
A Pathogenic Role of Non-Parenchymal Liver Cells in Alcohol-Associated Liver Disease of Infectious and Non-Infectious Origin

 ,
, 
 ,
, 
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Role of Liver Macrophages in ALD
3. The Role of T Cells in the Immunopathogenesis of ALD
3.1. ALD and HSC Activation
3.2. Alcohol Metabolites Affect the Crosstalk between Hepatocyte and Hepatic Stellate Cells to Facilitate Liver Fibrosis Progression: Potentiation by Infectious Agents (HIV)
3.3. Hepatic Apoptotic Bodies as Activators of Liver Fibrosis under HIV-Alcohol Exposure
3.4. Alcohol-Induced Endosomal Alkalinization Supports HIV Accumulation in Hepatocytes
3.5. Alcohol-Induced HIV Accumulation Triggers Reactive Oxygen Species Generation
3.6. Apoptotic Bodies Derived from HIV and Acetaldehyde-Exposed Hepatocytes Induce HSC Profibrotic Activation
3.7. Sex Differences in NPC Properties in ALD
3.8. Sex Differences in Macrophage Properties
3.9. Sex Differences in T-Cells Functions
3.10. Sex Differences in HSC Properties
4. Conclusions
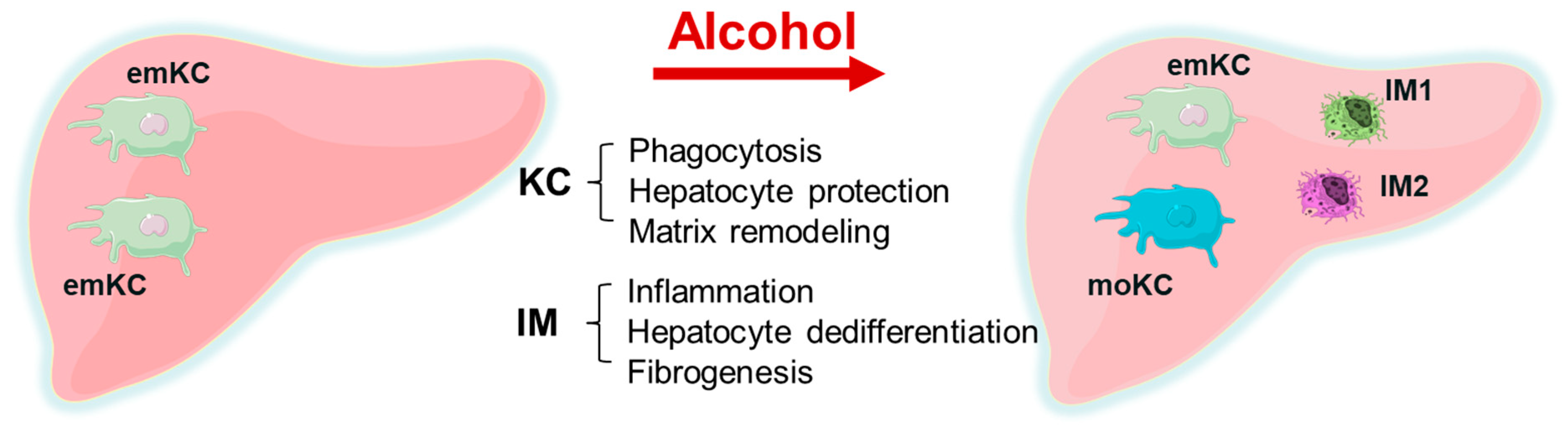
- There is a replacement of embryonic KC with monocyte-derived KC. KCs mainly play a hepatoprotective role and participate in phagocytosis and matrix remodeling, while a minor part is pro-inflammatory. Inflammation is related to infiltrating macrophages that cause hepatocyte dedifferentiation and fibrogenesis. In ALD, the selective removal of KCs without removing IMs from the alcohol-exposed liver can result in liver failure, while the removal of both KCs and IMs together reduces liver injury. This is illustrated by Figure 1.
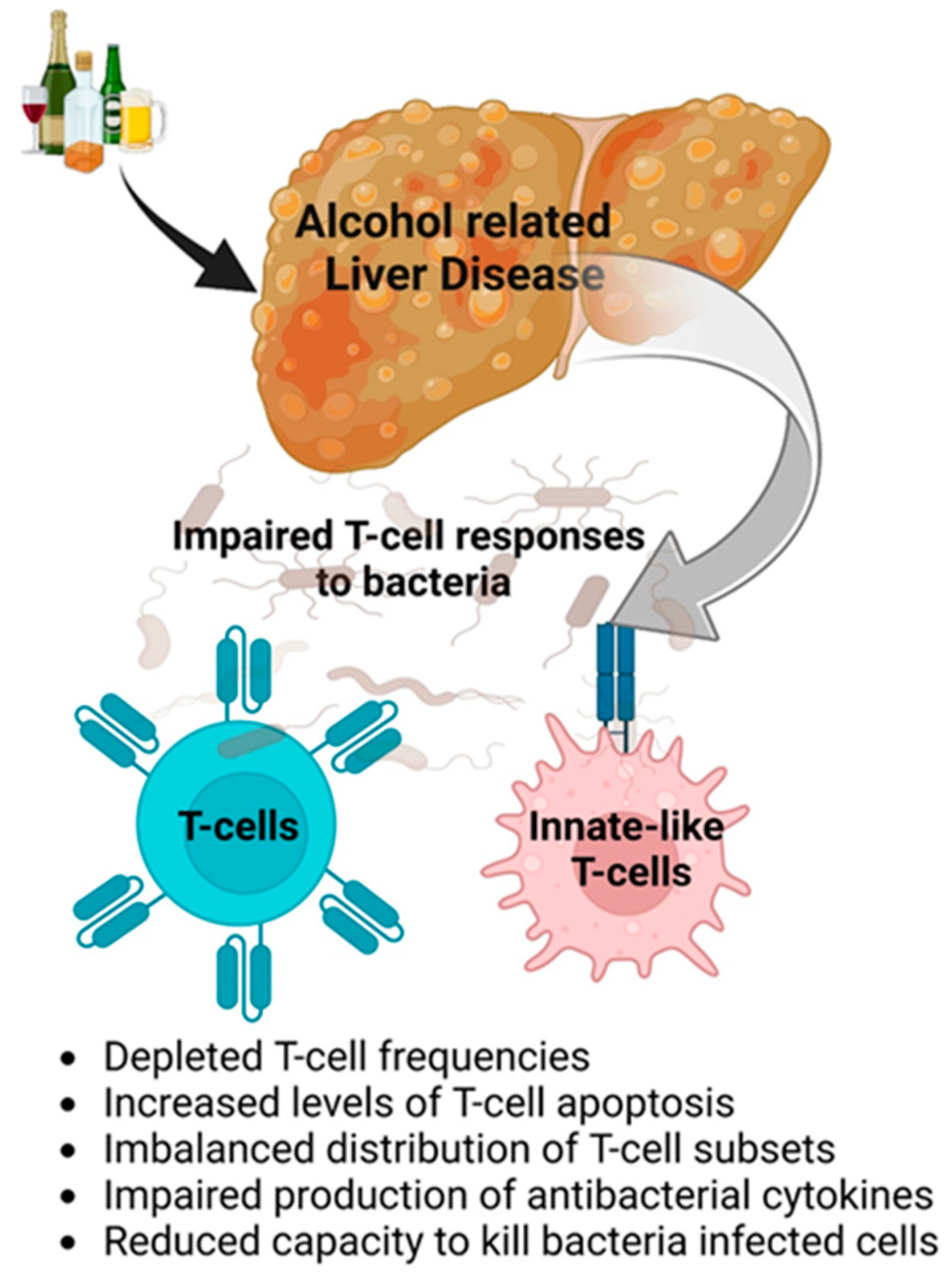
- Impaired anti-bacterial protection is due to dysfunctions in the innate immunity and T cells based on depleted T-cell frequencies, increased cell apoptosis, imbalanced T-cell subsets, impaired production of cytokines, and the reduced ability to kill bacteria (Figure 2).
