


Conservation Genetics of the Critically Endangered Southern River Terrapin (Batagur affinis) in Malaysia: Genetic Diversity and Novel Subspecies Distribution Ranges


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
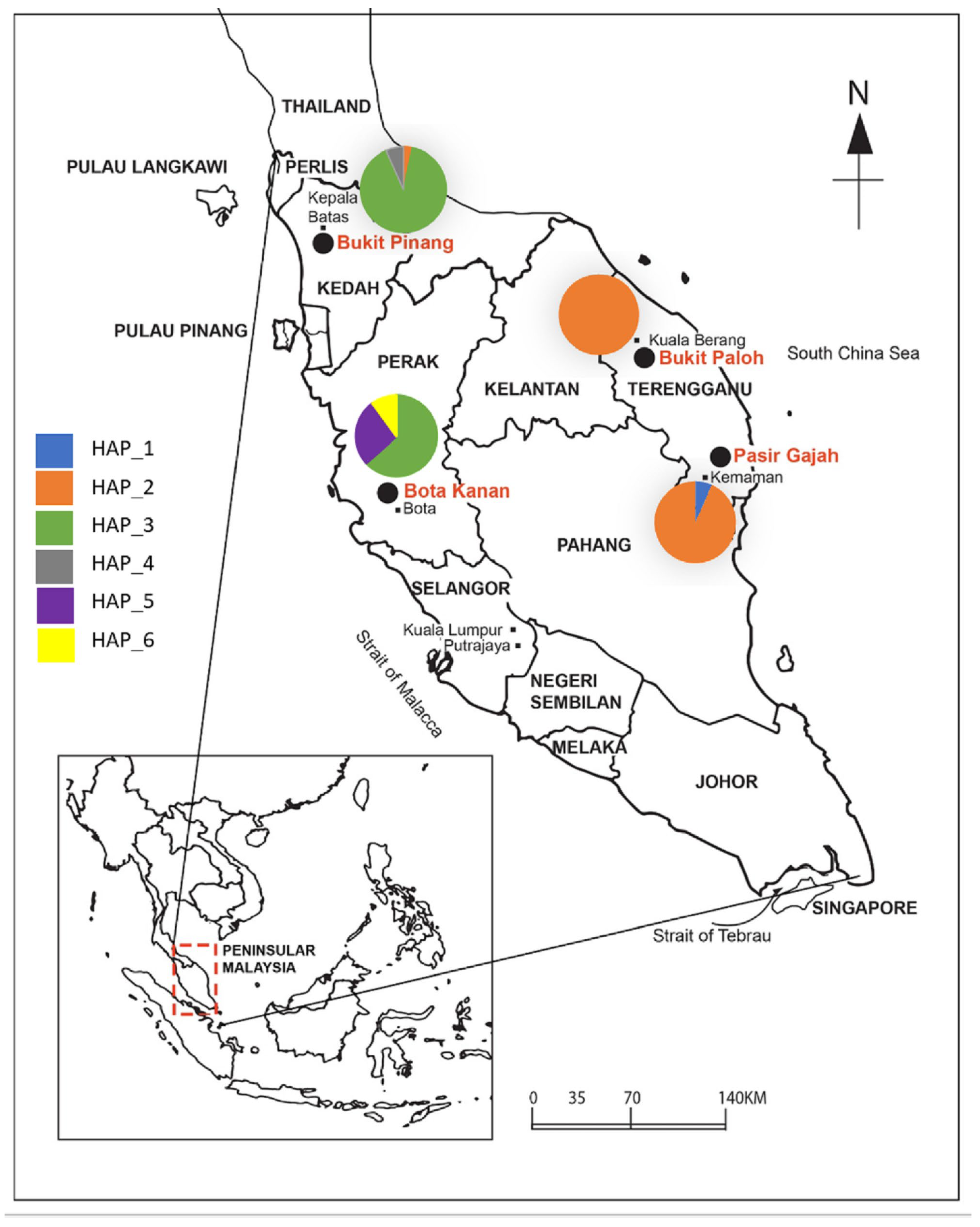
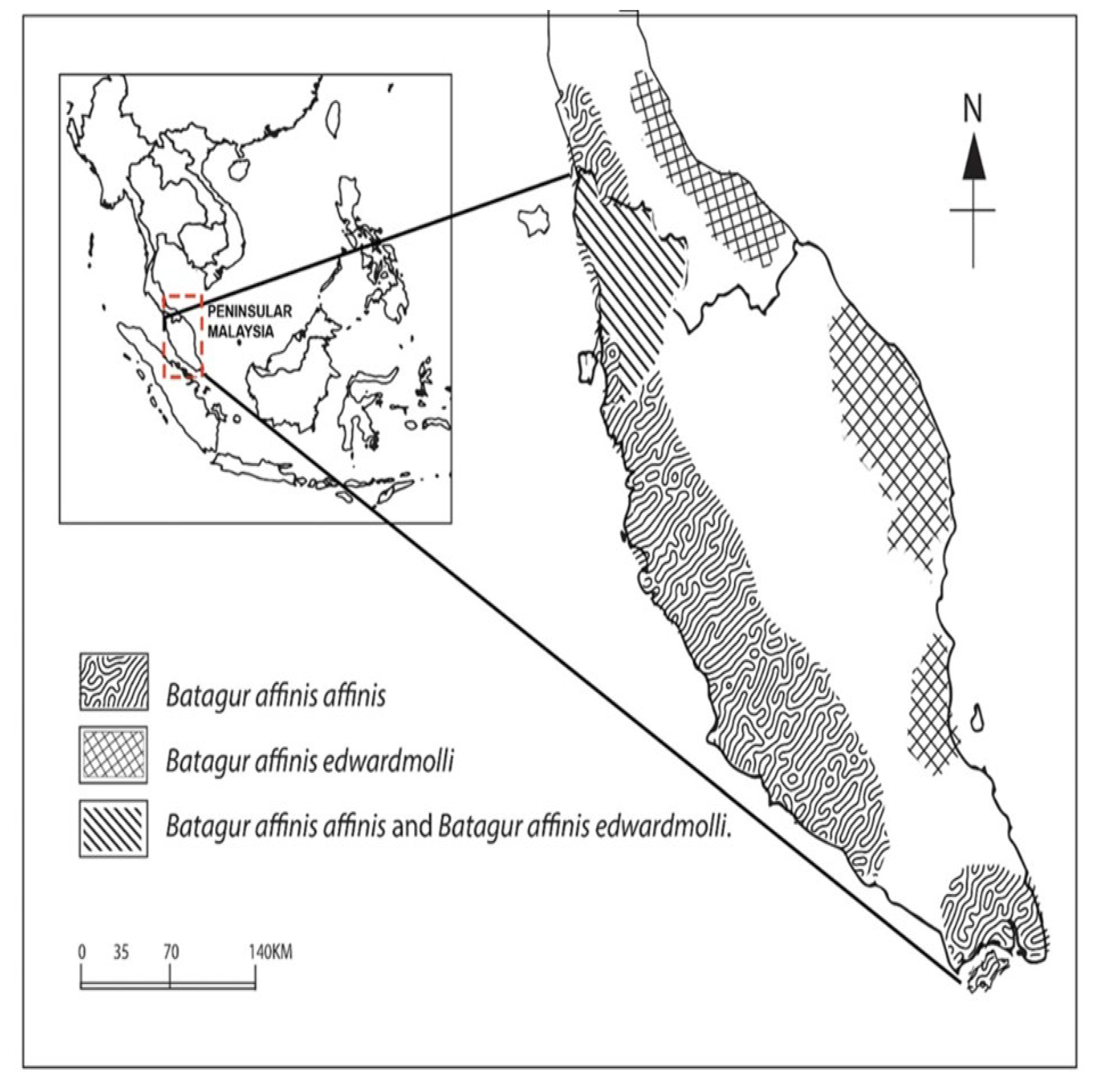
2.1. Sample Collection
2.2. DNA Extraction and PCR Amplification
2.3. Data Analysis
2.3.1. General Characterisation and Population Diversity
2.3.2. Population Structure and Demographic History
2.3.3. Phylogenetic Relationship
3. Results
3.1. Population Diversity
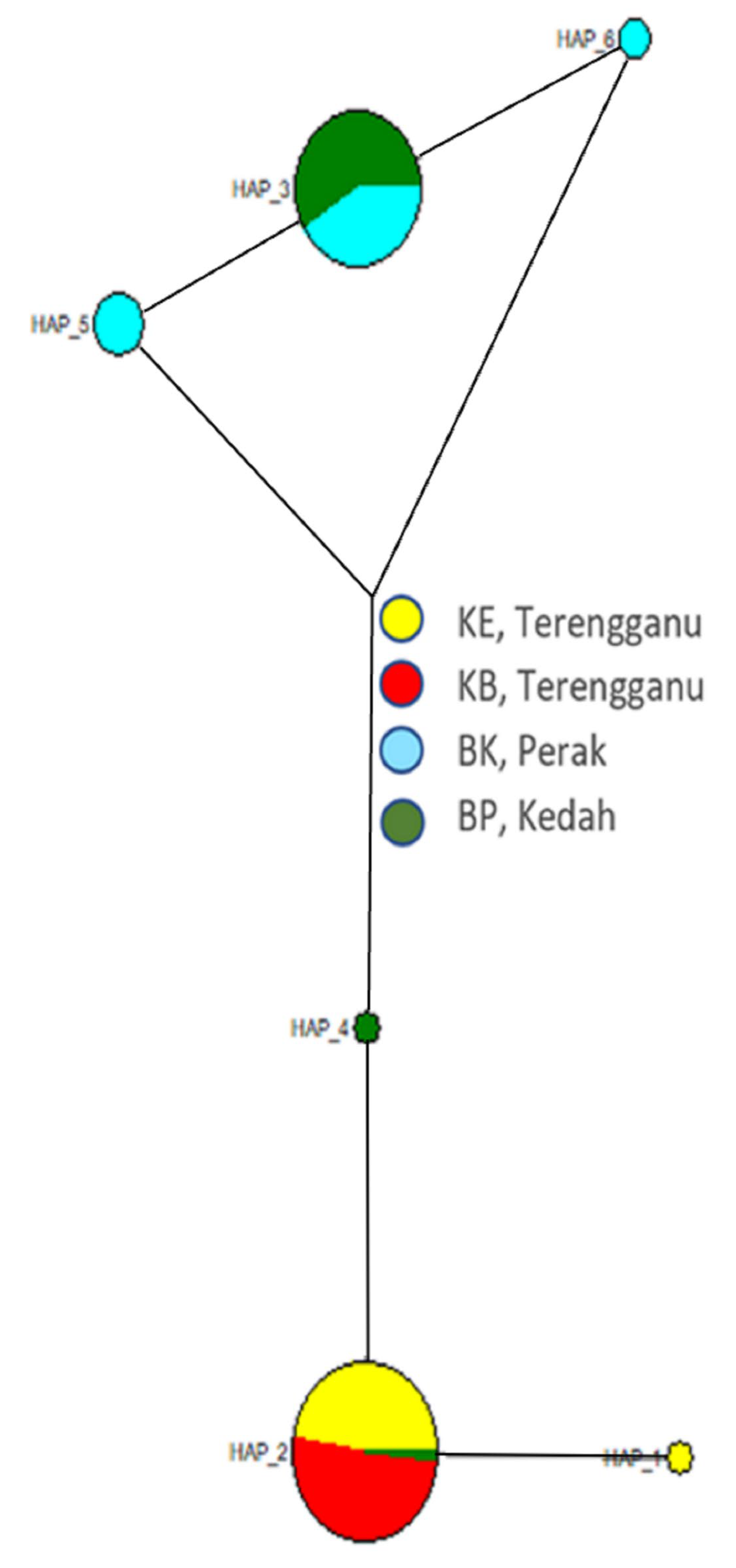
3.2. Population Structure
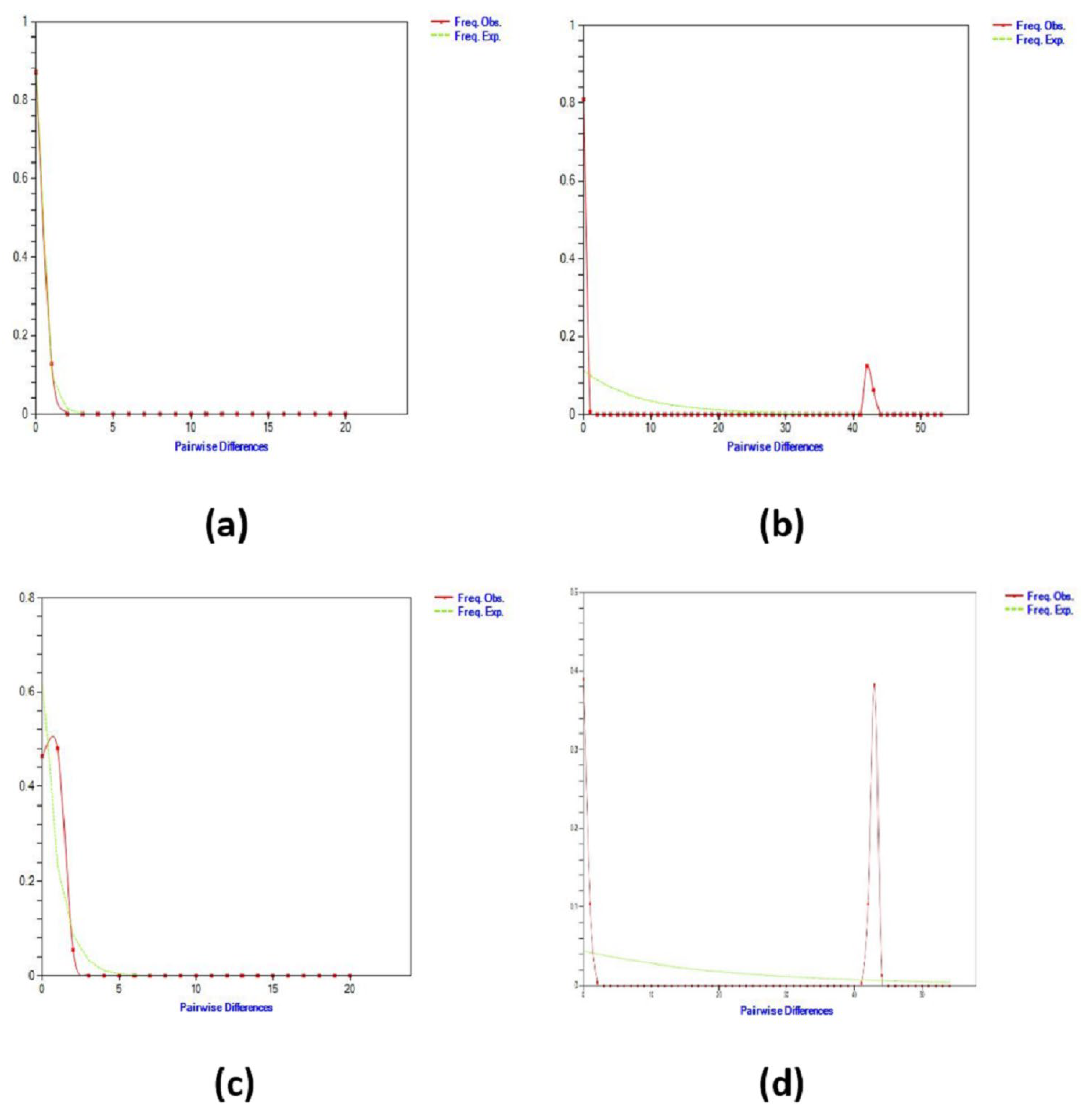
3.3. Population Demography History
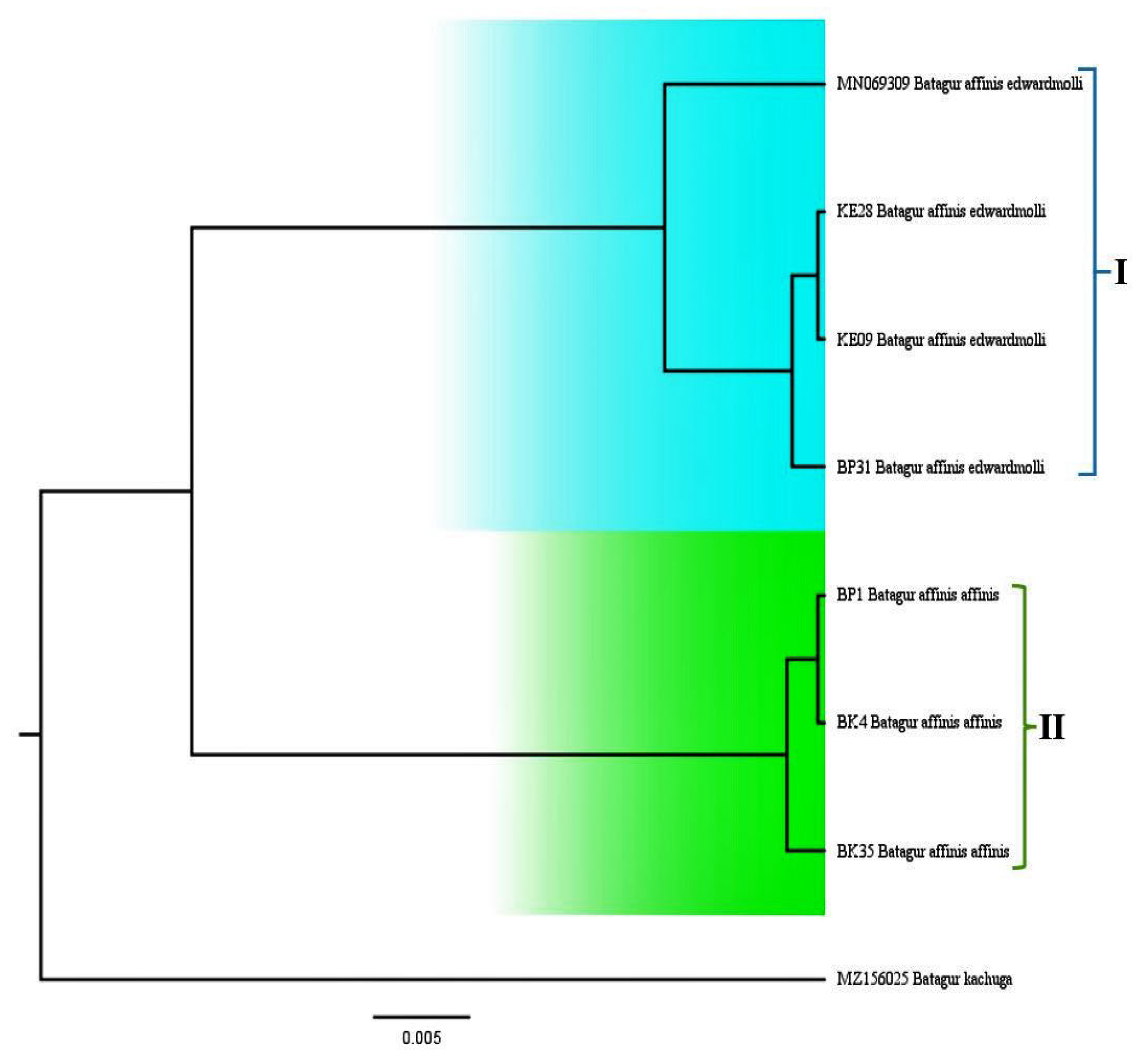
3.4. Phylogenetic Relationship
4. Discussion
4.1. Population Diversity
4.2. Population Structure
4.3. Population Demography History
4.4. Phylogenetic Relationship
4.5. The New Distribution Range of Batagur affinis edwardmolli Is a Novelty
4.6. Conservation Impact and Recommendation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cantor, T. Catalogue of reptiles inhabiting the Malayan peninsula and islands. J. Asiat. Soc. Bengal 1847, 16, 607–656, 897–952, 1026–1078. [Google Scholar]
- Moll, E.O.; Platt, S.G.; Chan, E.H.; Horne, B.D.; Platt, K.; Praschag, P.; Chen, P.N.; van Dijk, P.P. Batagur affinis (Cantor 1847) Southern river terrapin, tuntong. Chelonian Res. Monogr. 2015, 5, 090.1–090.17. [Google Scholar]
- Salleh, M.H.; Esa, Y. The mtDNA D-loop Marker Identifies the Genetic Variability of Indochina’s Batagur affinis. In Proceedings of the 1st Postgraduate Seminar on Agriculture and Forestry 2021 (PSAF 2021), Universiti Putra Malaysia, Bintulu, Sarawak, Malaysia, 6 October 2021; p. 82. [Google Scholar]
- IUCN/SSC Tortoise and Freshwater Turtle Specialist Group; Turtle Conservation Fund; Turtle Survival Alliance; Turtle Conservancy; Chelonian Research Foundation; Conservation International; Wildlife Conservation Society; San Diego Zoo Global. Turtles in Trouble: The World’s 25+ Most Endangered Tortoises and Freshwater Turtles-2011; Turtle Conservation Coalition: Lunenburg, MA, USA, 2011. [Google Scholar]
- Mohd Salleh, M.H.; Esa, Y.; Salleh, S.M.; Mohd Sah, S.A. Turtles in Malaysia: A Review of Conservation Status and a Call for Research. Animals 2022, 12, 2184. [Google Scholar] [CrossRef] [PubMed]
- Salleh, M.H.M.; Esa, Y.; Mohamed, R.; Nyok, C.P.; Ismail, M.H. Saving Freshwater Turtle-Batagur affinis. Res. Circ. Int. Inst. Knowl. Res. 2022, 161, 296–307. [Google Scholar]
