
Identification and Functional Analysis of Known and New Mutations in the Transcription Factor KLF1 Linked with β-Thalassemia-like Phenotypes
 ,
,  , , , and
, , , and
Abstract
Simple Summary
Abstract
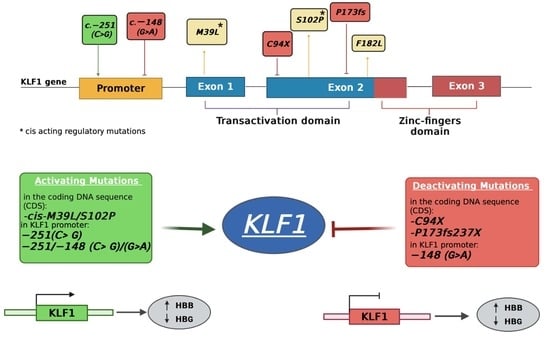
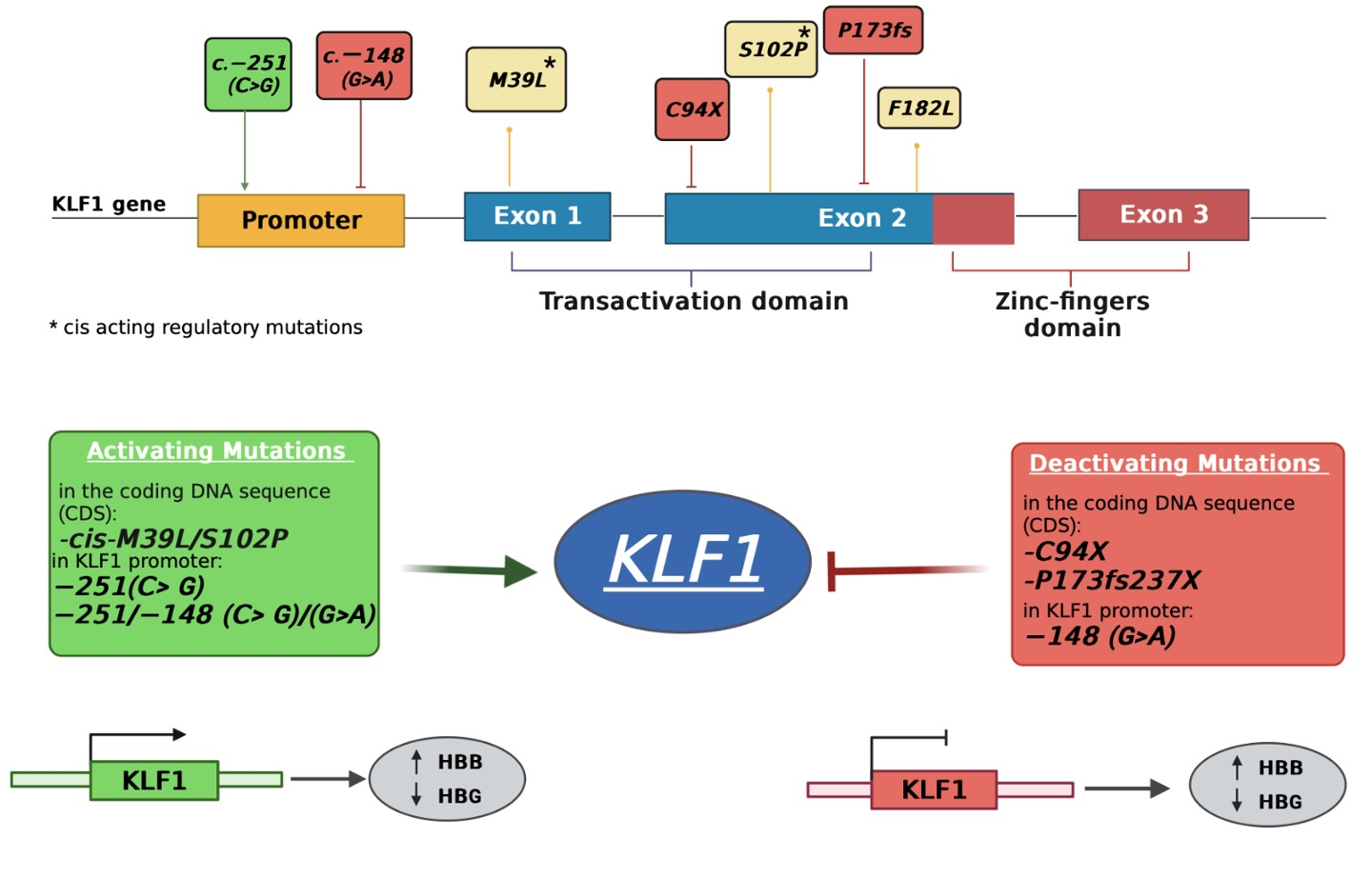
1. Introduction
2. Materials and Methods
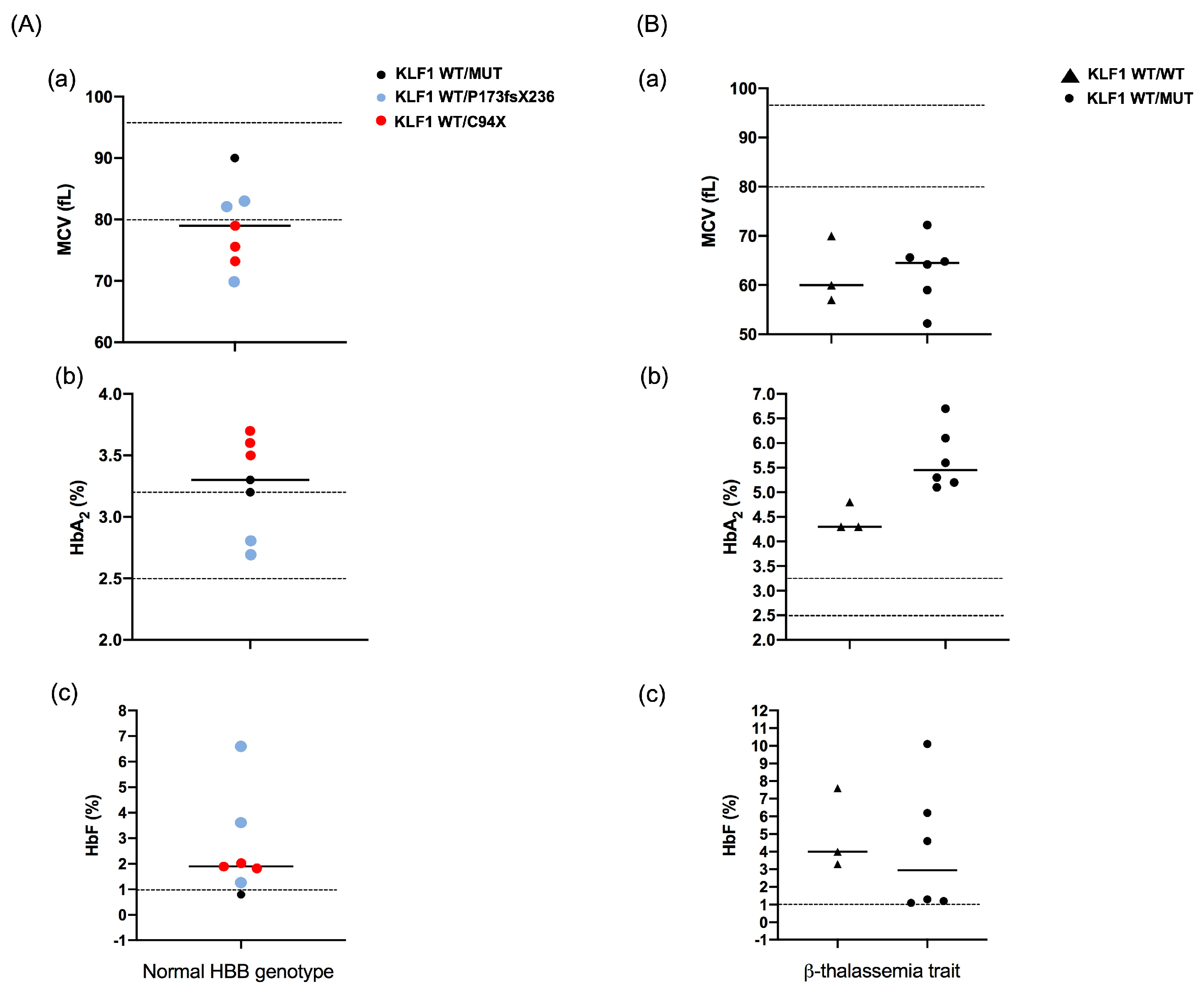
2.1. Patients and Hematological Data
2.2. Genetic Analysis of HBA and HBB Clusters
2.3. Cell Culture
2.3.1. Transient Transfection
2.3.2. RNA Extraction
2.3.3. Protein Extraction
2.4. Generation of KLF1 Mutants by Site-Specific Mutagenesis
2.5. Real-Time PCR Analysis
2.6. Western Blot Analysis
2.7. Luciferase Gene Reporter Assay
2.8. Statistical Analysis
3. Results
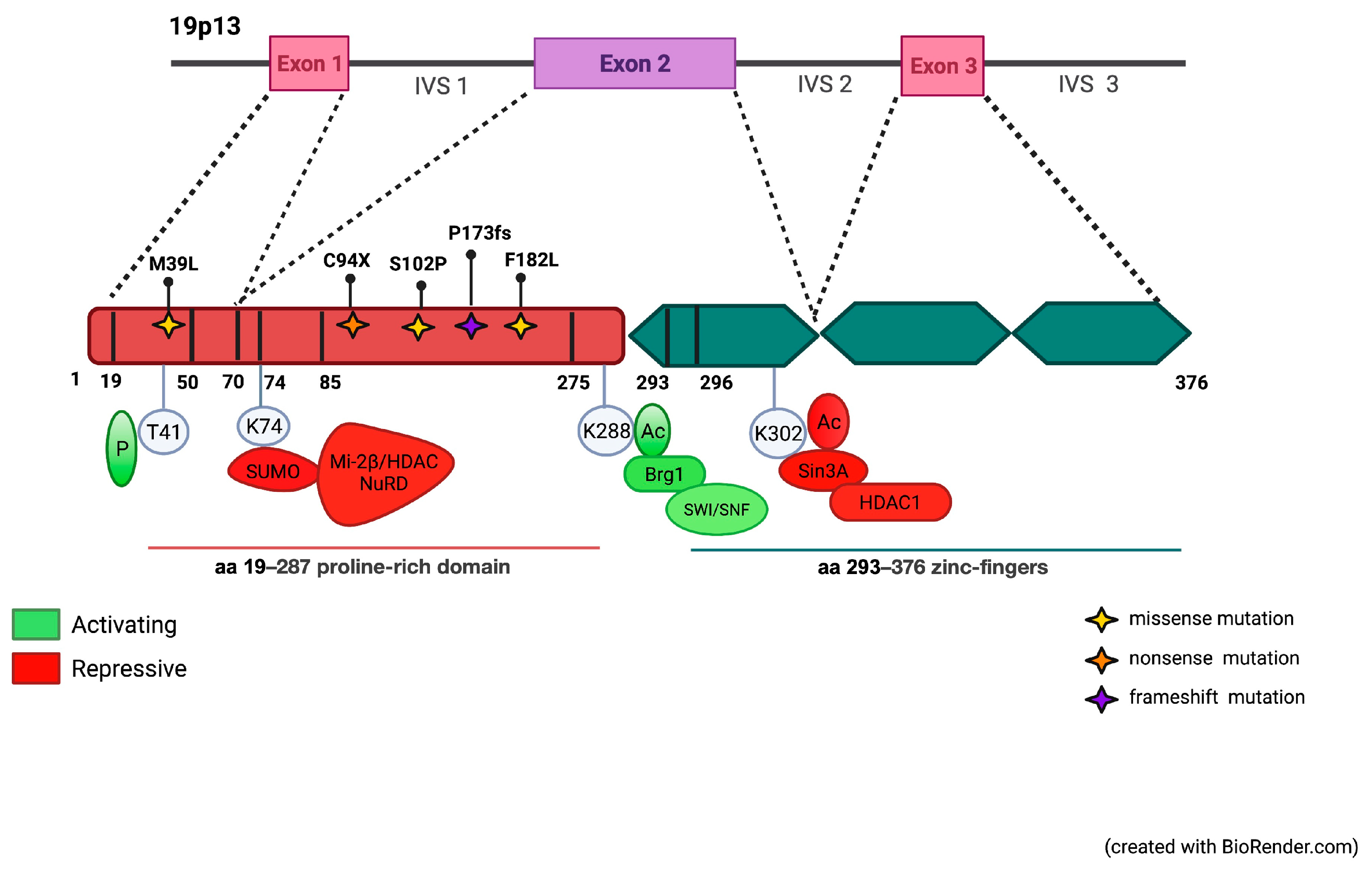
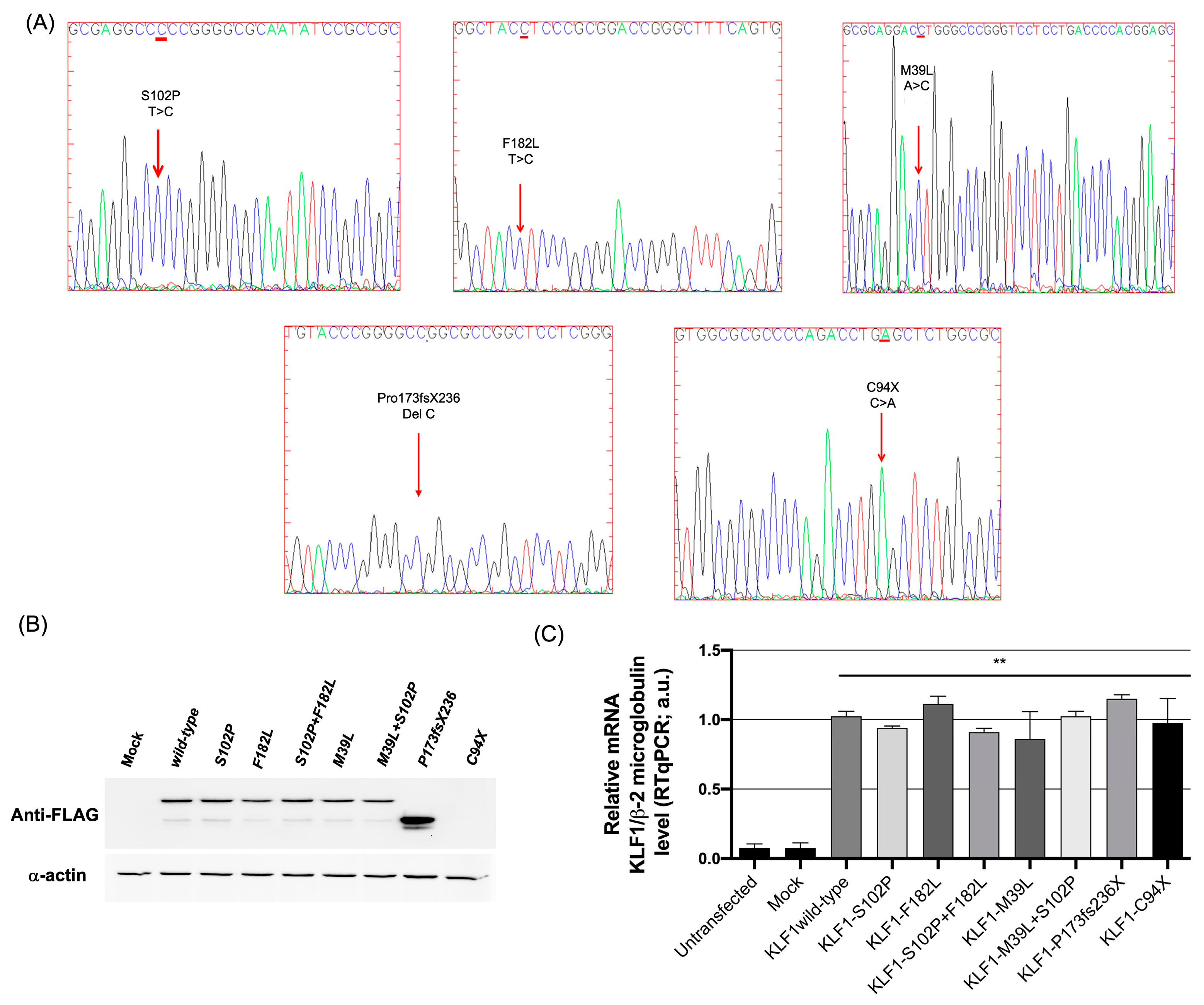
3.1. Identification of Mutations in the Coding Region of KLF1 Gene and Production of Mutant Constructs
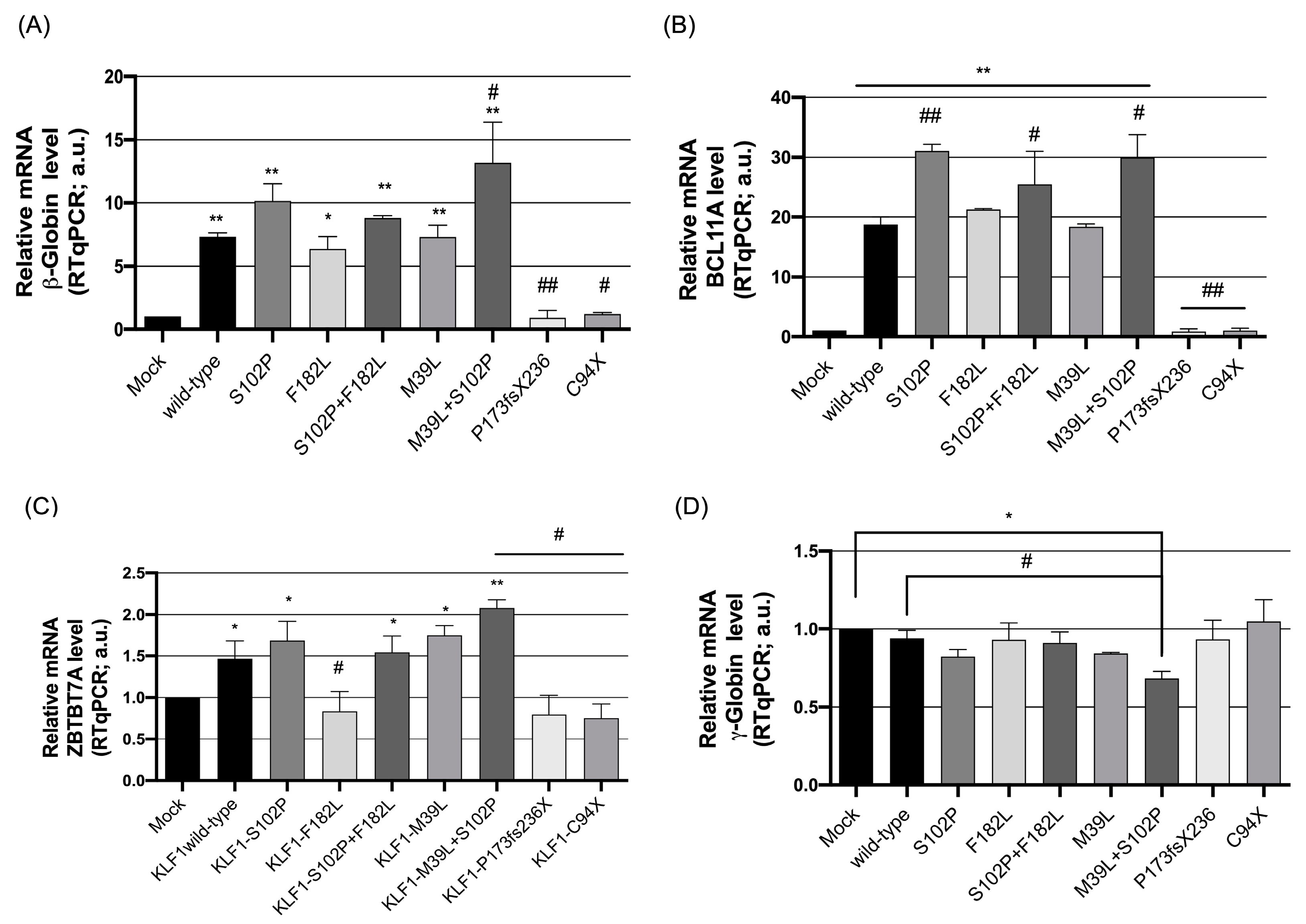
3.2. Functional Characterization of KLF1 Variants
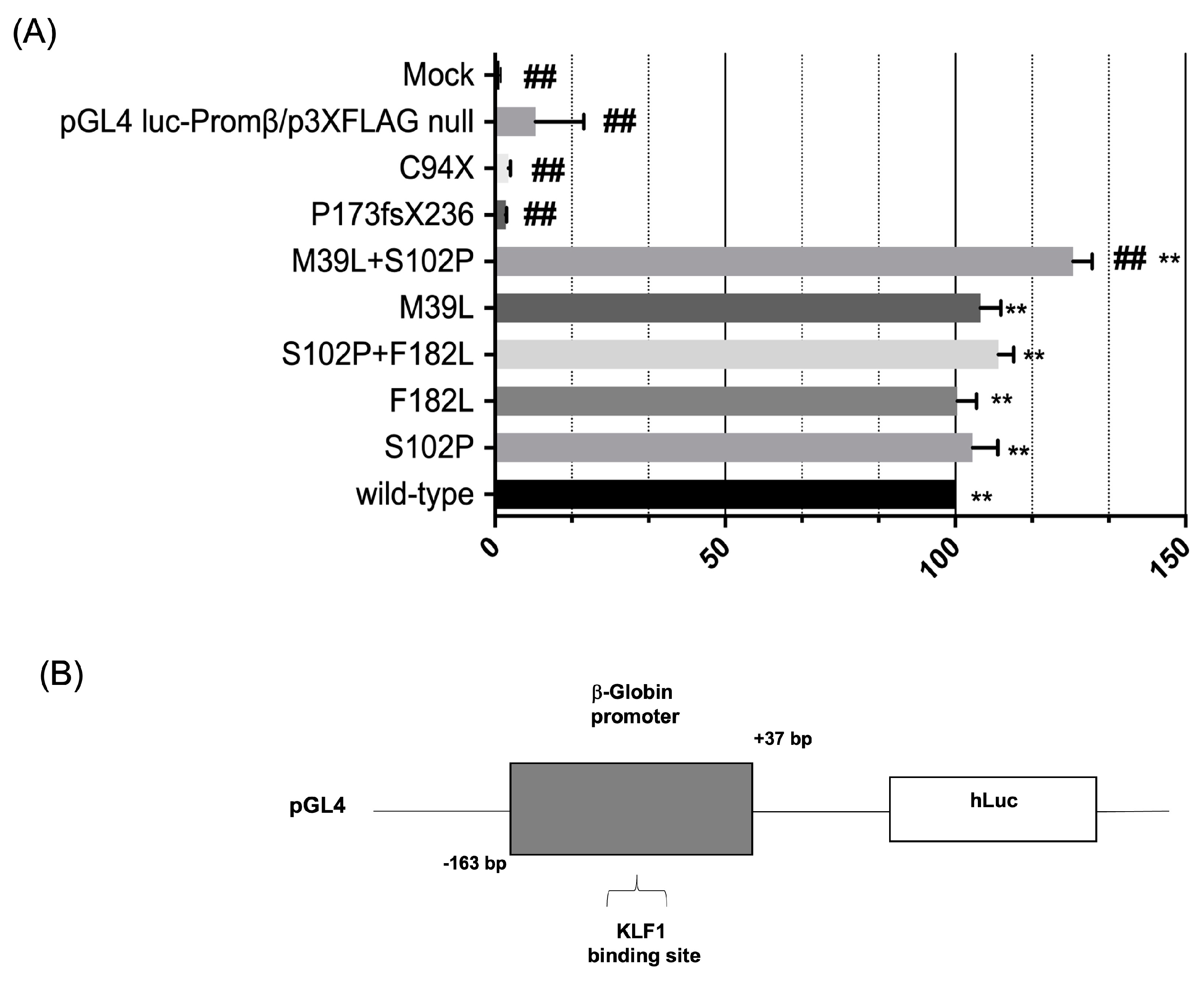
3.3. Evaluation of the Transcriptional Activity of KLF1 Variants on HBB Expression
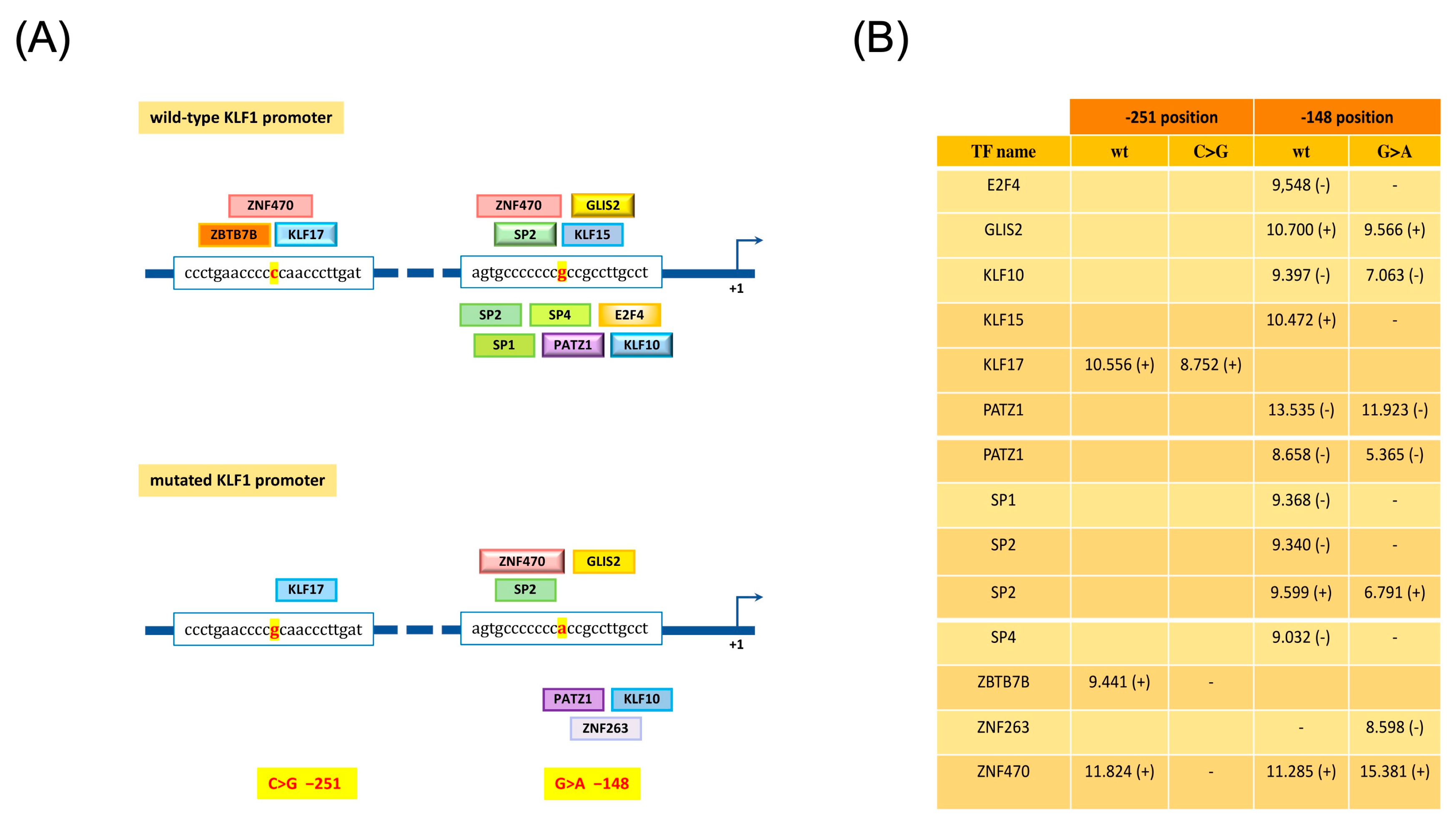
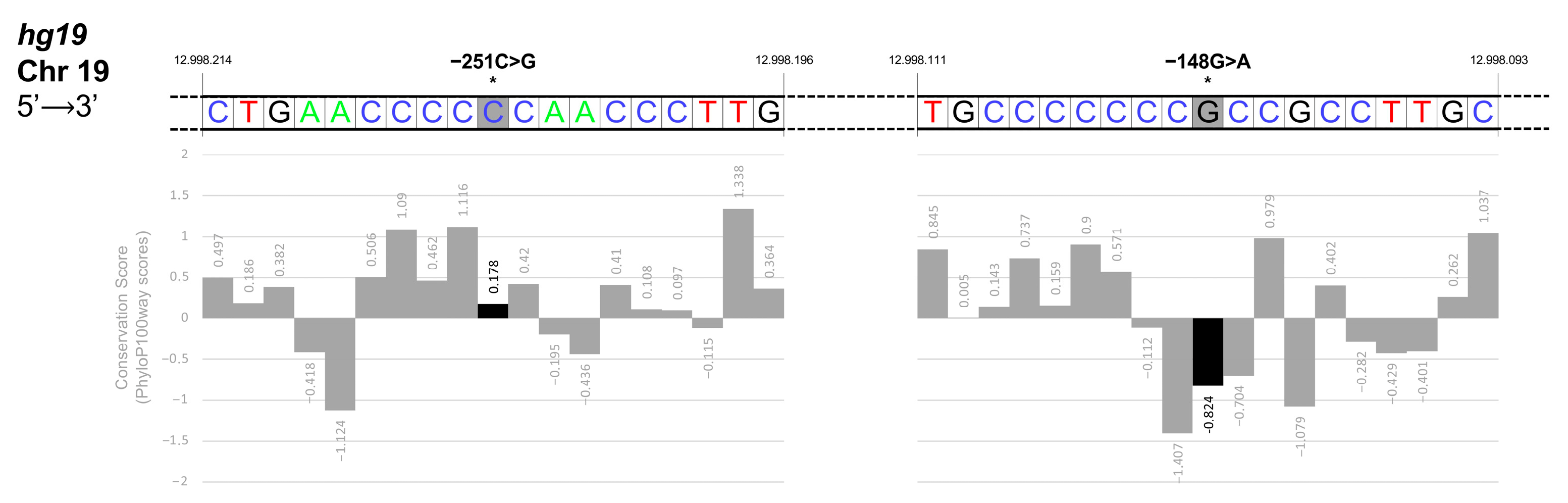
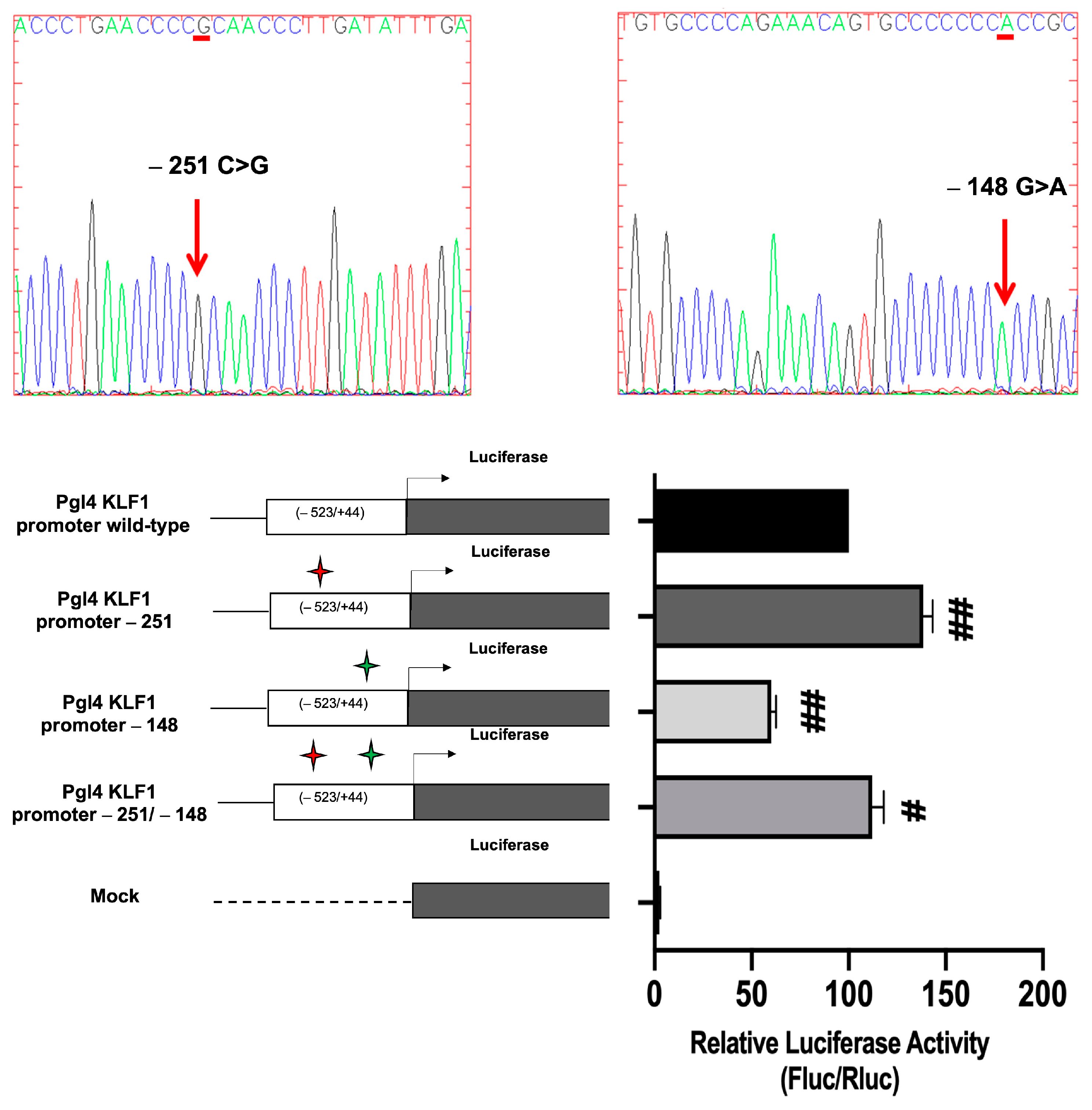
3.4. Identification and Functional Characterization of Sequence Variations in the KLF1 Promoter Region
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Origa, R. β-Thalassemia. Genet. Med. 2017, 19, 609–619. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Sessa, R.; Rosaria Storino, M.; Giuliano, M.; Trombetti, S.; Catapano, R.; Lo Bianco, A.; Izzo, P.; Grosso, M. Transcriptional Repressors of Fetal Globin Genes as Novel Therapeutic Targets in Beta-Thalassemia. In Beta Thalassemia; Zakaria, M., Hassan, T., Eds.; IntechOpen: London, UK, 2020; ISBN 978-1-83880-586-9. [Google Scholar]
- Suzuki, M.; Yamamoto, M.; Engel, J.D. Fetal Globin Gene Repressors as Drug Targets for Molecular Therapies to Treat the β-Globinopathies. Mol. Cell. Biol. 2014, 34, 3560–3569. [Google Scholar] [CrossRef]
- Steinberg, M.H. Fetal Hemoglobin in Sickle Hemoglobinopathies: High HbF Genotypes and Phenotypes. J. Clin. Med. 2020, 9, 3782. [Google Scholar] [CrossRef]
- Wilber, A.; Nienhuis, A.W.; Persons, D.A. Transcriptional Regulation of Fetal to Adult Hemoglobin Switching: New Therapeutic Opportunities. Blood 2011, 117, 3945–3953. [Google Scholar] [CrossRef]