- The significant trigger for profibrotic activation is the engulfment of apoptotic bodies by HSCs, which is induced by ethanol metabolites and can be further potentiated by viral infections, including HIV. This apoptotic body formation is regulated by the induction of oxidative stress due to the lysosome dysfunction-dependent accumulation of HIV proteins in ethanol-exposed hepatocytes.
- There is a sex difference in the regulation of liver NPCs number/functions in macrophages and HSC, while the sex-dependent regulation of endothelial and immune cells requires further clarification.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jones, A.L. Anatomy of the Normal Liver. Hepatol. A Textb. Liver Dis. 1996, 1, 3–32. [Google Scholar]
- Thurman, R.G., II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am. J. Physiol. 1998, 275, G605–G611. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Thurman, R.G. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001, 34, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, A.D.; Collins, P.L. Immune dysfunction in acute alcoholic hepatitis. World J. Gastroenterol. 2015, 21, 11904–11913. [Google Scholar] [CrossRef]
- Enomoto, N.; Ikejima, K.; Bradford, B.U.; Rivera, C.A.; Kono, H.; Goto, M.; Yamashina, S.; Schemmer, P.; Kitamura, T.; Oide, H.; et al. Role of Kupffer cells and gut-derived endotoxins in alcoholic liver injury. J. Gastroenterol. Hepatol. 2000, 15, 20–25. [Google Scholar] [CrossRef]
- Chen, P.; Schnabl, B. Host-microbiome interactions in alcoholic liver disease. Gut Liver 2014, 8, 237–241. [Google Scholar] [CrossRef]
- Choudhry, M.A.; Rana, S.N.; Kavanaugh, M.J.; Kovacs, E.J.; Gamelli, R.L.; Sayeed, M.M. Impaired intestinal immunity and barrier function: A cause for enhanced bacterial translocation in alcohol intoxication and burn injury. Alcohol 2004, 33, 199–208. [Google Scholar] [CrossRef]
- Gougol, A.; Clemente-Sanchez, A.; Argemi, J.; Bataller, R. Alcoholic Hepatitis. Clin. Liver Dis. 2021, 18, 90–95. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16. [Google Scholar] [CrossRef]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef]
- Ju, C.; Liangpunsakul, S. Role of hepatic macrophages in alcoholic liver disease. J. Investig. Med. 2016, 64, 1075–1077. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Ritz, T.; Krenkel, O.; Tacke, F. Dynamic plasticity of macrophage functions in diseased liver. Cell. Immunol. 2018, 330, 175–182. [Google Scholar] [CrossRef] [PubMed]
- de la, M.H.P.; Lieber, C.S.; DeCarli, L.M.; French, S.W.; Lindros, K.O.; Jarvelainen, H.; Bode, C.; Parlesak, A.; Bode, J.C. Models of alcoholic liver disease in rodents: A critical evaluation. Alcohol Clin. Exp. Res. 2001, 25, 254S–261S. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, J.; Zhang, S.; Weinman, S.A. FOXO3-dependent apoptosis limits alcohol-induced liver inflammation by promoting infiltrating macrophage differentiation. Cell Death Discov. 2018, 4, 16. [Google Scholar] [CrossRef]
- Li, Z.; Bridges, B.; Olson, J.; Weinman, S.A. The interaction between acetylation and serine-574 phosphorylation regulates the apoptotic function of FOXO3. Oncogene 2017, 36, 1887–1898. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, J.; Tikhanovich, I.; Kuravi, S.; Helzberg, J.; Dorko, K.; Roberts, B.; Kumer, S.; Weinman, S.A. Serine 574 phosphorylation alters transcriptional programming of FOXO3 by selectively enhancing apoptotic gene expression. Cell Death Differ. 2016, 23, 583–595. [Google Scholar] [CrossRef]
- Ju, C.; Mandrekar, P. Macrophages and Alcohol-Related Liver Inflammation. Alcohol Res. 2015, 37, 251–262. [Google Scholar]
- Wang, M.; You, Q.; Lor, K.; Chen, F.; Gao, B.; Ju, C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J. Leukoc. Biol. 2014, 96, 657–665. [Google Scholar] [CrossRef]
- Lee, J.; French, B.; Morgan, T.; French, S.W. The liver is populated by a broad spectrum of markers for macrophages. In alcoholic hepatitis the macrophages are M1 and M2. Exp. Mol. Pathol. 2014, 96, 118–125. [Google Scholar] [CrossRef]
- Kim, A.; Wu, X.; Allende, D.S.; Nagy, L.E. Gene Deconvolution Reveals Aberrant Liver Regeneration and Immune Cell Infiltration in Alcohol-Associated Hepatitis. Hepatology 2021, 74, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, M.; O’Neil, M.; Villar, M.T.; Artigues, A.; Averilla, J.; Gunewardena, S.; Weinman, S.A.; Tikhanovich, I. A Western diet with alcohol in drinking water recapitulates features of alcohol-associated liver disease in mice. Alcohol Clin. Exp. Res. 2021, 45, 1980–1993. [Google Scholar] [CrossRef] [PubMed]
- Buch, T.; Heppner, F.L.; Tertilt, C.; Heinen, T.J.; Kremer, M.; Wunderlich, F.T.; Jung, S.; Waisman, A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat. Methods 2005, 2, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Iwawaki, T.; Taya, C.; Yonekawa, H.; Noda, M.; Inui, Y.; Mekada, E.; Kimata, Y.; Tsuru, A.; Kohno, K. Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat. Biotechnol. 2001, 19, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e1057. [Google Scholar] [CrossRef]
- Sasaki, K.; Averilla, J.; Ghosh, P.; Tikhanovich, I.; Schonfeld, M.; O’Neil, M.; Pulido-Ruiz, I.A.; Wozniak, A.; Gunewardena, S.; Weinman, S.A. Selective ablation of kupffer cell subsets exacerbates alcohol-associated steatohepatitis in mice. Hepatology 2021, 74, 251A–252A. [Google Scholar]
- Keller, M. The First American Medical Work on the Effects of Alcohol: Benjamin Rush’s “An Inquiry Into the Effects of Ardent Spirits Upon the Human Body and Mind: With an Account of the Means of Preventing, and of the Remedies for Curing Them”. Q. J. Stud. Alcohol 1943, 4, 321–341. [Google Scholar]