- Platt, S.G.; Hendrie, D.; Chan, E.H.; Poynter, B.; Platt, K.; Sovannara, H.; Holloway, R.; Khin, M.M.; Chen, P.N.; Soh, C.L. Batagur baska: A Status Review and Conservation Action Plan; Unpublished report to Wildlife Conservation Society and Turtle Survival Alliance; 2006; p. 68. [Google Scholar]
- Praschag, P.; Holloway, R.; Georges, A.; Paeckert, M.; Hundsdoerfer, A.K.; Fritz, U. A new subspecies of Batagur affinis (Cantor, 1847), one of the world’s most critically endangered chelonians (Testudines: Geoemydidae). Zootaxa 2009, 2233, 57–68. [Google Scholar] [CrossRef]
- Mistar Siregar, A.J.; Singleton, I. Presence and Distribution of the Southern River Terrapin Batagur affinis and Painted Terrapin Batagur borneoensis in Eastern Coast of Sumatra; Unpublished Report to Auckland Zoo: Auckland, New Zealand, 2012; p. 25. [Google Scholar]
- Joshi, J.; Salar, R.K.; Banerjee, P. Genetic variation and phylogenetic relationships of Indian buffaloes of Uttar Pradesh. Asian-Australas. J. Anim. Sci. 2013, 26, 1229. [Google Scholar] [CrossRef] [Green Version]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Harrison, R.G. Animal mitochondrial DNA as a genetic marker in population biology and evolutionary biology. Trends Ecol. Evol. 1989, 4, 6–11. [Google Scholar] [CrossRef]
- Eduardoff, M.; Xavier, C.; Strobl, C.; Casas-Vargas, A.; Parson, W. Optimised mtDNA control region primer extension capture analysis for forensically relevant samples and highly compromised mtDNA of different age and origin. Genes 2017, 8, 237. [Google Scholar] [CrossRef] [Green Version]
- Cann, R.L.; Brown, W.M.; Wilson, A.C. Polymorphic sites and the mechanism of evolution in human mitochondrial DNA. Genetics 1984, 106, 479–499. [Google Scholar] [CrossRef]
- Kumar, S.; Nagarajan, M.; Sandhu, J.S.; Kumar, N.; Behl, V. Phylogeography and domestication of Indian River buffalo. BMC Evol. Biol. 2007, 7, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, D.G.; Loftus, R.T.; Cunningham, P.; MacHugh, D.E. Genetics and domestic cattle origins. Evol. Anthropol. Issues News Rev. Issues News Rev. 1998, 6, 79–86. [Google Scholar] [CrossRef]
- Troy, C.S.; MacHugh, D.E.; Bailey, J.F.; Magee, D.A.; Loftus, R.T.; Cunningham, P.; Chamberlain, A.T.; Sykes, B.C.; Bradley, D.G. Genetic evidence for near-eastern origins of european cattle. Nature 2001, 410, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Malau-Aduli, A.E.; Nishimura-Abe, A.; Niibayashi, T.; Yasuda, Y.; Kojima, T.; Abe, S.; Oshima, K.; Hasegawa, K.; Komatsu, M. Mitochondrial DNA polymorphism, maternal lineage and correlations with postnatal growth of Japanese black beef cattle to yearling age. Asian-Australas. J. Anim. Sci. 2004, 17, 1484–1490. [Google Scholar] [CrossRef]
