.A. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front. Biosci. Landmark 2021, 26, 206–237. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2017, 8, 1. [Google Scholar]
- Azzu, V.; Vacca, M.; Virtue, S.; Allison, M.; Vidal-Puig, A. Adipose Tissue-Liver Cross Talk in the Control of Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1899–1912. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gómez, R.; Mobasheri, A.; Gualillo, O. Adipokines and inflammation: Is it a question of weight? Br. J. Pharmacol. 2018, 175, 1569–1579. [Google Scholar] [CrossRef]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2020, 21, 157. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Leibel, R.L. Role of leptin in energy homeostasis in humans. J. Endocrinol. 2014, 223, T83–T96. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Uña, M.; López-Mancheño, Y.; Diéguez, C.; Fernández-Rojo, M.A.; Novelle, M.G. Unraveling the role of leptin in liver function and its relationship with liver diseases. Int. J. Mol. Sci. 2020, 21, 9368. [Google Scholar] [CrossRef]
- Jiménez-Cortegana, C.; García-Galey, A.; Tami, M.; Del Pino, P.; Carmona, I.; López, S.; Alba, G.; Sánchez-Margalet, V. Role of leptin in non-alcoholic fatty liver disease. Biomedicines 2021, 9, 762. [Google Scholar] [CrossRef] [PubMed]
- Heydari, M.; Cornide-Petronio, M.E.; Jiménez-Castro, M.B.; Peralta, C. Data on adiponectin from 2010 to 2020: Therapeutic target and prognostic factor for liver diseases? Int. J. Mol. Sci. 2020, 21, 5242. [Google Scholar] [CrossRef]
- Nguyen, T.M.D. Adiponectin: Role in Physiology and Pathophysiology. Int. J. Prev. Med. 2020, 11, 136. [Google Scholar] [CrossRef]
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef]
- Quiñones, M.; Fernø, J.; Al-Massadi, O. Ghrelin and liver disease. Rev. Endocr. Metab. Disord. 2020, 21, 45–56. [Google Scholar] [CrossRef]
- Acquarone, E.; Monacelli, F.; Borghi, R.; Nencioni, A.; Odetti, P. Resistin: A reappraisal. Mech. Ageing Dev. 2019, 178, 46–63. [Google Scholar] [CrossRef]
- Nono Nankam, P.A.; Blüher, M. Retinol-binding protein 4 in obesity and metabolic dysfunctions. Mol. Cell. Endocrinol. 2021, 531, 111312. [Google Scholar] [CrossRef]
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef]
- Carbone, F.; Liberale, L.; Bonaventura, A.; Vecchié, A.; Casula, M.; Cea, M.; Monacelli, F.; Caffa, I.; Bruzzone, S.; Montecucco, F.; et al. Regulation and Function of Extracellular Nicotinamide Phosphoribosyltransferase/Visfatin. Compr. Physiol. 2017, 7, 603–621. [Google Scholar] [PubMed]
- Helfer, G.; Wu, Q.F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Wu, X.; Xu, A.; Hoo, R.L.C. A-FABP in Metabolic Diseases and the Therapeutic Implications: An Update. Int. J. Mol. Sci. 2021, 22, 9386. [Google Scholar] [CrossRef] [PubMed]
- Micioni Di Bonaventura, E.; Botticelli, L.; Del Bello, F.; Giorgioni, G.; Piergentili, A.; Quaglia, W.; Cifani, C.; Micioni Di Bonaventura, M.V. Assessing the role of ghrelin and the enzyme ghrelin O-acyltransferase (GOAT) system in food reward, food motivation, and binge eating behavior. Pharmacol. Res. 2021, 172, 105847. [Google Scholar] [CrossRef]
- Li, Y.; Hai, J.; Li, L.; Chen, X.; Peng, H.; Cao, M.; Zhang, Q. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine 2013, 43, 376–386. [Google Scholar] [CrossRef]
- Mao, Y.; Cheng, J.; Yu, F.; Li, H.; Guo, C.; Fan, X. Ghrelin Attenuated Lipotoxicity via Autophagy Induction and Nuclear Factor-κB Inhibition. Cell. Physiol. Biochem. 2015, 37, 563–576. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, Q.; Qi, M.; Zhang, C.; Li, Z.; Zhang, W. Ghrelin ameliorates nonalcoholic steatohepatitis induced by chronic low-grade inflammation via blockade of Kupffer cell M1 polarization. J. Cell. Physiol. 2021, 236, 5121–5133. [Google Scholar] [CrossRef]
- Guillory, B.; Jawanmardi, N.; Iakova, P.; Anderson, B.; Zang, P.; Timchenko, N.A.; Garcia, J.M. Ghrelin deletion protects against age-associated hepatic steatosis by downregulating the C/EBPα-p300/DGAT1 pathway. Aging Cell 2018, 17, e12688. [Google Scholar] [CrossRef]
- Ezquerro, S.; Becerril, S.; Tuero, C.; Méndez-Giménez, L.; Mocha, F.; Moncada, R.; Valentí, V.; Cienfuegos, J.A.; Catalán, V.; Gómez-Ambrosi, J.; et al. Role of ghrelin isoforms in the mitigation of hepatic inflammation, mitochondrial dysfunction, and endoplasmic reticulum stress after bariatric surgery in rats. Int. J. Obes. 2020, 44, 475–487. [Google Scholar] [CrossRef]
- Ezquerro, S.; Méndez-Giménez, L.; Becerril, S.; Moncada, R.; Valentí, V.; Catalán, V.; Gómez-Ambrosi, J.; Frühbeck, G.; Rodríguez, A. Acylated and desacyl ghrelin are associated with hepatic lipogenesis, β-oxidation and autophagy: Role in NAFLD amelioration after sleeve gastrectomy in obese rats. Sci. Rep. 2016, 6, 39942. [Google Scholar] [CrossRef]
- Dallak, M.A. Acylated ghrelin induces but deacylated ghrelin prevents hepatic steatosis and insulin resistance in lean rats: Effects on DAG/PKC/JNK pathway. Biomed. Pharmacother. 2018, 105, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Muse, E.D.; Obici, S.; Bhanot, S.; Monia, B.P.; McKay, R.A.; Rajala, M.W.; Scherer, P.E.; Rossetti, L. Role of resistin in diet-induced hepatic insulin resistance. J. Clin. Investig. 2004, 114, 232–239. [Google Scholar] [CrossRef]