- Szabo, G. Consequences of alcohol consumption on host defense. Alcohol Alcohol. 1999, 34, 830–841. [Google Scholar] [CrossRef]
- Kaur, B.; Rosenblatt, R.; Sundaram, V. Infections in Alcoholic Hepatitis. J. Clin. Transl. Hepatol. 2022, 10, 718–725. [Google Scholar] [CrossRef]
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242. [Google Scholar] [CrossRef]
- Nelson, S.; Kolls, J.K. Alcohol, host defence and society. Nat. Rev. Immunol. 2002, 2, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D. Innate immunity to bacterial infection: Toll receptors, professional phagocytes, intra-phagosomal killing, defensins and cytoplasmic muramyl dipeptide sensors. Curr. Opin. Infect. Dis. 2005, 18, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Cortes, K.; Villageliu, D.N.; Samuelson, D.R. Innate lymphocytes: Role in alcohol-induced immune dysfunction. Front. Immunol. 2022, 13, 934617. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Z. Innate lymphocytes: Pathogenesis and therapeutic targets of liver diseases and cancer. Cell. Mol. Immunol. 2021, 18, 57–72. [Google Scholar] [CrossRef]
- Shepherd, F.R.; McLaren, J.E. T Cell Immunity to Bacterial Pathogens: Mechanisms of Immune Control and Bacterial Evasion. Int. J. Mol. Sci. 2020, 21, 6144. [Google Scholar] [CrossRef] [PubMed]
- Balin, S.J.; Pellegrini, M.; Klechevsky, E.; Won, S.T.; Weiss, D.I.; Choi, A.W.; Hakimian, J.; Lu, J.; Ochoa, M.T.; Bloom, B.R.; et al. Human antimicrobial cytotoxic T lymphocytes, defined by NK receptors and antimicrobial proteins, kill intracellular bacteria. Sci. Immunol. 2018, 3, eaat7668. [Google Scholar] [CrossRef]
- Szabo, G.; Saha, B. Alcohol’s Effect on Host Defense. Alcohol Res. 2015, 37, 159–170. [Google Scholar] [PubMed]
- Liu, Y.K. Leukopenia in alcoholics. Am. J. Med. 1973, 54, 605–610. [Google Scholar] [CrossRef]
- Kapasi, A.A.; Patel, G.; Goenka, A.; Nahar, N.; Modi, N.; Bhaskaran, M.; Reddy, K.; Franki, N.; Patel, J.; Singhal, P.C. Ethanol promotes T cell apoptosis through the mitochondrial pathway. Immunology 2003, 108, 313–320. [Google Scholar] [CrossRef]
- McFarland, W.; Libre, E.P. Abnormal Leukocyte Response in Alcoholism. Ann. Intern. Med. 1963, 59, 865–877. [Google Scholar] [CrossRef]
- Cho, B.K.; Rao, V.P.; Ge, Q.; Eisen, H.N.; Chen, J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J. Exp. Med. 2000, 192, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.Y.; Wang, N.; Feng, Y.; Wang, X.; Feng, Y. Recent Insights into the Role of Immune Cells in Alcoholic Liver Disease. Front. Immunol. 2019, 10, 1328. [Google Scholar] [CrossRef]
- Markwick, L.J.; Riva, A.; Ryan, J.M.; Cooksley, H.; Palma, E.; Tranah, T.H.; Manakkat Vijay, G.K.; Vergis, N.; Thursz, M.; Evans, A.; et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology 2015, 148, 590–602.e510. [Google Scholar] [CrossRef]
- Kasten, K.R.; Muenzer, J.T.; Caldwell, C.C. Neutrophils are significant producers of IL-10 during sepsis. Biochem. Biophys. Res. Commun. 2010, 393, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, Z.; Ren, T.; Kim, S.J.; He, Y.; Seo, W.; Guillot, A.; Ding, Y.; Wu, R.; Shao, S.; et al. Alcohol inhibits T-cell glucose metabolism and hepatitis in ALDH2-deficient mice and humans: Roles of acetaldehyde and glucocorticoids. Gut 2019, 68, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- McTernan, P.M.; Levitt, D.E.; Welsh, D.A.; Simon, L.; Siggins, R.W.; Molina, P.E. Alcohol Impairs Immunometabolism and Promotes Naive T Cell Differentiation to Pro-Inflammatory Th1 CD4(+) T Cells. Front. Immunol. 2022, 13, 839390. [Google Scholar] [CrossRef]
- Katz, P.S.; Siggins, R.W.; Porretta, C.; Armstrong, M.L.; Zea, A.H.; Mercante, D.E.; Parsons, C.; Veazey, R.S.; Bagby, G.J.; Nelson, S.; et al. Chronic alcohol increases CD8+ T-cell immunosenescence in simian immunodeficiency virus-infected rhesus macaques. Alcohol 2015, 49, 759–765. [Google Scholar] [CrossRef]
- Riva, A.; Chokshi, S. Immune checkpoint receptors: Homeostatic regulators of immunity. Hepatol. Int. 2018, 12, 223–236. [Google Scholar] [CrossRef]
- Legat, A.; Speiser, D.E.; Pircher, H.; Zehn, D.; Fuertes Marraco, S.A. Inhibitory Receptor Expression Depends More Dominantly on Differentiation and Activation than “Exhaustion” of Human CD8 T Cells. Front. Immunol. 2013, 4, 455. [Google Scholar] [CrossRef]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef]
- Evans, A.; Riva, A.; Cooksley, H.; Phillips, S.; Puranik, S.; Nathwani, A.; Brett, S.; Chokshi, S.; Naoumov, N.V. Programmed death 1 expression during antiviral treatment of chronic hepatitis B: Impact of hepatitis B e-antigen seroconversion. Hepatology 2008, 48, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Cooksley, H.; Riva, A.; Katzarov, K.; Hadzhiolova-Lebeau, T.; Pavlova, S.; Simonova, M.; Williams, R.; Chokshi, S. Differential Expression of Immune Inhibitory Checkpoint Signatures on Antiviral and Inflammatory T Cell Populations in Chronic Hepatitis B. J. Interferon Cytokine Res. 2018, 38, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Xie, Y.; Xiang, Y.; Sheng, J.; Zhang, D.; Yao, X.; Yang, Y.; Zhang, X. Immunotherapy for Hepatocellular Carcinoma: Current Advances and Future Expectations. J. Immunol. Res. 2018, 2018, 8740976. [Google Scholar] [CrossRef]