- Odahara, S.; Chung, H.J.; Choi, S.H.; Yu, S.L.; Sasazaki, S.; Mannen, H.; Park, C.S.; Lee, J.H. Mitochondrial DNA diversity of Korean native goats. Asian-Australas. J. Anim. Sci. 2006, 19, 482–485. [Google Scholar] [CrossRef]
- Lee, Y.J.; Bhuiyan, M.S.; Chung, H.J.; Jung, W.Y.; Choi, K.D.; Jang, B.G.; Paek, W.K.; Jeon, J.T.; Park, C.S.; Lee, J.H. Mitochondrial DNA diversity of Korean Ogol chicken. Asian-Australas. J. Anim. Sci. 2007, 20, 477–481. [Google Scholar] [CrossRef]
- Lei, C.Z.; Zhang, W.; Chen, H.; Lu, F.; Ge, Q.L.; Liu, R.Y.; Dang, R.H.; Yao, Y.Y.; Yao, L.B.; Lu, Z.F.; et al. Two maternal lineages revealed by mitochondrial DNA Dloop sequences in Chinese native water buffaloes (Bubalus Bubalis). Asian-Australas. J. Anim. Sci. 2007, 20, 471–476. [Google Scholar] [CrossRef]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Reed, D.H.; Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Primack, R.B. Essentials of Conservation Biology; Sinauer Associates Inc.: Sunderland, MA, USA, 1993; p. 538. [Google Scholar]
- Frankham, R. Conservation genetics. In Encyclopedia of Ecology, 2nd ed.; Fath, B., Ed.; Elsevier: Edinburgh, UK, 1995; Volume 1, pp. 305–327. [Google Scholar]
- Frankham, R.; Ballou, J.; Briscoe, D.; McInnes, K. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Frankham, R. Genetic adaptation to captivity in species conservation programmes. Mol. Ecol. 2008, 17, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Ralls, K.; Ballou, J. Captive breeding programmes for populations with a small number of founders. Trends Ecol. Evol. 1986, 1, 19–22. [Google Scholar] [CrossRef]
- Robert, A. Captive breeding genetics and reintroduction success. Biol. Conserv. 2009, 142, 2915–2922. [Google Scholar] [CrossRef]
- Salleh, M.H.M.; Esa, Y. Minimally invasive blood collection techniques as a source of gDNA for genetic studies on turtles and tortoises. Int. J. Aquat. Biol. 2022, 10, 145–150. [Google Scholar]
- Çilingir, F.G.; Seah, A.; Horne, B.D.; Som, S.; Bickford, D.P.; Rheindt, F.E. Last exit before the brink: Conservation genomics of the Cambodian population of the critically endangered southern river terrapin. Ecol. Evol. 2019, 9, 9500–9510. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Sánchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 117693430500100003. [Google Scholar] [CrossRef] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics from the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Weir, S.T.; Mitchell, A.C.; Nellis, W.J. Metallisation of fluid molecular hydrogen at 140 GPa (1.4 Mbar). Phys. Rev. Lett. 1996, 76, 1860. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Smouse, P.E.; Long, J.C.; Sokal, R.R. Multiple regression and correlation extensions of the Mantel test of matrix correspondence. Syst. Zool. 1986, 35, 627–632. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programmes to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics, and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Dupanloup, I.; Schneider, S.; Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 2002, 11, 2571–2581. [Google Scholar] [CrossRef]