- Venkatesan, V.; Srinivasan, S.; Babu, P.; Thangavel, S. Manipulation of Developmental Gamma-Globin Gene Expression: An Approach for Healing Hemoglobinopathies. Mol. Cell. Biol. 2020, 41, e00253-20. [Google Scholar] [CrossRef] [PubMed]
- Grosso, M.; Amendolara, M.; Rescigno, G.; Danise, P.; Todisco, N.; Izzo, P.; Amendola, G. Delayed Decline of γ-Globin Expression in Infant Age Associated with the Presence of G g−158 (C→T) Polymorphism. Int. J. Lab. Haematol. 2008, 30, 191–195. [Google Scholar] [CrossRef]
- Petruzzelli, R.; Gaudino, S.; Amendola, G.; Sessa, R.; Puzone, S.; Di Concilio, R.; D’Urzo, G.; Amendolara, M.; Izzo, P.; Grosso, M. Research Paper: Role of the Cold Shock Domain Protein A in the Transcriptional Regulation of HBG Expression: Role of CSDA in Modulation of Globin Gene Expression. Br. J. Haematol. 2010, 150, 689–699. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; Van Handel, B.; Mikkola, H.K.A.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human Fetal Hemoglobin Expression Is Regulated by the Developmental Stage-Specific Repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, Z.; He, C.; Zhang, B.; Shi, Y.; Li, F. Structural Insights into the Recognition of γ-Globin Gene Promoter by BCL11A. Cell Res. 2019, 29, 960–963. [Google Scholar] [CrossRef]
- Martyn, G.E.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.M.; Funnell, A.P.W.; et al. Natural Regulatory Mutations Elevate the Fetal Globin Gene via Disruption of BCL11A or ZBTB7A Binding. Nat. Genet. 2018, 50, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Xu, X.; Higgs, D.R.; Patrinos, G.P.; Arnaud, L.; Bieker, J.J.; Philipsen, S.; the KLF1 Consensus Workgroup. Krüppeling Erythropoiesis: An Unexpected Broad Spectrum of Human Red Blood Cell Disorders Due to KLF1 Variants. Blood 2016, 127, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Liu, K.; Sun, C.-W.; Pawlik, K.M.; Townes, T.M. KLF1 Regulates BCL11A Expression and γ- to β-Globin Gene Switching. Nat. Genet. 2010, 42, 742–744. [Google Scholar] [CrossRef]
- Heshusius, S.; Grech, L.; Gillemans, N.; Brouwer, R.W.W.; den Dekker, X.T.; van IJcken, W.F.J.; Nota, B.; Felice, A.E.; van Dijk, T.B.; von Lindern, M.; et al. Epigenomic Analysis of KLF1 Haploinsufficiency in Primary Human Erythroblasts. Sci. Rep. 2022, 12, 336. [Google Scholar] [CrossRef] [PubMed]
- Siatecka, M.; Bieker, J.J. The Multifunctional Role of EKLF/KLF1 during Erythropoiesis. Blood 2011, 118, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.; Colah, R.; Ghosh, K.; Nadkarni, A. Differential Role of Kruppel like Factor 1 (KLF1) Gene in Red Blood Cell Disorders. Genomics 2019, 111, 1771–1776. [Google Scholar] [CrossRef]