- Guignant, C.; Lepape, A.; Huang, X.; Kherouf, H.; Denis, L.; Poitevin, F.; Malcus, C.; Cheron, A.; Allaouchiche, B.; Gueyffier, F.; et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit. Care 2011, 15, R99. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Colston, E.; Yende, S.; Crouser, E.D.; Martin, G.S.; Albertson, T.; Bartz, R.R.; Brakenridge, S.C.; Delano, M.J.; Park, P.K.; et al. Immune checkpoint inhibition in sepsis: A Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med. 2019, 45, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Colston, E.; Yende, S.; Angus, D.C.; Moldawer, L.L.; Crouser, E.D.; Martin, G.S.; Coopersmith, C.M.; Brakenridge, S.; Mayr, F.B.; et al. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized, Placebo-Controlled, Single Ascending Dose Study of Antiprogrammed Cell Death-Ligand 1 Antibody (BMS-936559). Crit. Care Med. 2019, 47, 632–642. [Google Scholar] [CrossRef]
- Riva, A.; Palma, E.; Devshi, D.; Corrigall, D.; Adams, H.; Heaton, N.; Menon, K.; Preziosi, M.; Zamalloa, A.; Miquel, R.; et al. Soluble TIM3 and Its Ligands Galectin-9 and CEACAM1 Are in Disequilibrium During Alcohol-Related Liver Disease and Promote Impairment of Anti-bacterial Immunity. Front. Physiol. 2021, 12, 632502. [Google Scholar] [CrossRef]
- Masina, N.; Bekiswa, A.; Shey, M. Mucosal-associated invariant T cells in natural immunity and vaccination against infectious diseases in humans. Curr. Opin. Immunol. 2021, 71, 1–5. [Google Scholar] [CrossRef]
- Riva, A.; Patel, V.; Kurioka, A.; Jeffery, H.C.; Wright, G.; Tarff, S.; Shawcross, D.; Ryan, J.M.; Evans, A.; Azarian, S.; et al. Mucosa-associated invariant T cells link intestinal immunity with antibacterial immune defects in alcoholic liver disease. Gut 2018, 67, 918–930. [Google Scholar] [CrossRef]
- Vergis, N.; Atkinson, S.R.; Thursz, M.R. Assessment and Management of Infection in Alcoholic Hepatitis. Semin. Liver Dis. 2020, 40, 11–19. [Google Scholar] [CrossRef]
- Van der Merwe, S.; Chokshi, S.; Bernsmeier, C.; Albillos, A. The multifactorial mechanisms of bacterial infection in decompensated cirrhosis. J. Hepatol. 2021, 75 (Suppl. S1), S82–S100. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Dhar, D.; Baglieri, J.; Kisseleva, T.; Brenner, D.A. Mechanisms of liver fibrosis and its role in liver cancer. Exp. Biol. Med. 2020, 245, 96–108. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef]
- Koo, J.H.; Lee, H.J.; Kim, W.; Kim, S.G. Endoplasmic Reticulum Stress in Hepatic Stellate Cells Promotes Liver Fibrosis via PERK-Mediated Degradation of HNRNPA1 and Up-regulation of SMAD2. Gastroenterology 2016, 150, 181–193.e188. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Zhang, F.; Little, A.; Zhang, H. Chronic alcohol consumption inhibits peripheral NK cell development and maturation by decreasing the availability of IL-15. J. Leukoc. Biol. 2017, 101, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Batey, R.G.; George, J. Role of ethanol in the regulation of hepatic stellate cell function. World J. Gastroenterol. 2006, 12, 6926–6932. [Google Scholar] [CrossRef] [PubMed]
- Winau, F.; Quack, C.; Darmoise, A.; Kaufmann, S.H. Starring stellate cells in liver immunology. Curr. Opin. Immunol. 2008, 20, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Saile, B.; Matthes, N.; El Armouche, H.; Neubauer, K.; Ramadori, G. The bcl, NFkappaB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-beta or TNF-alpha on activated hepatic stellate cells. Eur. J. Cell Biol. 2001, 80, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D.A. The Crosstalk between Hepatocytes, Hepatic Macrophages, and Hepatic Stellate Cells Facilitates Alcoholic Liver Disease. Cell Metab. 2019, 30, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Osei-Hyiaman, D.; Park, O.; Liu, J.; Batkai, S.; Mukhopadhyay, P.; Horiguchi, N.; Harvey-White, J.; Marsicano, G.; Lutz, B.; et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008, 7, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.M.; Kim, H.H.; Kim, M.H.; Cinar, R.; Yi, H.S.; Eun, H.S.; Kim, S.H.; Choi, Y.J.; Lee, Y.S.; Kim, S.Y.; et al. Glutamate Signaling in Hepatic Stellate Cells Drives Alcoholic Steatosis. Cell Metab. 2019, 30, 877–889.e7. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Eguchi, A.; Feldstein, A.E.; Tsukamoto, H.; Dagur, R.S.; Ganesan, M.; New-Aaron, M.; Arumugam, M.K.; Chava, S.; Ribeiro, M.; et al. Cell-to-Cell Communications in Alcohol-Associated Liver Disease. Front. Physiol. 2022, 13, 831004. [Google Scholar] [CrossRef]
- Canbay, A.; Friedman, S.; Gores, G.J. Apoptosis: The nexus of liver injury and fibrosis. Hepatology 2004, 39, 273–278. [Google Scholar] [CrossRef]
- Suh, Y.G.; Jeong, W.I. Hepatic stellate cells and innate immunity in alcoholic liver disease. World J. Gastroenterol. 2011, 17, 2543–2551. [Google Scholar] [CrossRef]
- Seo, W.; Jeong, W.I. Hepatic non-parenchymal cells: Master regulators of alcoholic liver disease? World J. Gastroenterol. 2016, 22, 1348–1356. [Google Scholar] [CrossRef]
- Khomich, O.; Ivanov, A.V.; Bartosch, B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 2019, 9, 24. [Google Scholar] [CrossRef]
- Lyu, H.; Tang, H.; Liang, Y.; Huang, S.; Wang, Y.; Huang, W.; Zhou, Y. Alcohol Consumption and Risk of Liver Fibrosis in People Living With HIV: A Systematic Review and Meta-Analysis. Front. Immunol. 2022, 13, 841314. [Google Scholar] [CrossRef]
- New-Aaron, M.; Kingi, H.; Meza, J.L.; Goedert, M.H.; Kibusi, S.M.; Mkhoi, M.L.; Mayengo, C.D.; Charles, J.; Shabani, S.; New-Aaron, T.O. Duration on ART, Alcohol Use and HIV Stage May Predict Risky Sexual Behavior in a Resource-limited Environment: A Cross-sectional Study. Curr. HIV Res. 2021, 19, 420–433. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS. Global HIV & AIDS Statistics—Fact Sheet. 2022. Available online: https://repository.gheli.harvard.edu/repository/12559/ (accessed on 11 November 2022).