- Wen, F.; Shi, Z.; Liu, X.; Tan, Y.; Wei, L.; Zhu, X.; Zhang, H.; Zhu, X.; Meng, X.; Ji, W.; et al. Acute Elevated Resistin Exacerbates Mitochondrial Damage and Aggravates Liver Steatosis through AMPK/PGC-1α Signaling Pathway in Male NAFLD Mice. Horm. Metab. Res. 2021, 53, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Wang, J.; Whiteman, E.L.; Birnbaum, M.J.; Lazar, M.A. Activation of SOCS-3 by resistin. Mol. Cell. Biol. 2005, 25, 1569–1575. [Google Scholar] [CrossRef]
- Banerjee, R.R.; Rangwala, S.M.; Shapiro, J.S.; Rich, A.S.; Rhoades, B.; Qi, Y.; Wang, J.; Rajala, M.W.; Pocai, A.; Scherer, P.E.; et al. Regulation of fasted blood glucose by resistin. Science 2004, 303, 1195–1198. [Google Scholar] [CrossRef]
- Singhal, N.S.; Patel, R.T.; Qi, Y.; Lee, Y.S.; Ahima, R.S. Loss of resistin ameliorates hyperlipidemia and hepatic steatosis in leptin-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E331–E338. [Google Scholar] [CrossRef]
- Song, R.; Wang, X.; Mao, Y.; Li, H.; Li, Z.; Xu, W.; Wang, R.; Guo, T.; Jin, L.; Zhang, X.; et al. Resistin disrupts glycogen synthesis under high insulin and high glucose levels by down-regulating the hepatic levels of GSK3β. Gene 2013, 529, 50–56. [Google Scholar] [CrossRef]
- Garcia, C.C.; Piotrkowski, B.; Baz, P.; Poncino, D.; Benavides, J.; Colombato, L.; Toso, M.L.R.; Yantorno, S.; Descalzi, V.; Gondolesi, G.E.; et al. A Decreased Response to Resistin in Mononuclear Leukocytes Contributes to Oxidative Stress in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2021, 1, 3006–3016. [Google Scholar] [CrossRef]
- Beier, J.I.; Guo, L.; Von Montfort, C.; Kaiser, J.P.; Joshi-Barve, S.; Arteel, G.E. New role of resistin in lipopolysaccharide-induced liver damage in mice. J. Pharmacol. Exp. Ther. 2008, 325, 801–808. [Google Scholar] [CrossRef]
- Saxena, N.K.; Anania, F.A. Adipocytokines and hepatic fibrosis. Trends Endocrinol. Metab. 2015, 26, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.X.; Su, L.; Brymora, J.; Bird, C.; Xie, Q.; George, J.; Wang, J.H. Resistin mediates the hepatic stellate cell phenotype. World J. Gastroenterol. 2013, 19, 4475–4485. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mu, D.; Chen, H.; Li, D.; Song, J.; Zhong, Y.; Xia, M. Retinol-Binding Protein 4 Induces Hepatic Mitochondrial Dysfunction and Promotes Hepatic Steatosis. J. Clin. Endocrinol. Metab. 2016, 101, 4338–4348. [Google Scholar] [CrossRef]
- Saeed, A.; Bartuzi, P.; Heegsma, J.; Dekker, D.; Kloosterhuis, N.; de Bruin, A.; Jonker, J.W.; van de Sluis, B.; Faber, K.N. Impaired Hepatic Vitamin A Metabolism in NAFLD Mice Leading to Vitamin A Accumulation in Hepatocytes. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 309–325.e3. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Yuen, J.J.; Jiang, H.; Kahn, B.B.; Blaner, W.S. Adipocyte-specific overexpression of retinol-binding protein 4 causes hepatic steatosis in mice. Hepatology 2016, 64, 1534–1546. [Google Scholar] [CrossRef]
- Cioffi, C.L.; Racz, B.; Varadi, A.; Freeman, E.E.; Conlon, M.P.; Chen, P.; Zhu, L.; Kitchen, D.B.; Barnes, K.D.; Martin, W.H.; et al. Design, Synthesis, and Preclinical Efficacy of Novel Nonretinoid Antagonists of Retinol-Binding Protein 4 in the Mouse Model of Hepatic Steatosis. J. Med. Chem. 2019, 62, 5470–5500. [Google Scholar] [CrossRef]
- Heo, Y.J.; Choi, S.E.; Lee, N.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Visfatin exacerbates hepatic inflammation and fibrosis in a methionine-choline-deficient diet mouse model. J. Gastroenterol. Hepatol. 2021, 36, 2592–2600. [Google Scholar] [CrossRef]
- Heo, Y.J.; Choi, S.E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Visfatin Induces Inflammation and Insulin Resistance via the NF- κ B and STAT3 Signaling Pathways in Hepatocytes. J. Diabetes Res. 2019, 2019, 4021623. [Google Scholar] [CrossRef]
- Wang, L.F.; Wang, X.N.; Huang, C.C.; Hu, L.; Xiao, Y.F.; Guan, X.H.; Qian, Y.S.; Deng, K.Y.; Xin, H.B. Inhibition of NAMPT aggravates high fat diet-induced hepatic steatosis in mice through regulating Sirt1/AMPKα/SREBP1 signaling pathway. Lipids Health Dis. 2017, 16, 82. [Google Scholar] [CrossRef]
- Ilbeigi, D.; Nourbakhsh, M.; Pasalar, P.; Meshkani, R.; Afra, H.S.; Panahi, G.; Borji, M.; Sharifi, R. Nicotinamide phosphoribosyltransferase knockdown leads to lipid accumulation in HepG2 cells through the SIRT1-AMPK pathway. Cell J. 2020, 22, 125–132. [Google Scholar]
- Dall, M.; Hassing, A.S.; Niu, L.; Nielsen, T.S.; Ingerslev, L.R.; Sulek, K.; Trammell, S.A.J.; Gillum, M.P.; Barrès, R.; Larsen, S.; et al. Hepatocyte-specific perturbation of NAD+ biosynthetic pathways in mice induces reversible nonalcoholic steatohepatitis-like phenotypes. J. Biol. Chem. 2021, 297, 101388. [Google Scholar] [CrossRef] [PubMed]
- Bondue, B.; Wittamer, V.; Parmentier, M. Chemerin and its receptors in leukocyte trafficking, inflammation and metabolism. Cytokine Growth Factor Rev. 2011, 22, 331–338. [Google Scholar] [CrossRef]
- Bozaoglu, K.; Bolton, K.; McMillan, J.; Zimmet, P.; Jowett, J.; Collier, G.; Walder, K.; Segal, D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 2007, 148, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Divoux, A.; Poitou, C.; Basdevant, A.; Bouillot, J.L.; Bedossa, P.; Tordjman, J.; Eckel, J.; Clément, K. Chemerin Correlates with Markers for Fatty Liver in Morbidly Obese Patients and Strongly Decreases after Weight Loss Induced by Bariatric Surgery. J. Clin. Endocrinol. Metab. 2010, 95, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Liu, J.; Li, Y.; Dou, Z.; Li, N.; Suo, Y.; Ma, Y.; Sun, M.; Tian, Z.; Xu, L. Chemerin/CMKLR1 ameliorates nonalcoholic steatohepatitis by promoting autophagy and alleviating oxidative stress through the JAK2-STAT3 pathway. Peptides 2021, 135, 170422. [Google Scholar] [CrossRef] [PubMed]