- Verhoeven, K.J.; Simonsen, K.L.; McIntyre, L.M. Implementing false discovery rate control: Increasing your power. OIKOS 2005, 108, 643–647. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Tajima, F. The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics 1996, 143, 1457–1465. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Harpending, H. Signature of ancient population growth in a lowresolution mitochondrial DNA mismatch distribution. Hum. Biol. 1994, 66, 591–600. [Google Scholar]
- Schneider, S.; Excoffier, L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: Application to human mitochondrial DNA. Genetics 1999, 152, 1079–1089. [Google Scholar] [CrossRef]
- Rogers, A.R. Genetic evidence for a Pleistocene population expansion. Evolution 1995, 49, 608–615. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA 5.10: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D.; et al. XSEDE: Accelerating Scientifific Discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Rambaut, A. FigTree v1.4.5, a Graphical Viewer of Phylogenetic Trees. 2014. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 January 2022).
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Bilderbeek, R.J.; Etienne, R.S. Babette: BEAUti 2, BEAST2 and Tracer for R. bioRxiv 2018, 271866. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, S.; Jayashankar, M.; Alexander, R.; Avinash, K. Testudines of India: A Review on Diversity, Threats and Conservation Initiatives. Int. J. Pharm. Life Sci. 2014, 5, 3297–3304. [Google Scholar]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Briolay, J.; Galtier, N.; Brito, R.M.; Bouvet, Y. Molecular phylogeny of Cyprinidae inferred from cytochrome b DNA sequences. Mol. Phylogenetics Evol. 1998, 9, 100–108. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 7, 1193–1204. [Google Scholar]
- Kumazawa, Y.; Nishida, M. Complete mitochondrial DNA sequences of the green turtle and blue-tailed mole skink: Statistical evidence for archosaurian affinity of turtles. Mol. Biol. Evol. 1999, 16, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Heuertz, M.; Fineschi, S.; Anzidei, M.; Pastorelli, R.; Salvini, D.; Paule, L.; Frascaria-Lacoste, N.; Hardy, O.J.; Vekemans, X.; Vendramin, G. Chloroplast DNA variation and postglacial recolonisation of common ash (Fraxinus excelsior L.) in Europe. Mol. Ecol. 2004, 13, 3437–3452. [Google Scholar] [CrossRef]