- Caria, C.A.; Faà, V.; Ristaldi, M.S. Krüppel-Like Factor 1: A Pivotal Gene Regulator in Erythropoiesis. Cells 2022, 11, 3069. [Google Scholar] [CrossRef]
- Iolascon, A.; Heimpel, H.; Wahlin, A.; Tamary, H. Congenital Dyserythropoietic Anemias: Molecular Insights and Diagnostic Approach. Blood 2013, 122, 2162–2166. [Google Scholar] [CrossRef] [PubMed]
- Belgemen-Ozer, T.; Gorukmez, O. A Very Rare Congenital Dyserythropoietic Anemia Variant—Type IV in a Patient with a Novel Mutation in the KLF1 Gene: A Case Report and Review of the Literature. J. Pediatr. Hematol. Oncol. 2020, 42, e536–e540. [Google Scholar] [CrossRef]
- Kulczynska, K.; Bieker, J.J.; Siatecka, M. A Krüppel-like Factor 1 (KLF1) Mutation Associated with Severe Congenital Dyserythropoietic Anemia Alters Its DNA-Binding Specificity. Mol. Cell. Biol. 2020, 40, e00444-19. [Google Scholar] [CrossRef] [PubMed]
- Waye, J.S.; Eng, B. Krüppel-like Factor 1: Hematologic Phenotypes Associated with KLF1 Gene Mutations. Int. J. Lab. Hematol. 2015, 37, 78–84. [Google Scholar] [CrossRef]
- Borg, J.; Patrinos, G.P.; Felice, A.E.; Philipsen, S. Erythroid Phenotypes Associated with KLF1 Mutations. Haematologica 2011, 96, 635–638. [Google Scholar] [CrossRef]
- Perseu, L.; Satta, S.; Moi, P.; Demartis, F.R.; Manunza, L.; Sollaino, M.C.; Barella, S.; Cao, A.; Galanello, R. KLF1 Gene Mutations Cause Borderline HbA2. Blood 2011, 118, 4454–4458. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, X.; Yu, L.; Cai, R.; Ma, X.; Zheng, C.; Zhou, Y.; Liu, Q.; Wei, X.; Lin, L.; et al. KLF1 Mutations Are Relatively More Common in a Thalassemia Endemic Region and Ameliorate the Severity of β-Thalassemia. Blood 2014, 124, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Fanis, P.; Kousiappa, I.; Phylactides, M.; Kyrri, A.; Hadjigavriel, M.; Christou, S.; Sitarou, M.; Kleanthous, M. A Novel Mutation in the Erythroid Transcription Factor KLF1 Is Likely Responsible for Ameliorating β-thalassemia Major. Hum. Mutat. 2019, 40, 1768–1780. [Google Scholar] [CrossRef] [PubMed]
- Gallienne, A.E.; Dreau, H.M.P.; Schuh, A.; Old, J.M.; Henderson, S. Ten Novel Mutations in the Erythroid Transcription Factor KLF1 Gene Associated with Increased Fetal Hemoglobin Levels in Adults. Haematologica 2012, 97, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-M.; Ma, S.; Tang, X.-L.; Yang, J.-Y.; Yang, Z.-L. Diagnosis of the Accurate Genotype of HKαα Carriers in Patients with Thalassemia Using Multiplex Ligation-Dependent Probe Amplification Combined with Nested Polymerase Chain Reaction. Chin. Med. J. 2020, 133, 1175–1181. [Google Scholar] [CrossRef]