- Pohl, R.; Rein-Fischboeck, L.; Meier, E.M.; Eisinger, K.; Krautbauer, S.; Buechler, C. Resolvin E1 and chemerin C15 peptide do not improve rodent non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2015, 98, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Gruben, N.; Vergara, M.A.; Kloosterhuis, N.J.; Van Der Molen, H.; Stoelwinder, S.; Youssef, S.; De Bruin, A.; Delsing, D.J.; Kuivenhoven, J.A.; Van De Sluis, B.; et al. Chemokine-like receptor 1 deficiency does not affect the development of insulin resistance and nonalcoholic fatty liver disease in mice. PLoS ONE 2014, 9, e96345. [Google Scholar] [CrossRef]
- Krautbauer, S.; Wanninger, J.; Eisinger, K.; Hader, Y.; Beck, M.; Kopp, A.; Schmid, A.; Weiss, T.S.; Dorn, C.; Buechler, C. Chemerin is highly expressed in hepatocytes and is induced in non-alcoholic steatohepatitis liver. Exp. Mol. Pathol. 2013, 95, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Hao, J.; Yi, Y.; Sauter, E.; Li, B. Regulation of macrophage functions by FABP-mediated inflammatory and metabolic pathways. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158964. [Google Scholar] [CrossRef]
- Hoo, R.L.C.; Lee, I.P.C.; Zhou, M.; Wong, J.Y.L.; Hui, X.; Xu, A.; Lam, K.S.L. Pharmacological inhibition of adipocyte fatty acid binding protein alleviates both acute liver injury and non-alcoholic steatohepatitis in mice. J. Hepatol. 2013, 58, 358–364. [Google Scholar] [CrossRef]
- Wu, X.; Shu, L.; Zhang, Z.; Li, J.; Zong, J.; Cheong, L.Y.; Ye, D.; Lam, K.S.L.; Song, E.; Wang, C.; et al. Adipocyte Fatty Acid Binding Protein Promotes the Onset and Progression of Liver Fibrosis via Mediating the Crosstalk between Liver Sinusoidal Endothelial Cells and Hepatic Stellate Cells. Adv. Sci. 2021, 8, 2003721. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, D.H.; Karelis, A.D.; Coderre, L.; Malita, F.; Fontaine, J.; Mignault, D.; Brochu, M.; Bastard, J.P.; Cianflone, K.; Doucet, E.; et al. Association of acylated and nonacylated ghrelin with insulin sensitivity in overweight and obese postmenopausal women. J. Clin. Endocrinol. Metab. 2007, 92, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Barazzoni, R.; Zanetti, M.; Ferreira, C.; Vinci, P.; Pirulli, A.; Mucci, M.P.; Dore, F.; Fonda, M.; Ciocchi, B.; Cattin, L.; et al. Relationships between desacylated and acylated ghrelin and insulin sensitivity in the metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 3935–3940. [Google Scholar] [CrossRef]
- Pacifico, L.; Poggiogalle, E.; Costantino, F.; Anania, C.; Ferraro, F.; Chiarelli, F.; Chiesa, C. Acylated and nonacylated ghrelin levels and their associations with insulin resistance in obese and normal weight children with metabolic syndrome. Eur. J. Endocrinol. 2009, 161, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Arslan, N.; Sayin, O.; Tokgoz, Y. Evaluation of serum xenin and ghrelin levels and their relationship with nonalcoholic fatty liver disease and insulin resistance in obese adolescents. Acad. Psychiatry 2014, 37, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- MacHado, M.V.; Coutinho, J.; Carepa, F.; Costa, A.; Proença, H.; Cortez-Pinto, H. How adiponectin, leptin, and ghrelin orchestrate together and correlate with the severity of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1166–1172. [Google Scholar] [CrossRef]
- Marchesini, G.; Pagotto, U.; Bugianesi, E.; De Iasio, R.; Manini, R.; Vanni, E.; Pasquali, R.; Melchionda, N.; Rizzetto, M. Low Ghrelin Concentrations in Nonalcoholic Fatty Liver Disease Are Related to Insulin Resistance. J. Clin. Endocrinol. Metab. 2003, 88, 5674–5679. [Google Scholar] [CrossRef]
- Estep, M.; Abawi, M.; Jarrar, M.; Wang, L.; Stepanova, M.; Elariny, H.; Moazez, A.; Goodman, Z.; Chandhoke, V.; Baranova, A.; et al. Association of obestatin, ghrelin, and inflammatory cytokines in obese patients with non-alcoholic fatty liver disease. Obes. Surg. 2011, 21, 1750–1757. [Google Scholar] [CrossRef]
- Tabaeian, S.P.; Mahmoudi, T.; Sabzikarian, M.; Rezamand, G.; Dabiri, R.; Nobakht, H.; Asadi, A.; Farahani, H.; Mansour-Ghanaei, F.; Zali, M.R. The leu72met (Rs696217 g>t) polymorphism of the ghrelin gene might be a protective factor for nonalcoholic fatty liver disease. J. Gastrointest. Liver Dis. 2021, 30, 233–239. [Google Scholar] [CrossRef]
- Rezamand, G.; Mahmoudi, T.; Tabaeian, S.P.; Farahani, H.; Shahinmehr, F.; Nobakht, H.; Dabiri, R.; Asadi, A.; Mansour-Ghanaei, F.; Zali, M.R. The “GG” genotype of rs26802 variant in the ghrelin gene is a potential protective factor against nonalcoholic fatty liver disease. Physiol. Int. 2021, 108, 342–352. [Google Scholar] [CrossRef]
- Bekaert, M.; Verhelst, X.; Geerts, A.; Lapauw, B.; Calders, P. Association of recently described adipokines with liver histology in biopsy-proven non-alcoholic fatty liver disease: A systematic review. Obes. Rev. 2016, 17, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, C.; Ardicoglu, Y.; Bulut, S.; Boyacioglu, S. Serum retinol-binding protein 4 in patients with nonalcoholic fatty liver disease: Does it have a significant impact on pathogenesis? Eur. J. Gastroenterol. Hepatol. 2010, 22, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; De Luis, D.A.; Fernandez, L.; Calle, F.; Velayos, B.; Olcoz, J.L.; Izaola, O.; Sagrado, M.G.; Conde, R.; Gonzalez, J.M. Influence of insulin resistance and adipokines in the grade of steatosis of nonalcoholic fatty liver disease. Dig. Dis. Sci. 2008, 53, 1088–1092. [Google Scholar] [CrossRef]