- Tsukagoshi, H.; Yokoyama, R.; Goto, A. Mitochondrial DNA analysis reveals a unique population structure of the amphidromous sculpin Cottus pollux middle-egg type (Teleostei: Cottidae). Mol. Phylogenet. Evol. 2011, 60, 265–270. [Google Scholar] [CrossRef]
- Ruiz-Pesini, E.; Mishmar, D.; Brandon, M.; Procaccio, V.; Wallace, D.C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 2004, 303, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar]
- Ray, N.; Currat, M.; Excoffier, L. Intra-deme molecular diversity in spatially expanding populations. Mol. Biol. Evol. 2003, 20, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L. Patterns of DNA sequence diversity and genetic structure after a range expansion: Lessons from the infinite-islandmodel. Mol. Ecol. 2004, 13, 853–864. [Google Scholar] [CrossRef]
- Halim, S.A.; Othman, A.S.; Akib, N.A.; Jamaludin, N.A.; Esa, Y.; Nor, S.A. Mitochondrial Markers Identify a Genetic Boundary of the Green Tiger Prawn (Penaeus semisulcatus) in the Indo-Pacific Ocean. Zool. Stud. 2021, 60, 1–19. [Google Scholar]
- Sanjuán, R. Mutational fitness effects in RNA and single-stranded DNA viruses: Common patterns revealed by site-directed mutagenesis studies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1975–1982. [Google Scholar] [CrossRef] [Green Version]
- Eyre-Walker, A.; Keightley, P.D. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 2007, 8, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Hietpas, R.T.; Jensen, J.D.; Bolon, D.N. Experimental illumination of a fitness landscape. Proc. Natl. Acad. Sci. USA 2011, 108, 7896–7901. [Google Scholar] [CrossRef] [Green Version]
- Praschag, P.; Sommer, R.S.; Mccarthy, C.; Gemel, R. Naming one of the world’s rarest chelonians, the southern Batagur. Zootaxa 2008, 1758, 61–68. [Google Scholar] [CrossRef]
- Guntoro, J.; Riyanto, A. The very low genetic variability on Aceh Tamiang’s (Indonesia) population of Painted Terrapin (Batagur borneoensis) inferred by cytochrome oxidase I (CO I) and D-loop (control region). Biodiversitas J. Biol. Divers. 2020, 21, 2514–2520. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000; p. 447. [Google Scholar]
- De Jong, M.A.; Wahlberg, N.; Van Eijk, M.; Brakefield, P.M.; Zwaan, B.J. Mitochondrial DNA signature for range-wide populations of Bicyclus anynana suggests a rapid expansion from recent refugia. PLoS ONE 2011, 6, e21385. [Google Scholar] [CrossRef] [Green Version]