- Makis, A.; Georgiou, I.; Traeger-Synodinos, J.; Storino, M.R.; Giuliano, M.; Andolfo, I.; Hatzimichael, E.; Chaliasos, N.; Giapros, V.; Izzo, P.; et al. A Novel εγδβ-Thalassemia Deletion Associated with Severe Anemia at Birth and a β-Thalassemia Intermedia Phenotype Later in Life in Three Generations of a Greek Family. Hemoglobin 2021, 45, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Grosso, M.; Puzone, S.; Storino, M.R.; Sessa, R.; Izzo, P. Prenatal Diagnosis of Haemoglobinopathies: Our Experience of 523 Cases. Clin. Chem. Lab. Med. CCLM 2013, 51, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; Puzone, S.; Ammirabile, M.; Piscopo, C.; Pagano, L.; Colucci, S.; Izzo, P.; Grosso, M. Identification and Molecular Characterization of the CAMPANIA Deletion, a Novel α0-Thalassemic Defect, in Two Unrelated Italian Families. Am. J. Hematol. 2010, 85, 143–144. [Google Scholar] [CrossRef]
- Esposito, G.; Ruggiero, R.; Savarese, M.; Savarese, G.; Tremolaterra, M.R.; Salvatore, F.; Carsana, A. Prenatal Molecular Diagnosis of Inherited Neuromuscular Diseases: Duchenne/Becker Muscular Dystrophy, Myotonic Dystrophy Type 1 and Spinal Muscular Atrophy. Clin. Chem. Lab. Med. CCLM 2013, 51, 2239–2245. [Google Scholar] [CrossRef]
- Trombetti, S.; Sessa, R.; Catapano, R.; Rinaldi, L.; Bianco, A.L.; Feliciello, A.; Izzo, P.; Grosso, M. Exploring the Leukemogenic Potential of GATA-1S, the Shorter Isoform of GATA-1: Novel Insights into Mechanisms Hampering Respiratory Chain Complex II Activity and Limiting Oxidative Phosphorylation Efficiency. Antioxidants 2021, 10, 1603. [Google Scholar] [CrossRef] [PubMed]
- Riccio, P.; Sessa, R.; de Nicola, S.; Petruzziello, F.; Trombetti, S.; Menna, G.; Pepe, G.; Maddalena, P.; Izzo, P.; Grosso, M. GATA-1 Isoforms Differently Contribute to the Production and Compartmentation of Reactive Oxygen Species in the Myeloid Leukemia Cell Line K562. J. Cell. Physiol. 2019, 234, 20829–20846. [Google Scholar] [CrossRef] [PubMed]
- Lamsfus-Calle, A.; Daniel-Moreno, A.; Antony, J.S.; Epting, T.; Heumos, L.; Baskaran, P.; Admard, J.; Casadei, N.; Latifi, N.; Siegmund, D.M.; et al. Comparative Targeting Analysis of KLF1, BCL11A, and HBG1/2 in CD34+ HSPCs by CRISPR/Cas9 for the Induction of Fetal Hemoglobin. Sci. Rep. 2020, 10, 10133. [Google Scholar] [CrossRef]