- Debes, J.D.; Bohjanen, P.R.; Boonstra, A. Mechanisms of accelerated liver fibrosis progression during HIV infection. J. Clin. Transl. Hepatol. 2016, 4, 328. [Google Scholar] [PubMed]
- Rein, S.M.; Lampe, F.C.; Chaloner, C.; Stafford, A.; Rodger, A.J.; Johnson, M.A.; McDonnell, J.; Burns, F.; Madge, S.; Miners, A. Causes of hospitalisation among a cohort of people with HIV from a London centre followed from 2011 to 2018. BMC Infect. Dis. 2021, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- New-Aaron, M.; Ganesan, M.; Dagur, R.S.; Kharbanda, K.K.; Poluektova, L.Y.; Osna, N.A. Pancreatogenic Diabetes: Triggering Effects of Alcohol and HIV. Biology 2021, 10, 108. [Google Scholar] [CrossRef]
- Leake, I. Liver disease: Alcohol causes epigenetic changes in hepatic stellate cells. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 704. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr. 2014, 3, 344–363. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; Todero, S.L.; Shubert, K.A.; Sorrell, M.F.; Tuma, D.J. Malondialdehyde-acetaldehyde-protein adducts increase secretion of chemokines by rat hepatic stellate cells. Alcohol 2001, 25, 123–128. [Google Scholar] [CrossRef]
- Dagur, R.S.; New-Aaron, M.; Ganesan, M.; Wang, W.; Romanova, S.; Kidambi, S.; Kharbanda, K.K.; Poluektova, L.Y.; Osna, N.A. Alcohol-and-HIV-Induced Lysosomal Dysfunction Regulates Extracellular Vesicles Secretion in Vitro and in Liver-Humanized Mice. Biology 2021, 10, 29. [Google Scholar] [CrossRef]
- New-Aaron, M.; Dagur, R.S.; Koganti, S.S.; Ganesan, M.; Wang, W.; Makarov, E.; Ogunnaike, M.; Kharbanda, K.K.; Poluektova, L.Y.; Osna, N.A. Alcohol and HIV-Derived Hepatocyte Apoptotic Bodies Induce Hepatic Stellate Cell Activation. Biology 2022, 11, 1059. [Google Scholar] [CrossRef]
- Ribeiro, P.S.; Cortez-Pinto, H.; Sola, S.; Castro, R.E.; Ramalho, R.M.; Baptista, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol. 2004, 99, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Sagnelli, E.; Stroffolini, T.; Mele, A.; Imparato, M.; Sagnelli, C.; Coppola, N.; Almasio, P.L. Impact of comorbidities on the severity of chronic hepatitis B at presentation. World J. Gastroenterol. 2012, 18, 1616–1621. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; Natarajan, S.K.; Zhang, J.; Mott, J.L.; Poluektova, L.I.; McVicker, B.L.; Kharbanda, K.K.; Tuma, D.J.; Osna, N.A. Role of apoptotic hepatocytes in HCV dissemination: Regulation by acetaldehyde. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G930–G940. [Google Scholar] [CrossRef] [PubMed]
- Llamosas-Falcon, L.; Shield, K.D.; Gelovany, M.; Manthey, J.; Rehm, J. Alcohol use disorders and the risk of progression of liver disease in people with hepatitis C virus infection—A systematic review. Subst. Abus. Treat. Prev. Policy 2020, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; New-Aaron, M.; Dagur, R.S.; Makarov, E.; Wang, W.; Kharbanda, K.K.; Kidambi, S.; Poluektova, L.Y.; Osna, N.A. Alcohol metabolism potentiates HIV-induced hepatotoxicity: Contribution to end-stage liver disease. Biomolecules 2019, 9, 851. [Google Scholar] [CrossRef]
- Blackard, J.T.; Ma, G.; Martin, C.M.; Rouster, S.D.; Shata, M.T.; Sherman, K.E. HIV variability in the liver and evidence of possible compartmentalization. AIDS Res. Hum. Retrovir. 2011, 27, 1117–1126. [Google Scholar] [CrossRef]
- Cao, Y.; Dieterich, D.; Thomas, P.A.; Huang, Y.; Mirabile, M.; Ho, D.D. Identification and quantitation of HIV-1 in the liver of patients with AIDS. AIDS 1992, 6, 65–70. [Google Scholar] [CrossRef]
- Donaldson, Y.; Simmonds, P.; Busuttil, A.; Bell, J.; Ironside, J.; Brettle, R.; Robertson, J. Redistribution of HIV outside the lymphoid system with onset of AIDS. Lancet 1994, 343, 382–385. [Google Scholar] [CrossRef]
- Crispe, I.N.; Dao, T.; Klugewitz, K.; Mehal, W.Z.; Metz, D.P. The liver as a site of T-cell apoptosis: Graveyard, or killing field? Immunol. Rev. 2000, 174, 47–62. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Khan, S.; Telwatte, S.; Trapecar, M.; Yukl, S.; Sanjabi, S. Differentiating Immune Cell Targets in Gut-Associated Lymphoid Tissue for HIV Cure. AIDS Res. Hum. Retrovir. 2017, 33, S40–S58. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV Enters Cells via Endocytosis and Dynamin-Dependent Fusion with Endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Cardona Maya, W.; Moreno-Fernandez, M.E.; Ma, G.; Shata, M.T.; Sherman, K.E.; Chougnet, C.; Blackard, J.T. Low-level HIV infection of hepatocytes. Virol. J. 2012, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Casey, C.A.; Wiegert, R.L.; Tuma, D.J. Chronic ethanol administration impairs ATP-dependent acidification of endosomes in rat liver. Biochem. Biophys. Res. Commun. 1993, 195, 1127–1133. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; McVicker, D.L.; Zetterman, R.K.; MacDonald, R.G.; Donohue, T.M., Jr. Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology 1997, 26, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, H.; Hieshima, K.; Nakayama, T.; Sakaguchi, T.; Fujisawa, R.; Tanase, S.; Nishiura, H.; Matsuno, K.; Takamori, H.; Tabira, Y.; et al. Human CC chemokine liver-expressed chemokine/CCL16 is a functional ligand for CCR1, CCR2 and CCR5, and constitutively expressed by hepatocytes. Int. Immunol. 2001, 13, 1021–1029. [Google Scholar] [CrossRef]