- Saccone, C.; Attimonelli, M.; Sbisa, E. Structural elements highly preserved during the evolution of the D-loopcontaining region in vertebrate mitochondrial DNA. J. Mol. Evol. 1987, 26, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Tikochinski, Y.; Bendelac, R.; Barash, A.; Daya, A.; Levy, Y.; Friedmann, A. Mitochondrial DNA STR analysis as a tool for studying the green sea turtle (Chelonia mydas) populations: The Mediterranean Sea case study. Mar. Genom. 2012, 6, 17–24. [Google Scholar] [CrossRef]
- Kincaid, H.L. Effects of inbreeding on rainbow trout populations. Trans. Am. Fish. Soc. 1976, 105, 273–280. [Google Scholar] [CrossRef]
- Kincaid, H.L. Inbreeding in rainbow trout (Salmo gairdneri). J. Fish. Board Can. 1976, 33, 2420–2426. [Google Scholar] [CrossRef]
- Saura, M.; Fernández, A.; Varona, L.; Fernández, A.I.; de Cara, M.Á.R.; Barragán, C.; Villanueva, B. Detecting inbreeding depression for reproductive traits in Iberian pigs using genome-wide data. Genet. Sel. Evol. 2015, 47, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.E.; Crist, A.B.; Young, E.M.; Willis, J.H.; Phillips, P.C.; Fierst, J.L. Slow recovery from inbreeding depression generated by the complex genetic architecture of segregating deleterious mutations. Mol. Biol. Evol. 2022, 39, msab330. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ingram, B.; Sungan, S.; Gooley, G.; Sim, S.Y.; Tinggi, D.; De Silva, S.S. Mitochondrial DNA diversity of broodstock of two indigenous Mahseer species, Tor tambroides and Tor douronensis (Cyprinidae) cultured in Sarawak. Aquaculture 2006, 253, 259–269. [Google Scholar] [CrossRef]
- Dutton, P.H.; Bowen, B.W.; Owens, D.W.; Barragan, A.; Davis, S.K. Global phylogeography of the leatherback turtle (Dermochelys coriacea). J. Zool. 1999, 248, 397–409. [Google Scholar] [CrossRef]
- Ashfaq, M.; Hebert, P.D.; Mirza, M.S.; Khan, A.M.; Mansoor, S.; Shah, G.S.; Zafar, Y. DNA Barcoding of Bemisia tabaci Complex (Hemiptera: Aleyrodidae) Reveals Southerly Expansion of the Dominant Whitefly Species on Cotton in Pakistan. PLoS ONE 2014, 9, e104485. [Google Scholar] [CrossRef] [Green Version]
- Pichler, F.B. Genetic Assessment of Population Boundaries and Gene Exchange in Hector’s Dolphin; DOC Science Internal Series; Department of Conservation: Wellington, New Zealand, 2002; Volume 44, pp. 1–37. [Google Scholar]
- Kusza, S.; Podgorski, T.; Scandura, M.; Borowik, T.; Javor, A.; Sidorovich, V.E.; Bunevich, A.N.; Kolesnikov, M.; Jędrzejewska, B. Contemporary genetic structure, phylogeography and past demographic processes of wild boar Sus scrofa population in Central and Eastern Europe. PLoS ONE 2014, 9, e91401. [Google Scholar] [CrossRef] [Green Version]
- Slatkin, M.; Hudson, R.R. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 1991, 129, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Spitzweg, C.; Praschag, P.; DiRuzzo, S.; Fritz, U. Conservation genetics of the northern river terrapin (Batagur baska) breeding project using a microsatellite marker system. Salamandra 2018, 54, 63–70. [Google Scholar]