- Hojo, N.; Tatsumi, N.; Moriguchi, N.; Matsumura, A.; Morimoto, S.; Nakata, J.; Fujiki, F.; Nishida, S.; Nakajima, H.; Tsuboi, A.; et al. A Zbtb7a Proto-Oncogene as a Novel Target for MiR-125a: Zbtb7a AS A TARGET FOR MiR-125a. Mol. Carcinog. 2016, 55, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Grech, L. Control of Globin Gene Expression by Kruppel-like Factors; Malta Chamber of Scientists: Msida, Malta, 2014; pp. 64–71. [Google Scholar] [CrossRef]
- Sarnelli, G.; Grosso, M.; Palumbo, I.; Pesce, M.; D’Alessandro, A.; Zaninotto, G.; Annese, V.; Petruzzelli, R.; Izzo, P.; Sepulveres, R.; et al. Allele-specific Transcriptional Activity of the Variable Number of Tandem Repeats of the Inducible Nitric Oxide Synthase Gene Is Associated with Idiopathic Achalasia. United Eur. Gastroenterol. J. 2017, 5, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Mikołajczyk, K.; Kaczmarek, R.; Czerwiński, M. Can mutations in the gene encoding transcription factor EKLF (Erythroid Krüppel-Like Factor) protect us against infectious and parasitic diseases? Adv. Hyg. Exp. Med. 2016, 70, 1068–1086. [Google Scholar] [CrossRef] [PubMed]
- Zaker-Kandjani, B.; Namdar-Aligoodarzi, P.; Azarkeivan, A.; Najmabadi, H.; Banan, M. Mutation Screening of the Krüppel-Like Factor 1 Gene Using Single-Strand Conformational Polymorphism in a Cohort of Iranian β-Thalassemia Patients. Hemoglobin 2015, 39, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Tepakhan, W.; Kanjanaopas, S.; Srewaradachpisal, K. Association Between Genetic Polymorphisms and Hb F Levels in Heterozygous β-Thalassemia 3.5 Kb Deletions. Hemoglobin 2020, 44, 338–343. [Google Scholar] [CrossRef]
- Hariharan, P. Insight of Fetal to Adult Hemoglobin Switch: Genetic Modulators and Therapeutic Targets. Blood Rev. 2021, 49, 100823. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, J.A.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E. Nonsense-Mediated Decay Approaches the Clinic. Nat. Genet. 2004, 36, 801–808. [Google Scholar] [CrossRef]
- Kuroha, K.; Tatematsu, T.; Inada, T. Upf1 Stimulates Degradation of the Product Derived from Aberrant Messenger RNA Containing a Specific Nonsense Mutation by the Proteasome. EMBO Rep. 2009, 10, 1265–1271. [Google Scholar] [CrossRef]
- Kuroha, K.; Ando, K.; Nakagawa, R.; Inada, T. The Upf Factor Complex Interacts with Aberrant Products Derived from MRNAs Containing a Premature Termination Codon and Facilitates Their Proteasomal Degradation. J. Biol. Chem. 2013, 288, 28630–28640. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ren, R.; Ly, L.C.; Horton, J.R.; Li, F.; Quinlan, K.G.R.; Crossley, M.; Shi, Y.; Cheng, X. Structural Basis for Human ZBTB7A Action at the Fetal Globin Promoter. Cell Rep. 2021, 36, 109759. [Google Scholar] [CrossRef]