- Marechal, V.; Prevost, M.C.; Petit, C.; Perret, E.; Heard, J.M.; Schwartz, O. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J. Virol. 2001, 75, 11166–11177. [Google Scholar] [CrossRef]
- Pauza, C.D.; Price, T.M. Human immunodeficiency virus infection of T cells and monocytes proceeds via receptor-mediated endocytosis. J. Cell Biol. 1988, 107, 959–968. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Wei, B.L.; Yao, J.; Luo, T.; Garcia, J.V. Inhibition of endosomal/lysosomal degradation increases the infectivity of human immunodeficiency virus. J. Virol. 2002, 76, 11440–11446. [Google Scholar] [CrossRef]
- Elbim, C.; Pillet, S.; Prevost, M.H.; Preira, A.; Girard, P.M.; Rogine, N.; Matusani, H.; Hakim, J.; Israel, N.; Gougerot-Pocidalo, M.A. Redox and Activation Status of Monocytes from Human Immunodeficiency Virus-Infected Patients: Relationship with Viral Load. J. Virol. 1999, 73, 4561–4566. [Google Scholar] [CrossRef] [PubMed]
- Masutani, H.; Naito, M.; Takahashi, K.; Hattori, T.; Koito, A.; Takatsuki, K.; Go, T.; Nakamura, H.; Fujii, S.; Yoshida, Y. Dysregulation of adult T-cell leukemia-derived factor (ADF)/thioredoxin in HIV infection: Loss of ADF high-producer cells in lymphoid tissues of AIDS patients. AIDS Res. Hum. Retrovir. 1992, 8, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Ven, V.D.; Blom; Peters; Jacobs; Verver; Koopmans; Demacker; Meer, V.D. Glutathione homeostasis is disturbed in CD4-positive lymphocytes of HIV-seropositive individuals. Eur. J. Clin. Investig. 1998, 28, 187–193. [Google Scholar]
- Staal, F.; Roederer, M.; Herzenberg, L.A.; Herzenberg, L.A. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1990, 87, 9943–9947. [Google Scholar] [CrossRef] [PubMed]
- Couret, J.; Chang, T.L. Reactive Oxygen Species in HIV Infection. EC Microbiol. 2016, 3, 597–604. [Google Scholar]
- Shah, A.; Kumar, S.; Simon, S.D.; Singh, D.P.; Kumar, A. HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis. 2013, 4, e850. [Google Scholar] [CrossRef]
- Samikkannu, T.; Ranjith, D.; Rao, K.V.; Atluri, V.S.; Pimentel, E.; El-Hage, N.; Nair, M.P. HIV-1 gp120 and morphine induced oxidative stress: Role in cell cycle regulation. Front. Microbiol. 2015, 6, 614. [Google Scholar] [CrossRef]
- Stromájer-Rácz, T.; Gazdag, Z.; Belágyi, J.; Vágvölgyi, C.; Zhao, R.Y.; Pesti, M. Oxidative stress induced by HIV-1 F34IVpr in Schizosaccharomyces pombe is one of its multiple functions. Exp. Mol. Pathol. 2010, 88, 38–44. [Google Scholar] [CrossRef]
- Gu, Y.; Wu, R.F.; Xu, Y.C.; Flores, S.C.; Terada, L.S. HIV Tat Activates c-Jun Amino-terminal Kinase through an Oxidant-Dependent Mechanism. Virology 2001, 286, 62–71. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef]
- El-Amine, R.; Germini, D.; Zakharova, V.V.; Tsfasman, T.; Sheval, E.V.; Louzada, R.A.N.; Dupuy, C.; Bilhou-Nabera, C.; Hamade, A.; Najjar, F.; et al. HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. 2018, 15, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.; Byrnes, S.; Cochrane, C.; Roche, M.; Estes, J.D.; Selemidis, S.; Angelovich, T.A.; Churchill, M.J. The role of oxidative stress in HIV-associated neurocognitive disorders. Brain Behav. Immun. Health 2021, 13, 100235. [Google Scholar] [CrossRef] [PubMed]
- Paiva, C.N.; Bozza, M.T. Are reactive oxygen species always detrimental to pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef] [PubMed]
- Buttke, T.M.; Sandstrom, P.A. Oxidative stress as a mediator of apoptosis. Immunol. Today 1994, 15, 7–10. [Google Scholar] [CrossRef]
- New-Aaron, M.; Thomes, P.G.; Ganesan, M.; Dagur, R.S.; Donohue, T.M.; Kusum, K.K.; Poluektova, L.Y.; Osna, N.A. Alcohol-Induced Lysosomal Damage and Suppression of Lysosome Biogenesis Contribute to Hepatotoxicity in HIV-Exposed Liver Cells. Biomolecules 2021, 11, 1497. [Google Scholar] [CrossRef]
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Investig. 2003, 83, 655–663. [Google Scholar] [CrossRef]
- Watanabe, A.; Hashmi, A.; Gomes, D.A.; Town, T.; Badou, A.; Flavell, R.A.; Mehal, W.Z. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 2007, 46, 1509–1518. [Google Scholar] [CrossRef]
- Gentile, M.; Latonen, L.; Laiho, M. Cell cycle arrest and apoptosis provoked by UV radiation-induced DNA damage are transcriptionally highly divergent responses. Nucleic Acids Res. 2003, 31, 4779–4790. [Google Scholar] [CrossRef]
- Fadok, V.A.; De Cathelineau, A.; Daleke, D.L.; Henson, P.M.; Bratton, D.L. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem. 2001, 276, 1071–1077. [Google Scholar] [CrossRef]
- Parola, M.; Robino, G.; Marra, F.; Pinzani, M.; Bellomo, G.; Leonarduzzi, G.; Chiarugi, P.; Camandola, S.; Poli, G.; Waeg, G. HNE interacts directly with JNK isoforms in human hepatic stellate cells. J. Clin. Investig. 1998, 102, 1942–1950. [Google Scholar] [CrossRef]