- Pagano, C.; Soardo, G.; Pilon, C.; Milocco, C.; Basan, L.; Milan, G.; Donnini, D.; Faggian, D.; Mussap, M.; Plebani, M.; et al. Increased serum resistin in nonalcoholic fatty liver disease is related to liver disease severity and not to insulin resistance. J. Clin. Endocrinol. Metab. 2006, 91, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Şenateş, E.; Çolak, Y.; Yeşil, A.; Coşkunpinar, E.; Şahin, Ö.; Kahraman, Ö.T.; Erkalma Şenateş, B.; Tuncer, I. Circulating resistin is elevated in patients with non-alcoholic fatty liver disease and is associated with steatosis, portal inflammation, insulin resistance and nonalcoholic steatohepatitis scores. Minerva Med. 2012, 103, 369–376. [Google Scholar] [PubMed]
- Younossi, Z.M.; Jarrar, M.; Nugent, C.; Randhawa, M.; Afendy, M.; Stepanova, M.; Rafiq, N.; Goodman, Z.; Chandhoke, V.; Baranova, A. A novel diagnostic biomarker panel for obesity-related nonalcoholic steatohepatitis (NASH). Obes. Surg. 2008, 18, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Page, S.; Rafiq, N.; Birerdinc, A.; Stepanova, M.; Hossain, N.; Afendy, A.; Younoszai, Z.; Goodman, Z.; Baranova, A. A biomarker panel for non-alcoholic steatohepatitis (NASH) and NASH-related fibrosis. Obes. Surg. 2011, 21, 431–439. [Google Scholar] [CrossRef]
- Argentou, M.; Tiniakos, D.G.; Karanikolas, M.; Melachrinou, M.; Makri, M.G.; Kittas, C.; Kalfarentzos, F. Adipokine serum levels are related to liver histology in severely obese patients undergoing bariatric surgery. Obes. Surg. 2009, 19, 1313–1323. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Durazzo, M.; Biroli, G.; Carello, M.; Fagà, E.; Pacini, G.; De Michieli, F.; Rabbione, L.; Premoli, A.; et al. Adipokines in NASH: Postprandial lipid metabolism as a link between adiponectin and liver disease. Hepatology 2005, 42, 1175–1183. [Google Scholar] [CrossRef]
- Jarrar, M.H.; Baranova, A.; Collantes, R.; Ranard, B.; Stepanova, M.; Bennett, C.; Fang, Y.; Elariny, H.; Goodman, Z.; Chandhoke, V.; et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 27, 412–421. [Google Scholar] [CrossRef]
- Wong, V.W.S.; Hui, A.Y.; Tsang, S.W.C.; Chan, J.L.Y.; Tse, A.M.L.; Chan, K.F.; So, W.Y.; Cheng, A.Y.S.; Ng, W.F.; Wong, G.L.H.; et al. Metabolic and adipokine profile of Chinese patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2006, 4, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Koehler, E.; Swain, J.; Sanderson, S.; Krishnan, A.; Watt, K.; Charlton, M. Growth hormone, dehydroepiandrosterone and adiponectin levels in non-alcoholic steatohepatitis: An endocrine signature for advanced fibrosis in obese patients. Liver Int. 2012, 32, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Baltieri, L.; Chaim, E.A.; Chaim, F.D.M.; Utrini, M.P.; Gestic, M.A.; Cazzo, E. Correlation between nonalcoholic fatty liver disease features and levels of adipokines and inflammatory cytokines among morbidly obese individuals. Arq. Gastroenterol. 2018, 55, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Ajmera, V.; Perito, E.R.; Bass, N.M.; Terrault, N.A.; Yates, K.P.; Gill, R.; Loomba, R.; Diehl, A.M.; Aouizerat, B.E. Novel plasma biomarkers associated with liver disease severity in adults with nonalcoholic fatty liver disease. Hepatology 2017, 65, 65–77. [Google Scholar] [CrossRef]
- Shen, C.; Zhao, C.Y.; Wang, W.; Wang, Y.D.; Sun, H.; Cao, W.; Yu, W.Y.; Zhang, L.; Ji, R.; Li, M.; et al. The relationship between hepatic resistin overexpression and inflammation in patients with nonalcoholic steatohepatitis. BMC Gastroenterol. 2014, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Carpino, G.; Alisi, A.; Franchitto, A.; Alpini, G.; De Vito, R.; Onori, P.; Alvaro, D.; Gaudio, E. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012, 56, 2142–2153. [Google Scholar] [CrossRef]
- Gierej, P.; Gierej, B.; Kalinowski, P.; Wróblewski, T.; Paluszkiewicz, R.; Kobryń, K.; Ziarkiewicz-Wróblewska, B. Expression of resistin in the liver of patients with Non-Alcoholic fatty liver disease. Pol. J. Pathol. 2017, 68, 225–233. [Google Scholar] [CrossRef]
- Kohan, L.; Safarpur, M.; Abdollahi, H. Omentin-1 rs2274907 and resistin rs1862513 polymorphisms influence genetic susceptibility to nonalcoholic fatty liver disease. Mol. Biol. Res. Commun. 2016, 5, 11. [Google Scholar]
- Balagopal, P.; Graham, T.E.; Kahn, B.B.; Altomare, A.; Funanage, V.; George, D. Reduction of elevated serum retinol binding protein in obese children by lifestyle intervention: Association with subclinical inflammation. J. Clin. Endocrinol. Metab. 2007, 92, 1971–1974. [Google Scholar] [CrossRef]
- Haider, D.G.; Schindler, K.; Prager, G.; Bohdjalian, A.; Luger, A.; Wolzt, M.; Ludvik, B. Serum retinol-binding protein 4 is reduced after weight loss in morbidly obese subjects. J. Clin. Endocrinol. Metab. 2007, 92, 1168–1171. [Google Scholar] [CrossRef]
- Stefan, N.; Hennige, A.M.; Staiger, H.; Machann, J.; Schick, F.; Schleicher, E.; Fritsche, A.; Häring, H.U. High circulating retinol-binding protein 4 is associated with elevated liver fat but not with total, subcutaneous, visceral, or intramyocellular fat in humans. Diabetes Care 2007, 30, 1173–1178. [Google Scholar] [CrossRef]