- Satta, S.; Perseu, L.; Maccioni, L.; Giagu, N.; Galanello, R. Delayed Fetal Hemoglobin Switching in Subjects with KLF1 Gene Mutation. Blood Cells Mol. Dis. 2012, 48, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Radmilovic, M.; Zukic, B.; Petrovic, M.S.; Bartsakoulia, M.; Stankovic, B.; Kotur, N.; Dokmanovic, L.; Georgitsi, M.; Patrinos, G.P.; Pavlovic, S. Functional Analysis of a Novel KLF1 Gene Promoter Variation Associated with Hereditary Persistence of Fetal Hemoglobin. Ann. Hematol. 2013, 92, 53–58. [Google Scholar] [CrossRef]
- Kumar, R. Krüppel-like Factor 1 (KLF1) Gene Single Nucleotide Polymorphisms in Sickle Cell Disease and Its Association with Disease-Related Morbidities. Ann. Hematol. 2021, 100, 365–373. [Google Scholar] [CrossRef]
- Nicolau, M.; Vargas, S.; Silva, M.; Coelho, A.; Ferreira, E.; Mendonça, J.; Vieira, L.; Kjöllerström, P.; Maia, R.; Silva, R.; et al. Genetic Modulators of Fetal Hemoglobin Expression and Ischemic Stroke Occurrence in African Descendant Children with Sickle Cell Anemia. Ann. Hematol. 2019, 98, 2673–2681. [Google Scholar] [CrossRef]
- Khamphikham, P.; Sripichai, O.; Munkongdee, T.; Fucharoen, S.; Tongsima, S.; Smith, D.R. Genetic Variation of Krüppel-like Factor 1 (KLF1) and Fetal Hemoglobin (HbF) Levels in β0-Thalassemia/HbE Disease. Int. J. Hematol. 2018, 107, 297–310. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Huang, Y.; Yang, K.; Xiao, T.; Xiong, J.; Wang, K.; Liu, C.; He, T.; Yu, Y.; et al. IRF-1 Promotes Renal Fibrosis by Downregulation of Klotho. FASEB J. 2020, 34, 4415–4429. [Google Scholar] [CrossRef] [PubMed]
- Tepakhan, W.; Yamsri, S.; Sanchaisuriya, K.; Fucharoen, G.; Xu, X.; Fucharoen, S. Nine Known and Five Novel Mutations in the Erythroid Transcription Factor KLF1 Gene and Phenotypic Expression of Fetal Hemoglobin in Hemoglobin E Disorder. Blood Cells Mol. Dis. 2016, 59, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Eernstman, J.; Veldhuisen, B.; Ligthart, P.; von Lindern, M.; van der Schoot, C.E.; van den Akker, E. Novel Variants in Krueppel like Factor 1 That Cause Persistence of Fetal Hemoglobin in In(Lu) Individuals. Sci. Rep. 2021, 11, 18557. [Google Scholar] [CrossRef] [PubMed]