- Jiang, J.X.; Mikami, K.; Venugopal, S.; Li, Y.; Török, N.J. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-κB-dependent pathways. J. Hepatol. 2009, 51, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.X.; Ross, E.; Borman, M.A.; Zimmer, S.; Kaplan, G.G.; Heitman, S.J.; Swain, M.G.; Burak, K.; Quan, H.; Myers, R.P. Risk factors for mortality in patients with alcoholic hepatitis and assessment of prognostic models: A population-based study. Can. J. Gastroenterol. Hepatol. 2015, 29, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.Q.; Karatayev, O.; Boorgu, D.; Leibowitz, S.F. CCL2/CCR2 Chemokine System in Embryonic Hypothalamus: Involvement in Sexually Dimorphic Stimulatory Effects of Prenatal Ethanol Exposure on Peptide-Expressing Neurons. Neuroscience 2020, 424, 155–171. [Google Scholar] [CrossRef]
- Eagon, P.K.; Willett, J.E.; Seguiti, S.M.; Appler, M.L.; Gavaler, J.S.; Van Thiel, D.H. Androgen-responsive functions of male rat liver. Effect of chronic alcohol ingestion. Gastroenterology 1987, 93, 1162–1169. [Google Scholar] [CrossRef]
- Penaloza, C.G.; Cruz, M.; Germain, G.; Jabeen, S.; Javdan, M.; Lockshin, R.A.; Zakeri, Z. Higher sensitivity of female cells to ethanol: Methylation of DNA lowers Cyp2e1, generating more ROS. Cell Commun. Signal. 2020, 18, 111. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.R.; Fortune, J.; Iwahashi, M.; Sutherland, E. Sexual dimorphic expression of ADH in rat liver: Importance of the hypothalamic-pituitary-liver axis. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G646–G655. [Google Scholar] [CrossRef][Green Version]
- Lourens, S.; Sunjaya, D.B.; Singal, A.; Liangpunsakul, S.; Puri, P.; Sanyal, A.; Ren, X.; Gores, G.J.; Radaeva, S.; Chalasani, N.; et al. Acute Alcoholic Hepatitis: Natural History and Predictors of Mortality Using a Multicenter Prospective Study. Mayo Clin. Proc. Innov. Qual. Outcomes 2017, 1, 37–48. [Google Scholar] [CrossRef]
- Muller, M.F.; Kendall, T.J.; Adams, D.J.; Zhou, Y.; Arends, M.J. The murine hepatic sequelae of long-term ethanol consumption are sex-specific and exacerbated by Aldh1b1 loss. Exp. Mol. Pathol. 2018, 105, 63–70. [Google Scholar] [CrossRef]
- Li, S.Q.; Wang, P.; Wang, D.M.; Lu, H.J.; Li, R.F.; Duan, L.X.; Zhu, S.; Wang, S.L.; Zhang, Y.Y.; Wang, Y.L. Molecular mechanism for the influence of gender dimorphism on alcoholic liver injury in mice. Hum. Exp. Toxicol. 2019, 38, 65–81. [Google Scholar] [CrossRef]
- Yin, M.; Ikejima, K.; Wheeler, M.D.; Bradford, B.U.; Seabra, V.; Forman, D.T.; Sato, N.; Thurman, R.G. Estrogen is involved in early alcohol-induced liver injury in a rat enteral feeding model. Hepatology 2000, 31, 117–123. [Google Scholar] [CrossRef]
- Kurt, Z.; Barrere-Cain, R.; LaGuardia, J.; Mehrabian, M.; Pan, C.; Hui, S.T.; Norheim, F.; Zhou, Z.; Hasin, Y.; Lusis, A.J.; et al. Tissue-specific pathways and networks underlying sexual dimorphism in non-alcoholic fatty liver disease. Biol. Sex Differ. 2018, 9, 46. [Google Scholar] [CrossRef]
- Kono, H.; Wheeler, M.D.; Rusyn, I.; Lin, M.; Seabra, V.; Rivera, C.A.; Bradford, B.U.; Forman, D.T.; Thurman, R.G. Gender differences in early alcohol-induced liver injury: Role of CD14, NF-kappaB, and TNF-alpha. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G652–G661. [Google Scholar] [CrossRef] [PubMed]
- Eagon, P.K. Alcoholic liver injury: Influence of gender and hormones. World J. Gastroenterol. 2010, 16, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, M.; Averilla, J.; Gunewardena, S.; Weinman, S.A.; Tikhanovich, I. Male-Specific Activation of Lysine Demethylases 5B and 5C Mediates Alcohol-Induced Liver Injury and Hepatocyte Dedifferentiation. Hepatol. Commun. 2022, 6, 1373–1391. [Google Scholar] [CrossRef] [PubMed]
- Marcos, R.; Correia-Gomes, C.; Miranda, H.; Carneiro, F. Liver gender dimorphism--insights from quantitative morphology. Histol. Histopathol. 2015, 30, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Marcos, R.; Lopes, C.; Malhao, F.; Correia-Gomes, C.; Fonseca, S.; Lima, M.; Gebhardt, R.; Rocha, E. Stereological assessment of sexual dimorphism in the rat liver reveals differences in hepatocytes and Kupffer cells but not hepatic stellate cells. J. Anat. 2016, 228, 996–1005. [Google Scholar] [CrossRef]
- Thurman, R.G. Sex-related liver injury due to alcohol involves activation of Kupffer cells by endotoxin. Can. J. Gastroenterol. 2000, 14, 129D–135D. [Google Scholar] [CrossRef]
- Wagnerberger, S.; Fiederlein, L.; Kanuri, G.; Stahl, C.; Millonig, G.; Mueller, S.; Bischoff, S.C.; Bergheim, I. Sex-specific differences in the development of acute alcohol-induced liver steatosis in mice. Alcohol Alcohol. 2013, 48, 648–656. [Google Scholar] [CrossRef]
- Muller, C. Liver, alcohol and gender. Wien. Med. Wochenschr. 2006, 156, 523–526. [Google Scholar] [CrossRef]
- Mandrekar, P.; Ambade, A.; Lim, A.; Szabo, G.; Catalano, D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: Regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology 2011, 54, 2185–2197. [Google Scholar] [CrossRef]