- Duli, N.B. Morphometric and Genetic Variability of River Terrapin (Batagur baska) and Painted Terrapin (Batagur borneoensis). Master’s Thesis, Universiti Sains Malaysia, Pulau Pinang, Malaysia, 2009. [Google Scholar]
- Waples, R.S. Testing for Hardy-Weinberg proportions: Have we lost the plot? J. Hered. 2014, 106, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Shafer, A.B.; Wolf, J.B.; Alves, P.C.; Bergström, L.; Bruford, M.W.; Brännström, I.; Colling, G.; Dalén, L.; De Meester, L.; Ekblom, R.; et al. Genomics and the challenging translation into conservation practice. Trends Ecol. Evol. 2015, 30, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Garner, B.A.; Hand, B.K.; Amish, S.J.; Bernatchez, L.; Foster, J.T.; Miller, K.M.; Morin, P.A.; Narum, S.R.; O’Brien, S.J.; Roffler, G.; et al. Genomics in conservation: Case studies and bridging the gap between data and application. Trends Ecol. Evol. 2016, 31, 81–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafer, A.B.; Wolf, J.B.; Alves, P.C.; Bergström, L.; Colling, G.; Dalén, L.; De Meester, L.; Ekblom, R.; Fior, S.; Hajibabaei, M.; et al. Reply to Garner et al. Trends Ecol. Evol. 2016, 31, 83–84. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Haplotype | Single Nucleotide Polymorphisms (SNPs) | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 4 | 4 | 4 | 5 | 5 | 5 | 5 | |||||||||||||||
| 1 | 1 | 2 | 3 | 4 | 4 | 7 | 7 | 1 | 2 | 2 | 3 | 3 | 4 | 4 | 4 | 5 | 5 | 5 | 5 | 9 | 0 | 1 | 1 | 1 | 7 | 9 | 9 | 3 | 5 | 5 | 5 | 6 | 6 | 7 | 8 | 8 | 2 | 3 | 4 | 1 | 4 | 6 | 7 | |||||||
| 1 | 2 | 1 | 9 | 3 | 4 | 1 | 7 | 2 | 3 | 9 | 5 | 8 | 5 | 7 | 2 | 7 | 8 | 2 | 4 | 5 | 7 | 3 | 9 | 1 | 7 | 9 | 5 | 5 | 6 | 7 | 4 | 5 | 6 | 0 | 1 | 8 | 0 | 5 | 7 | 7 | 2 | 6 | 4 | 4 | 3 | |||||
| Batagur affinis edwardmolli KE28 | C | A | T | A | A | G | G | G | A | T | A | A | T | G | G | T | G | G | A | C | T | G | G | C | T | T | T | A | C | T | G | C | C | T | C | T | A | T | C | G | A | A | C | T | C | T | ||||
| Batagur affinis edwardmolli KE09 | . | . | . | . | C | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | ||||
| Batagur affinis edwardmolli BP31 | . | . | . | . | C | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | ||||
| Batagur affinis affinis BP1 | A | G | G | G | C | A | A | A | G | C | G | G | C | A | A | C | A | A | G | T | C | A | A | T | C | C | C | G | T | C | A | T | T | C | T | C | G | C | T | A | G | G | T | C | G | C | ||||
| Batagur affinis affinis BK4 | . | . | G | G | C | A | A | A | . | C | G | G | C | A | A | C | A | A | G | T | C | A | A | T | C | C | C | G | T | C | A | T | T | C | T | C | G | C | T | A | G | G | T | C | G | C | ||||