- Satta, S.; Paglietti, M.E.; Sollaino, M.C.; Barella, S.; Moi, P.; Desogus, M.F.; Demartis, F.R.; Manunza, L.; Origa, R. Changes in HbA2 and HbF in Alpha Thalassemia Carriers with KLF1 Mutation. Blood Cells Mol. Dis. 2017, 64, 30–32. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer | Sequence (5′ > 3′) | Amplicon Size (bp) |
|---|---|---|---|
| KLF1 gene (NG_013087.1) | |||
| Exon 1 | Forward | GATAGCAGCCTCCAACGTCTG | 731 bp |
| Reverse | CGTACCTCAGTCCTGGTTAAG | ||
| Intron 1 | Forward | CAGGGCCACAGTCACAGATC | 885 bp |
| Reverse | TCTTCGGAGCGCCACCACTG | ||
| Exon 2 | Forward | TCTGCGTCAGAGTGTCCAGC | 768 bp |
| Reverse | CACCTGGATCCTCTGCAGTC | ||
| Exon 3 | Forward | CAGTACCAAGGGCACTTCCAG | 794 bp |
| Reverse | CGTCCGTGTGAAGAGACCAC | ||
| Exon 4 | Forward | CTTGCACATGAAGCGCCACC | 604 bp |
| Reverse | TGTCACTGTAGTTTAGGAGATG | ||
| Exon 5 | Forward | GAAGGTCCCTTATTGTGGCTG | 765 bp |
| Reverse | CAGCCATTATTCTGGAGGTCA | ||
| KLF1 cDNA (NM_006563.5) | |||
| EcoR1 KLF1 Forward | GCGAATTCC/ATGGCCACAGCCGAGACCGCCTTGCCCTC | 1111 bp | |
| BglII KLF1 Reverse | GGAAGATCTTCCGC/TCAAAGGTGGCGCTTCATGTGCAAGGC | ||
| KLF1promoter (NG_013087.1) | |||
| XhoI Prom KLF1 Forward | ATACTCGAG/GATAGCAGCCTCCAACGTCTG | 524 bp | |
| BglII Prom KLF1 Reverse | GGAAGATCTTCC/TGGGCCTCAAGCCTCCTCTTC | ||
| HBBpromoter (NG_059281.1) | |||
| Bgl II-HBB Forward | GGAAGATCTTC/ACTCCTAAGCCAGTGCCAG | 200 bp | |
| HindIII HBB Reverse | CCCAAGCTTG/GGTTGCTAGTGAACACAGTTG | ||
| Transcript | Accession Number | Primer | Sequence 5′-3′ | Amplicon Size |
|---|---|---|---|---|
| HBB | NM_000518.5 | Forward | ACACAACTGTGTTCACTAGCA | 306 bp |
| Reverse | ACTCAGTGTGGCAAAGGTG | |||
| HBG1/HBG2 | NM_000559.3 | Forward | GACAGCTTTGGCAACCTGT | 192 bp |
| NM_000184.3 | Reverse | CCAGGAGCTTGAAGTTCTC | ||
| BCL11A | NM_022893.4 | Forward | CGAGCACAAACGGAAACAATG | 212 bp |
| Reverse | CTCCAGTGCAGAAGTTTATCTG | |||
| ZBTB7A | NM_015898.4 | Forward | ACGAGTGCAACATCTGCAAG | 128 bp |
| Reverse | GGTCGTAGTTGTGGGCAAAG | |||
| KLF1 | NM_006563.5 | Forward | CACACAGGGGAGAAGCCATA | 149 bp |
| Reverse | GTCAGAGCGCGAAAAAGC | |||
| β2-microglobulin | NM_004048.4 | Forward | CCGTGGCCTTAGCTGTGCT | 150 bp |
| Reverse | TCGGATGGATGAAACCGAGA |
| Case | Age-Sex | Hb (g/dL) | MCV (fL) | HbF (%) | HbA2 (%) | HBB Genotype | KLF1 Genotype | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Promoter | Coding Region | ||||||||||
| Mutation 1 | Mutation 2 | Mutation 1 | Mutation 2 | Mutation 3 | |||||||
| 1 | 13 y-M | 12.2 | 52.2 | 1.1 | 6.1 | β039/N | -- | -- | M39L * | S102P * | -- |
| 2 | 20 y-F | 8.1 | 72.2 | 4.6 | 5.3 | β0IVSII-1/N | -- | -- | S102P | -- | -- |