- Seo, J.A.; Kim, N.H.; Park, S.Y.; Kim, H.Y.; Ryu, O.H.; Lee, K.W.; Lee, J.; Kim, D.L.; Choi, K.M.; Baik, S.H.; et al. Serum retinol-binding protein 4 levels are elevated in non-alcoholic fatty liver disease. Clin. Endocrinol. 2008, 68, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Yang, Y.J. Serum retinol-binding protein 4 is independently associated with pediatric NAFLD and fasting triglyceride level. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chen, H.; Ju, H.; Sun, M. Circulating retinol binding protein 4 levels in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Lipids Health Dis. 2017, 16, 180. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.; Zhang, H.; Pang, J.; Lin, J.; Xu, X.; Yang, L.; Ma, J.; Ling, W.; Chen, Y. Circulating retinol-binding protein 4 is associated with the development and regression of non-alcoholic fatty liver disease. Diabetes Metab. 2020, 46, 119–128. [Google Scholar] [CrossRef]
- Karamfilova, V.; Gateva, A.; Alexiev, A.; Zheleva, N.; Velikova, T.; Ivanova-Boyanova, R.; Ivanova, R.; Cherkezov, N.; Kamenov, Z.; Mateva, L. The association between retinol-binding protein 4 and prediabetes in obese patients with nonalcoholic fatty liver disease. Arch. Physiol. Biochem. 2022, 128, 217–222. [Google Scholar] [CrossRef]
- Ismaiel, A.; Leucuta, D.C.; Popa, S.L.; Dumitrascu, D.L. Serum visfatin levels in nonalcoholic fatty liver disease and liver fibrosis: Systematic review and meta-analysis. J. Clin. Med. 2021, 10, 3029. [Google Scholar] [CrossRef]
- Genc, H.; Dogru, T.; Kara, M.; Tapan, S.; Ercin, C.N.; Acikel, C.; Karslioglu, Y.; Bagci, S. Association of plasma visfatin with hepatic and systemic inflammation in nonalcoholic fatty liver disease. Ann. Hepatol. 2013, 12, 380–387. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Papatheodorou, A.; Katsiki, E.; Patsiaoura, K.; Zafeiriadou, E.; Zavos, C.; Papadopoulou, E.; Terpos, E. Adipocytokines and cytokeratin-18 in patients with nonalcoholic fatty liver disease: Introduction of CHA index. Ann. Hepatol. 2013, 12, 749–757. [Google Scholar] [CrossRef]
- Yoon, M.Y.; Sung, J.M.; Song, C.S.; Lee, W.Y.; Rhee, E.J.; Shin, J.H.; Yoo, C.H.; Chae, S.W.; Kim, J.Y.; Jin, W.; et al. Enhanced A-FABP expression in visceral fat: Potential contributor to the progression of NASH. Clin. Mol. Hepatol. 2012, 18, 279–286. [Google Scholar] [CrossRef]
- Dahl, T.B.; Haukeland, J.W.; Yndestad, A.; Ranheim, T.; Gladhaug, I.P.; Damås, J.K.; Haaland, T.; Løberg, E.M.; Arntsen, B.; Birkeland, K.; et al. Intracellular Nicotinamide Phosphoribosyltransferase Protects against Hepatocyte Apoptosis and Is Down-Regulated in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2010, 95, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Kukla, M.; Ciupińska-Kajor, M.; Kajor, M.; Wylezoł, M.; Zwirska-Korczala, K.; Hartleb, M.; Berdowska, A.; Mazur, W. Liver visfatin expression in morbidly obese patients with nonalcoholic fatty liver disease undergoing bariatric surgery. Polish J. Pathol. 2010, 61, 147–153. [Google Scholar]
- Auguet, T.; Terra, X.; Porras, J.A.; Orellana-Gavaldà, J.M.; Martinez, S.; Aguilar, C.; Lucas, A.; Pellitero, S.; Hernández, M.; Del Castillo, D.; et al. Plasma visfatin levels and gene expression in morbidly obese women with associated fatty liver disease. Clin. Biochem. 2013, 46, 202–208. [Google Scholar] [CrossRef]
- Gaddipati, R.; Mitnala, S.; Padaki, N.; Mukherjee, R.M.; Sekaran, A.; Jayaraj-Mansard, M.; Rabella, P.; Rao-Guduru, V.; Reddy-Duwuru, N. Visceral adipose tissue visfatin in nonalcoholic fatty liver disease. Ann. Hepatol. 2010, 9, 266–270. [Google Scholar] [CrossRef]
- Amirkalali, B.; Sohrabi, M.R.; Esrafily, A.; Jalali, M.; Gholami, A.; Hosseinzadeh, P.; Keyvani, H.; Shidfar, F.; Zamani, F. Association between Nicotinamide Phosphoribosyltransferase and de novo Lipogenesis in Nonalcoholic Fatty Liver Disease. Med. Princ. Pract. 2017, 26, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; De Luis, D.A.; Izaola, O.; Sagrado, M.G.; Conde, R.; Velasco, M.C.; Alvarez, T.; Pacheco, D.; González, J.M. Influence of visfatin on histopathological changes of non-alcoholic fatty liver disease. Dig. Dis. Sci. 2009, 54, 1772–1777. [Google Scholar] [CrossRef] [PubMed]
- Elkabany, Z.A.; Hamza, R.T.; Ismail, E.A.R.; Elsharkawy, A.; Yosry, A.; Musa, S.; Khalaf, M.A.; Elgawesh, R.M.; Esmat, G. Serum visfatin level as a noninvasive marker for nonalcoholic fatty liver disease in children and adolescents with obesity: Relation to transient elastography with controlled attenuation parameter. Eur. J. Gastroenterol. Hepatol. 2020, 32, 1008–1016. [Google Scholar] [CrossRef]
- Johannsen, K.; Flechtner-Mors, M.; Kratzer, W.; Koenig, W.; Boehm, B.O.; Schmidberger, J. Association Between Visfatin and Hepatic Steatosis in the General Population During Long-Term Follow-Up. Horm. Metab. Res. 2019, 51, 602–607. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Wang, H. Correlation of blood glucose, serum chemerin and insulin resistance with NAFLD in patients with type 2 diabetes mellitus. Exp. Ther. Med. 2018, 15, 2936. [Google Scholar] [CrossRef]
- Kłusek-Oksiuta, M.; Bialokoz-Kalinowska, I.; Tarasów, E.; Wojtkowska, M.; Werpachowska, I.; Lebensztejn, D.M. Chemerin as a novel non-invasive serum marker of intrahepatic lipid content in obese children. Ital. J. Pediatr. 2014, 40, 84. [Google Scholar] [CrossRef]