- Olubadewo, J.O.; Spitzer, J.A. Immune response modulation in acutely ethanol-intoxicated, acutely diabetic male and female rats. Alcohol 2003, 31, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Nanji, A.A.; Jokelainen, K.; Fotouhinia, M.; Rahemtulla, A.; Thomas, P.; Tipoe, G.L.; Su, G.L.; Dannenberg, A.J. Increased severity of alcoholic liver injury in female rats: Role of oxidative stress, endotoxin, and chemokines. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1348–G1356. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, E.; Chinetti-Gbaguidi, G.; Staels, B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Houben, T.; Bitorina, A.V.; Oligschlaeger, Y.; Jeurissen, M.L.; Rensen, S.; Kohler, S.E.; Westerterp, M.; Lutjohann, D.; Theys, J.; Romano, A.; et al. Sex-opposed inflammatory effects of 27-hydroxycholesterol are mediated via differences in estrogen signaling. J. Pathol. 2020, 251, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Fulham, M.A.; Mandrekar, P. Sexual Dimorphism in Alcohol Induced Adipose Inflammation Relates to Liver Injury. PLoS ONE 2016, 11, e0164225. [Google Scholar] [CrossRef]
- Zuluaga, P.; Sanvisens, A.; Teniente, A.; Fuster, D.; Tor, J.; Martinez-Caceres, E.; Muga, R. Wide array of T-cell subpopulation alterations in patients with alcohol use disorders. Drug Alcohol Depend. 2016, 162, 124–129. [Google Scholar] [CrossRef]
- Kasztelan-Szczerbinska, B.; Surdacka, A.; Celinski, K.; Rolinski, J.; Zwolak, A.; Miacz, S.; Szczerbinski, M. Prognostic Significance of the Systemic Inflammatory and Immune Balance in Alcoholic Liver Disease with a Focus on Gender-Related Differences. PLoS ONE 2015, 10, e0128347. [Google Scholar] [CrossRef]
- Thompson, M.G.; Navarro, F.; Chitsike, L.; Ramirez, L.; Kovacs, E.J.; Watkins, S.K. Alcohol exposure differentially effects anti-tumor immunity in females by altering dendritic cell function. Alcohol 2016, 57, 1–8. [Google Scholar] [CrossRef]
- Xu, J.; Ma, H.Y.; Liu, X.; Rosenthal, S.; Baglieri, J.; McCubbin, R.; Sun, M.; Koyama, Y.; Geoffroy, C.G.; Saijo, K.; et al. Blockade of IL-17 signaling reverses alcohol-induced liver injury and excessive alcohol drinking in mice. JCI Insight 2020, 5, e131277. [Google Scholar] [CrossRef]
- Harrison, C.A.; Laubitz, D.; Midura-Kiela, M.T.; Jamwal, D.R.; Besselsen, D.G.; Ghishan, F.K.; Kiela, P.R. Sexual Dimorphism in the Response to Broad-spectrum Antibiotics During T Cell-mediated Colitis. J. Crohns Colitis 2019, 13, 115–126. [Google Scholar] [CrossRef]
- Fuseini, H.; Newcomb, D.C. Mechanisms Driving Gender Differences in Asthma. Curr. Allergy Asthma Rep. 2017, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Schwinge, D.; Carambia, A.; Quaas, A.; Krech, T.; Wegscheid, C.; Tiegs, G.; Prinz, I.; Lohse, A.W.; Herkel, J.; Schramm, C. Testosterone suppresses hepatic inflammation by the downregulation of IL-17, CXCL-9, and CXCL-10 in a mouse model of experimental acute cholangitis. J. Immunol. 2015, 194, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Montefusco, D.; Jamil, M.; Maczis, M.A.; Schroeder, W.; Levi, M.; Ranjit, S.; Allegood, J.; Bandyopadhyay, D.; Retnam, R.; Spiegel, S.; et al. Sphingosine kinase 1 mediates sexual dimorphism in fibrosis in a mouse model of NASH. Mol. Metab. 2022, 62, 101523. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.W.; Gong, J.; Chang, X.M.; Luo, J.Y.; Dong, L.; Hao, Z.M.; Jia, A.; Xu, G.P. Estrogen reduces CCL4- induced liver fibrosis in rats. World J. Gastroenterol. 2002, 8, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wu, Z.Y. Estrogen derivatives: Novel therapeutic agents for liver cirrhosis and portal hypertension. Eur. J. Gastroenterol. Hepatol. 2013, 25, 263–270. [Google Scholar] [CrossRef]
- Shimizu, I.; Mizobuchi, Y.; Yasuda, M.; Shiba, M.; Ma, Y.R.; Horie, T.; Liu, F.; Ito, S. Inhibitory effect of oestradiol on activation of rat hepatic stellate cells in vivo and in vitro. Gut 1999, 44, 127–136. [Google Scholar] [CrossRef]
- Farooq, M.O.; Bataller, R. Pathogenesis and Management of Alcoholic Liver Disease. Dig. Dis. 2016, 34, 347–355. [Google Scholar] [CrossRef]
- Kamath, P.S.; Kim, W.R.; Advanced Liver Disease Study, G. The model for end-stage liver disease (MELD). Hepatology 2007, 45, 797–805. [Google Scholar] [CrossRef]
- Schonfeld, M.; Averilla, J.; Gunewardena, S.; Weinman, S.A.; Tikhanovich, I. Alcohol-associated fibrosis in females is mediated by female-specific activation of lysine demethylases KDM5B and KDM5C. Hepatol. Commun. 2022, 6, 2042–2057. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kharbanda, K.K.; Chokshi, S.; Tikhanovich, I.; Weinman, S.A.; New-Aaron, M.; Ganesan, M.; Osna, N.A. A Pathogenic Role of Non-Parenchymal Liver Cells in Alcohol-Associated Liver Disease of Infectious and Non-Infectious Origin. Biology 2023, 12, 255. https://doi.org/10.3390/biology12020255
Kharbanda KK, Chokshi S, Tikhanovich I, Weinman SA, New-Aaron M, Ganesan M, Osna NA. A Pathogenic Role of Non-Parenchymal Liver Cells in Alcohol-Associated Liver Disease of Infectious and Non-Infectious Origin. Biology. 2023; 12(2):255. https://doi.org/10.3390/biology12020255
Chicago/Turabian StyleKharbanda, Kusum K., Shilpa Chokshi, Irina Tikhanovich, Steven A. Weinman, Moses New-Aaron, Murali Ganesan, and Natalia A. Osna. 2023. "A Pathogenic Role of Non-Parenchymal Liver Cells in Alcohol-Associated Liver Disease of Infectious and Non-Infectious Origin" Biology 12, no. 2: 255. https://doi.org/10.3390/biology12020255
APA StyleKharbanda, K. K., Chokshi, S., Tikhanovich, I., Weinman, S. A., New-Aaron, M., Ganesan, M., & Osna, N. A. (2023). A Pathogenic Role of Non-Parenchymal Liver Cells in Alcohol-Associated Liver Disease of Infectious and Non-Infectious Origin. Biology, 12(2), 255. https://doi.org/10.3390/biology12020255