| 3 | 28 y-F | 10.1 | 59.0 | 6.2 | 5.2 | β0IVSI-1/N | -- | -- | S102P * | F182L * | -- |
| 4 | 28 y-F | 8.9 | 64.8 | 1.2 | 5.1 | β039/N | −251 (C/G) | -- | S102P | -- | -- |
| 5 | 35 y-F | 7.3 | 65.6 | 10.1 | 6.7 | β039/N | −251 (C/G) | −148 (G/A) | S102P | -- | -- |
| 6 | 38 y-F | 13.3 | 64.2 | 1.3 | 5.6 | β039/N | -- | -- | M39L * | S102P * | -- |
| 7 | 7 y-M | 6.7 | 70.0 | 7.6 | 4.3 | β+IVSI-110/N | -- | -- | -- | -- | -- |
| 8 | 15 y-M | 11.6 | 57.0 | 3.3 | 4.8 | β0IVSI-1/N | -- | -- | -- | -- | -- |
| 9 | 40 y-M | 10.5 | 60.0 | 4.0 | 4.3 | β0IVSII-1/N | -- | -- | -- | -- | -- |
| 10 | 9 y-F | 13.2 | 73.3 | 1.8 | 3.7 | N/N | -- | -- | C94X | -- | -- |
| 11 | 13 y-F | 12.5 | 75.6 | 1.9 | 3.5 | N/N | -- | -- | C94X | -- | -- |
| 12 | 30 y-M | 14.9 | 90.0 | 0.8 | 3.2 | N/N | −251 (C/G) | -- | F182L | -- | -- |
| 13 | 32 y-F | 12.0 | 82.0 | 1.3 | 3.3 | N/N | -- | -- | P173fsX236 | -- | -- |
| 14 | 34 y-F | 11.8 | 79.0 | 2.0 | 3.6 | N/N | -- | -- | C94X | -- | -- |
| 15 | 56 y-M | 14.5 | 83.0 | 6.6 | 2.7 | N/N | -- | -- | S102P * | F182L * | P173fsX236 |
| 16 | 73 y-M | 10.7 | 70.0 | 3.6 | 2.8 | N/N | −251 (C/G) * | −148 (G/A) * | P173fsX236 | -- | -- |
| 17 | 30 y-F | 15.5 | 88.3 | 0.5 | 3.5 | N/N | -- | -- | -- | -- | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catapano, R.; Sessa, R.; Trombetti, S.; Cesaro, E.; Russo, F.; Izzo, P.; Makis, A.; Grosso, M. Identification and Functional Analysis of Known and New Mutations in the Transcription Factor KLF1 Linked with β-Thalassemia-like Phenotypes. Biology 2023, 12, 510. https://doi.org/10.3390/biology12040510
Catapano R, Sessa R, Trombetti S, Cesaro E, Russo F, Izzo P, Makis A, Grosso M. Identification and Functional Analysis of Known and New Mutations in the Transcription Factor KLF1 Linked with β-Thalassemia-like Phenotypes. Biology. 2023; 12(4):510. https://doi.org/10.3390/biology12040510
Chicago/Turabian StyleCatapano, Rosa, Raffaele Sessa, Silvia Trombetti, Elena Cesaro, Filippo Russo, Paola Izzo, Alexandros Makis, and Michela Grosso. 2023. "Identification and Functional Analysis of Known and New Mutations in the Transcription Factor KLF1 Linked with β-Thalassemia-like Phenotypes" Biology 12, no. 4: 510. https://doi.org/10.3390/biology12040510
APA StyleCatapano, R., Sessa, R., Trombetti, S., Cesaro, E., Russo, F., Izzo, P., Makis, A., & Grosso, M. (2023). Identification and Functional Analysis of Known and New Mutations in the Transcription Factor KLF1 Linked with β-Thalassemia-like Phenotypes. Biology, 12(4), 510. https://doi.org/10.3390/biology12040510