- Ren, Q.; Wang, H.; Zeng, Y.; Fang, X.; Wang, M.; Li, D.; Huang, W.; Xu, Y. Circulating chemerin levels in metabolic-associated fatty liver disease: A systematic review and meta-analysis. Lipids Health Dis. 2022, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, M.; Ouwens, D.M.; Hörbelt, T.; Van de Velde, F.; Fahlbusch, P.; Herzfeld de Wiza, D.; Van Nieuwenhove, Y.; Calders, P.; Praet, M.; Hoorens, A.; et al. Reduced expression of chemerin in visceral adipose tissue associates with hepatic steatosis in patients with obesity. Obesity 2016, 24, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- Pohl, R.; Haberl, E.M.; Rein-Fischboeck, L.; Zimny, S.; Neumann, M.; Aslanidis, C.; Schacherer, D.; Krautbauer, S.; Eisinger, K.; Weiss, T.S.; et al. Hepatic chemerin mRNA expression is reduced in human nonalcoholic steatohepatitis. Eur. J. Clin. Investig. 2017, 47, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kajor, M.; Kukla, M.; Waluga, M.; Liszka, Ł.; Dyaczyński, M.; Kowalski, G.; Żądło, D.; Berdowska, A.; Chapuła, M.; Kostrząb-Zdebel, A.; et al. Hepatic chemerin mRNA in morbidly obese patients with nonalcoholic fatty liver disease. Pol. J. Pathol. 2017, 68, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Döcke, S.; Lock, J.F.; Birkenfeld, A.L.; Hoppe, S.; Lieske, S.; Rieger, A.; Raschzok, N.; Sauer, I.M.; Florian, S.; Osterhoff, M.A.; et al. Elevated hepatic chemerin mRNA expression in human non-alcoholic fatty liver disease. Eur. J. Endocrinol. 2013, 169, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.B.; Kim, S.M.; Cho, G.J.; Choi, K.M. Serum AFBP levels are elevated in patients with nonalcoholic fatty liver disease. Scand. J. Gastroenterol. 2014, 49, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, X.; Pan, X.; He, X.; Wang, Y.; Bao, Y. Serum adipocyte fatty acid-binding protein levels: An indicator of non-alcoholic fatty liver disease in Chinese individuals. Liver Int. 2019, 39, 568–574. [Google Scholar] [CrossRef]
- Milner, K.L.; van der Poorten, D.; Xu, A.; Bugianesi, E.; Kench, J.G.; Lam, K.S.L.; Chisholm, D.J.; George, J. Adipocyte fatty acid binding protein levels relate to inflammation and fibrosis in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1926–1934. [Google Scholar] [CrossRef]
- Shen, J.; Chan, H.L.Y.; Wong, G.L.H.; Choi, P.C.L.; Chan, A.W.H.; Chan, H.Y.; Chim, A.M.L.; Yeung, D.K.W.; Chan, F.K.L.; Woo, J.; et al. Non-invasive diagnosis of non-alcoholic steatohepatitis by combined serum biomarkers. J. Hepatol. 2012, 56, 1363–1370. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef]
- Salman, A.A.; Sultan, A.A.E.A.; Abdallah, A.; Abdelsalam, A.; Mikhail, H.M.S.; Tourky, M.; Omar, M.G.; Youssef, A.; Ahmed, R.A.; Elkassar, H.; et al. Effect of weight loss induced by laparoscopic sleeve gastrectomy on liver histology and serum adipokine levels. J. Gastroenterol. Hepatol. 2020, 35, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, V.; Wills, R.; DeLany, J.P.; Kershaw, E.E.; Behari, J. Differential Impact of Weight Loss on Nonalcoholic Fatty Liver Resolution in a North American Cohort with Obesity. Obesity 2017, 25, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Kader, S.M.; Al-Shreef, F.M.; Al-Jiffri, O.H. Biochemical parameters response to weight loss in patients with non-alcoholic steatohepatitis. Afr. Health Sci. 2016, 16, 242. [Google Scholar] [CrossRef]
- Boutari, C.; Perakakis, N.; Mantzoros, C.S. Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol. Metab. 2018, 33, 33–43. [Google Scholar] [CrossRef]
- Meir, A.Y.; Rinott, E.; Tsaban, G.; Zelicha, H.; Kaplan, A.; Rosen, P.; Shelef, I.; Youngster, I.; Shalev, A.; Blüher, M.; et al. Effect of green-Mediterranean diet on intrahepatic fat: The DIRECT PLUS randomised controlled trial. Gut 2021, 70, 2085–2095. [Google Scholar] [CrossRef]
- Deibert, P.; Lazaro, A.; Schaffner, D.; Berg, A.; Koenig, D.; Kreisel, W.; Baumstark, M.W.; Steinmann, D.; Buechert, M.; Lange, T. Comprehensive lifestyle intervention vs soy protein-based meal regimen in non-alcoholic steatohepatitis. World J. Gastroenterol. 2019, 25, 1116. [Google Scholar] [CrossRef]
- Nanjan, M.J.; Mohammed, M.; Prashantha Kumar, B.R.; Chandrasekar, M.J.N. Thiazolidinediones as antidiabetic agents: A critical review. Bioorg. Chem. 2018, 77, 548–567. [Google Scholar] [CrossRef]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients with Nonalcoholic Steatohepatitis with vs Without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. 2018, 16, 558–566.e2. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Mantzoros, C.S. Adiponectin as a target for the treatment of nonalcoholic steatohepatitis with thiazolidinediones: A systematic review. Metabolism 2016, 65, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Sabatini, S.; Carli, F.; Gaggini, M.; Bril, F.; Belfort-DeAguiar, R.; Positano, V.; Barb, D.; Kadiyala, S.; Harrison, S.; et al. PPAR-γ-induced changes in visceral fat and adiponectin levels are associated with improvement of steatohepatitis in patients with NASH. Liver Int. 2021, 41, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Lutchman, G.; Modi, A.; Kleiner, D.E.; Promrat, K.; Heller, T.; Ghany, M.; Borg, B.; Loomba, R.; Liang, T.J.; Premkumar, A.; et al. The effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitis. Hepatology 2007, 46, 424–429. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Rosiglitazone revisited: An updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch. Intern. Med. 2010, 170, 1191–1201. [Google Scholar] [CrossRef]
- Lewis, J.D.; Ferrara, A.; Peng, T.; Hedderson, M.; Bilker, W.B.; Quesenberry, C.P.; Vaughn, D.J.; Nessel, L.; Selby, J.; Strom, B.L. Risk of Bladder Cancer Among Diabetic Patients Treated With PioglitazoneInterim report of a longitudinal cohort study. Diabetes Care 2011, 34, 916–922. [Google Scholar] [CrossRef]