| Batagur affinis affinis BK35 | A | G | G | G | C | A | A | A | G | . | G | G | C | A | A | C | A | A | G | T | C | A | A | T | C | C | C | G | T | C | A | T | T | C | T | C | G | C | T | A | G | G | T | C | G | C | ||||
| Population (ID) * | No. of Haplotypes | Sample Size | SNPs | Haplotype Diversity (Hd) | Nucleotide Diversity (π) |
|---|---|---|---|---|---|
| 1 (KE) | 2 | 30 | 1 | 0.1287 | 0.0002 |
| 2 (BP) | 3 | 30 | 43 | 0.1908 | 0.0123 |
| 3 (BK) | 3 | 30 | 2 | 0.5356 | 0.0009 |
| 4 (KB) | 1 | 30 | 0 | 0.0000 | 0.0000 |
| Source of Variation | df | Sum of Squares | Variance Components | % Total Variance |
|---|---|---|---|---|
| AMOVA | ||||
| Among populations within groups | 3 | 1162.850 | 12.885 | 92.29 |
| Within populations | 116 | 124.800 | 1.076 | 7.71 |
| Total | 119 | 1287.650 | 13.960 | 100 |
| SAMOVA, 2 groups | ||||
| Among groups | 1 | 1167.241 | 19.241 | 94.39 |
| Among populations within groups | 2 | 6.424 | 0.071 | 0.35 |
| Within populations | 117 | 125.641 | 1.074 | 5.27 |
| Total | 120 | 1299.306 | 1299.306 | 100 |
| Population (ID) * | 1 (KE) | 2 (BP) | 3 (BK) | 4 (KB) |
|---|---|---|---|---|
| 1 (KE) | - | 0.896 | 0.991 | 0.034 |
| 2 (BP) | 0.065 | - | 0.063 | 0.898 |
| 3 (BK) | 0.072 | 0.008 | - | 0.993 |
| 4 (KB) | 0.000 | 0.065 | 0.072 | - |
| Population (ID) * | Tajima’s D (p) | Fu’s FS (p) | SSD (p) | RI (p) | ||||
|---|---|---|---|---|---|---|---|---|
| 1 (KE) | −0.76373 | 0.229 | −0.43926 | 0.155 | 0.04026 | 0.090 | 0.56792 | 0.390 |
| 2 (BP) | −1.01094 | 0.154 | 13.82783 | 0.999 | 0.03459 | 0.060 | 0.67051 | 0.690 |
| 3 (BK) | 0.34880 | 0.698 | 0.39996 | 0.555 | 0.02202 | 0.080 | 0.18417 | 0.060 |
| 4 (KB) | 0.00000 | 1.000 | 0.00000 | NA | 0.00000 | 0.000 | 0.00000 | 0.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salleh, M.H.M.; Esa, Y.; Pau, S.-S.N. Conservation Genetics of the Critically Endangered Southern River Terrapin (Batagur affinis) in Malaysia: Genetic Diversity and Novel Subspecies Distribution Ranges. Biology 2023, 12, 520. https://doi.org/10.3390/biology12040520
Salleh MHM, Esa Y, Pau S-SN. Conservation Genetics of the Critically Endangered Southern River Terrapin (Batagur affinis) in Malaysia: Genetic Diversity and Novel Subspecies Distribution Ranges. Biology. 2023; 12(4):520. https://doi.org/10.3390/biology12040520
Chicago/Turabian StyleSalleh, Mohd Hairul Mohd, Yuzine Esa, and Suriyanti-Su Nyun Pau. 2023. "Conservation Genetics of the Critically Endangered Southern River Terrapin (Batagur affinis) in Malaysia: Genetic Diversity and Novel Subspecies Distribution Ranges" Biology 12, no. 4: 520. https://doi.org/10.3390/biology12040520