- Athyros, V.G.; Polyzos, S.A.; Kountouras, J.; Katsiki, N.; Anagnostis, P.; Doumas, M.; Mantzoros, C.S. Non-Alcoholic Fatty Liver Disease Treatment in Patients with Type 2 Diabetes Mellitus; New Kids on the Block. Curr. Vasc. Pharmacol. 2020, 18, 172–181. [Google Scholar] [CrossRef]
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef]
- Zhou, J.; Massey, S.; Story, D.; Li, L. Metformin: An Old Drug with New Applications. Int. J. Mol. Sci. 2018, 19, 2863. [Google Scholar] [CrossRef]
- Said, A.; Akhter, A. Meta-analysis of randomized controlled trials of pharmacologic agents in non-alcoholic steatohepatitis. Ann. Hepatol. 2017, 16, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Sun, F.; Li, L.; Jiang, D.; Li, X.; Sun, A.; Pan, Z.; Lou, N.; Zhang, L.; Lou, F. Therapeutic Effect of Metformin on Chemerin in Non-Obese Patients with Non-Alcoholic Fatty Liver Disease (NAFLD). Clin. Lab. 2015, 61, 1409–1414. [Google Scholar] [PubMed]
- Yang, L.; Song, M.Q.; Zhang, Q.L.; Shou, L.; Zang, S.F.; Yang, Y.L. Effect of piglitazone and metformin on retinol-binding protein-4 and adiponectin in patients with type 2 diabetes mellitus complicated with non-alcohol fatty acid liver diseases. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2014, 36, 309–312. [Google Scholar] [PubMed]
- Fan, H.; Pan, Q.; Xu, Y.; Yang, X. Exenatide improves type 2 diabetes concomitant with non-alcoholic fatty liver disease. Arq. Bras. Endocrinol. Metabol. 2013, 57, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Shargorodsky, M.; Omelchenko, E.; Matas, Z.; Boaz, M.; Gavish, D. Relation between augmentation index and adiponectin during one-year metformin treatment for nonalcoholic steatohepatosis: Effects beyond glucose lowering? Cardiovasc. Diabetol. 2012, 11, 61. [Google Scholar] [CrossRef]
- Wei, Q.; Xu, X.; Guo, L.; Li, J.; Li, L. Effect of SGLT2 Inhibitors on Type 2 Diabetes Mellitus With Non-Alcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. Front. Endocrinol. 2021, 12, 635556. [Google Scholar] [CrossRef]
- Mo, M.; Huang, Z.; Liang, Y.; Liao, Y.; Xia, N. The safety and efficacy evaluation of sodium-glucose co-transporter 2 inhibitors for patients with non-alcoholic fatty liver disease: An updated meta-analysis. Dig. Liver Dis. 2022, 54, 461–468. [Google Scholar] [CrossRef]
- Phrueksotsai, S.; Pinyopornpanish, K.; Euathrongchit, J.; Leerapun, A.; Phrommintikul, A.; Buranapin, S.; Chattipakorn, N.; Thongsawat, S. The effects of dapagliflozin on hepatic and visceral fat in type 2 diabetes patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2021, 36, 2952–2959. [Google Scholar] [CrossRef]
- Liu, M.; Liu, F. Regulation of adiponectin multimerization, signaling and function. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 25–31. [Google Scholar] [CrossRef]
- Narasimhan, M.L.; Coca, M.A.; Jin, J.; Yamauchi, T.; Ito, Y.; Kadowaki, T.; Kim, K.K.; Pardo, J.M.; Damsz, B.; Hasegawa, P.M.; et al. Osmotin Is a Homolog of Mammalian Adiponectin and Controls Apoptosis in Yeast through a Homolog of Mammalian Adiponectin Receptor. Mol. Cell 2005, 17, 171–180. [Google Scholar] [CrossRef]
- Ahmad, A.; Ali, T.; Kim, M.W.; Khan, A.; Jo, M.H.; Rehman, S.U.; Khan, M.S.; Abid, N.B.; Khan, M.; Ullah, R.; et al. Adiponectin homolog novel osmotin protects obesity/diabetes-induced NAFLD by upregulating AdipoRs/PPARα signaling in ob/ob and db/db transgenic mouse models. Metabolism 2019, 90, 31–43. [Google Scholar] [CrossRef]
- Meehan, C.A.; Cochran, E.; Kassai, A.; Brown, R.J.; Gorden, P. Metreleptin for injection to treat the complications of leptin deficiency in patients with congenital or acquired generalized lipodystrophy. Expert Rev. Clin. Pharmacol. 2016, 9, 59–68. [Google Scholar] [CrossRef]
- Akinci, B.; Subauste, A.; Ajluni, N.; Esfandiari, N.H.; Meral, R.; Neidert, A.H.; Eraslan, A.; Hench, R.; Rus, D.; Mckenna, B.; et al. Metreleptin therapy for nonalcoholic steatohepatitis: Open-label therapy interventions in two different clinical settings. Med 2021, 2, 814–835.e6. [Google Scholar] [CrossRef] [PubMed]
- Javor, E.D.; Ghany, M.G.; Cochran, E.K.; Oral, E.A.; DePaoli, A.M.; Premkumar, A.; Kleiner, D.E.; Gorden, P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology 2005, 41, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Safar Zadeh, E.; Lungu, A.O.; Cochran, E.K.; Brown, R.J.; Ghany, M.G.; Heller, T.; Kleiner, D.E.; Gorden, P. The liver diseases of lipodystrophy: The long-term effect of leptin treatment. J. Hepatol. 2013, 59, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Pei, H.; Ma, L.; Ran, Y.; Chen, J.; Wang, G.; Chen, L. Synthesis and biological evaluation of novel urea-and guanidine-based derivatives for the treatment of obesity-related hepatic steatosis. Molecules 2014, 19, 6163–6183. [Google Scholar] [PubMed]
- Marchesini, G.; Day, C.P.; Dufour, J.F.; Canbay, A.; Nobili, V.; Ratziu, V.; Tilg, H.; Roden, M.; Gastaldelli, A.; Yki-Jarvinen, H.; et al. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Hardy, T.; Wonders, K.; Younes, R.; Aithal, G.P.; Aller, R.; Allison, M.; Bedossa, P.; Betsou, F.; Boursier, J.; Brosnan, M.J.; et al. The European NAFLD Registry: A real-world longitudinal cohort study of nonalcoholic fatty liver disease. Contemp. Clin. Trials 2020, 98, 106175. [Google Scholar] [CrossRef]
- Krieg, L.; Schaffert, A.; Kern, M.; Landgraf, K.; Wabitsch, M.; Beck-Sickinger, A.G.; Koerner, A.; Blüher, M.; von Bergen, M.; Schubert, K. An MRM-Based Multiplexed Quantification Assay for Human Adipokines and Apolipoproteins. Molecules 2020, 25, 775. [Google Scholar] [CrossRef]
- Magni, P.; Liuzzi, A.; Ruscica, M.; Dozio, E.; Ferrario, S.; Bussi, I.; Minocci, A.; Castagna, A.; Motta, M.; Savia, G. Free and bound plasma leptin in normal weight and obese men and women: Relationship with body composition, resting energy expenditure, insulin-sensitivity, lipid profile and macronutrient preference. Clin. Endocrinol. 2005, 62, 189–196. [Google Scholar] [CrossRef]
- Francisco, V.; Tovar, S.; Conde, J.; Pino, J.; Mera, A.; Lago, F.; González-Gay, M.A.; Dieguez, C.; Gualillo, O. Levels of the novel endogenous antagonist of ghrelin receptor, liver-enriched antimicrobial peptide-2, in patients with rheumatoid arthritis. Nutrients 2020, 12, 1006. [Google Scholar] [CrossRef]
- Schultz, A.; Saville, B.R.; Marsh, J.A.; Snelling, T.L. An introduction to clinical trial design. Paediatr. Respir. Rev. 2019, 32, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Hollis, S.; Fletcher, C.; Lynn, F.; Urban, H.J.; Branson, J.; Burger, H.U.; Tudur Smith, C.; Sydes, M.R.; Gerlinger, C. Best practice for analysis of shared clinical trial data. BMC Med. Res. Methodol. 2016, 16, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Reimer, K.C.; Wree, A.; Roderburg, C.; Tacke, F. New drugs for NAFLD: Lessons from basic models to the clinic. Hepatol. Int. 2020, 14, 8–23. [Google Scholar] [CrossRef]
- Mazza, G.; Al-Akkad, W.; Rombouts, K. Engineering in vitro models of hepatofibrogenesis. Adv. Drug Deliv. Rev. 2017, 121, 147–157. [Google Scholar] [CrossRef]
- Abdolahi, A.; Vahabzadeh, Z.; Izadpanah, E.; Moloudi, M.R. Vaspin attenuates steatosis-induced fibrosis via GRP78 receptor by targeting AMPK signaling pathway. J. Physiol. Biochem. 2022, 78, 185–197. [Google Scholar] [CrossRef]
- Waluga, M.; Kukla, M.; Kotulski, R.; Zorniak, M.; Boryczka, G.; Kajor, M.; Ciupinska-Kajor, M.; Lekstan, A.; Olczyk, P.; Waluga, E. Omentin, vaspin and irisin in chronic liver diseases. J. Physiol. Pharmacol. 2019, 70, 277–285. [Google Scholar]
- Aliasghari, F.; Izadi, A.; Jabbari, M.; Imani, B.; Gargari, B.P.; Asjodi, F.; Ebrahimi, S. Are Vaspin and Omentin-1 Related to Insulin Resistance, Blood Pressure and Inflammation in NAFLD Patients? J. Med. Biochem. 2018, 37, 470–475. [Google Scholar] [CrossRef]
- Gu, Y.; Luo, J.; Chen, Q.; Qiu, Y.; Zhou, Y.; Wang, X.; Qian, X.; Liu, Y.; Xie, J.; Xu, Z.; et al. Inverse Association of Serum Adipsin with the Remission of Nonalcoholic Fatty-Liver Disease: A 3-Year Community-Based Cohort Study. Ann. Nutr. Metab. 2022, 78, 21–32. [Google Scholar] [CrossRef]
- Wang, Y.; Song, J.; Bian, H.; Bo, J.; Lv, S.; Pan, W.; Lv, X. Apelin promotes hepatic fibrosis through ERK signaling in LX-2 cells. Mol. Cell. Biochem. 2019, 460, 205–215. [Google Scholar] [CrossRef]
- Montazerifar, F.; Bakhshipour, A.R.; Karajibani, M.; Torki, Z.; Dashipour, A.R. Serum omentin-1, vaspin, and apelin levels and central obesity in patients with nonalcoholic fatty liver disease. J. Res. Med. Sci. 2017, 22, 70. [Google Scholar]
- Khaleel, E.F.; Abdel-Aleem, G.A. Obestatin protects and reverses nonalcoholic fatty liver disease and its associated insulin resistance in rats via inhibition of food intake, enhancing hepatic adiponectin signaling, and blocking ghrelin acylation. Arch. Physiol. Biochem. 2019, 125, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L.; Shao, W.-H.; Vanniasinghe, A.S.; Amon, M.A.; Holub, M.C.; Kovalszky, I.; Wade, J.D.; Doll, M.; Cohen, P.L.; Manolios, N.; et al. Toward understanding the role of leptin and leptin receptor antagonism in preclinical models of rheumatoid arthritis. Peptides 2011, 32, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- DePaoli, A.M. Leptin in common obesity and associated disorders of metabolism. J. Endocrinol. 2014, 223, T71–T81. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Adipokine | Description | Receptor/Signaling | Functions | Refs. |
| Leptin |
|
|
| [6,22,23] |
| Adiponectin (ACRP30, AdipoQ, GBP28 or apM1) |
|
|
| [24,25] |
| Ghrelin |
|
|
| [26,27] |
| Resistin (ADSF or FIZZ3) |
|
|
| [28] |
| RBP4 |
|
|
| [29] |
| Visfatin (PBEF or NAMPT) |
|
|
| [30,31] |
| Chemerin (TIG2 or RARRES2) |
|
|
| [32] |
| AFABP (ap2 or FABP4) |
|
|
| [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francisco, V.; Sanz, M.J.; Real, J.T.; Marques, P.; Capuozzo, M.; Ait Eldjoudi, D.; Gualillo, O. Adipokines in Non-Alcoholic Fatty Liver Disease: Are We on the Road toward New Biomarkers and Therapeutic Targets? Biology 2022, 11, 1237. https://doi.org/10.3390/biology11081237
Francisco V, Sanz MJ, Real JT, Marques P, Capuozzo M, Ait Eldjoudi D, Gualillo O. Adipokines in Non-Alcoholic Fatty Liver Disease: Are We on the Road toward New Biomarkers and Therapeutic Targets? Biology. 2022; 11(8):1237. https://doi.org/10.3390/biology11081237
Chicago/Turabian StyleFrancisco, Vera, Maria Jesus Sanz, José T. Real, Patrice Marques, Maurizio Capuozzo, Djedjiga Ait Eldjoudi, and Oreste Gualillo. 2022. "Adipokines in Non-Alcoholic Fatty Liver Disease: Are We on the Road toward New Biomarkers and Therapeutic Targets?" Biology 11, no. 8: 1237. https://doi.org/10.3390/biology11081237
APA StyleFrancisco, V., Sanz, M. J., Real, J. T., Marques, P., Capuozzo, M., Ait Eldjoudi, D., & Gualillo, O. (2022). Adipokines in Non-Alcoholic Fatty Liver Disease: Are We on the Road toward New Biomarkers and Therapeutic Targets? Biology, 11(8), 1237. https://doi.org/10.3390/biology11081237