
Integration of Chemoinformatics and Multi-Omics Analysis Defines ECT2 as a Potential Target for Cancer Drug Therapy

 , ,
, ,  , , , , ,
, , , , ,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
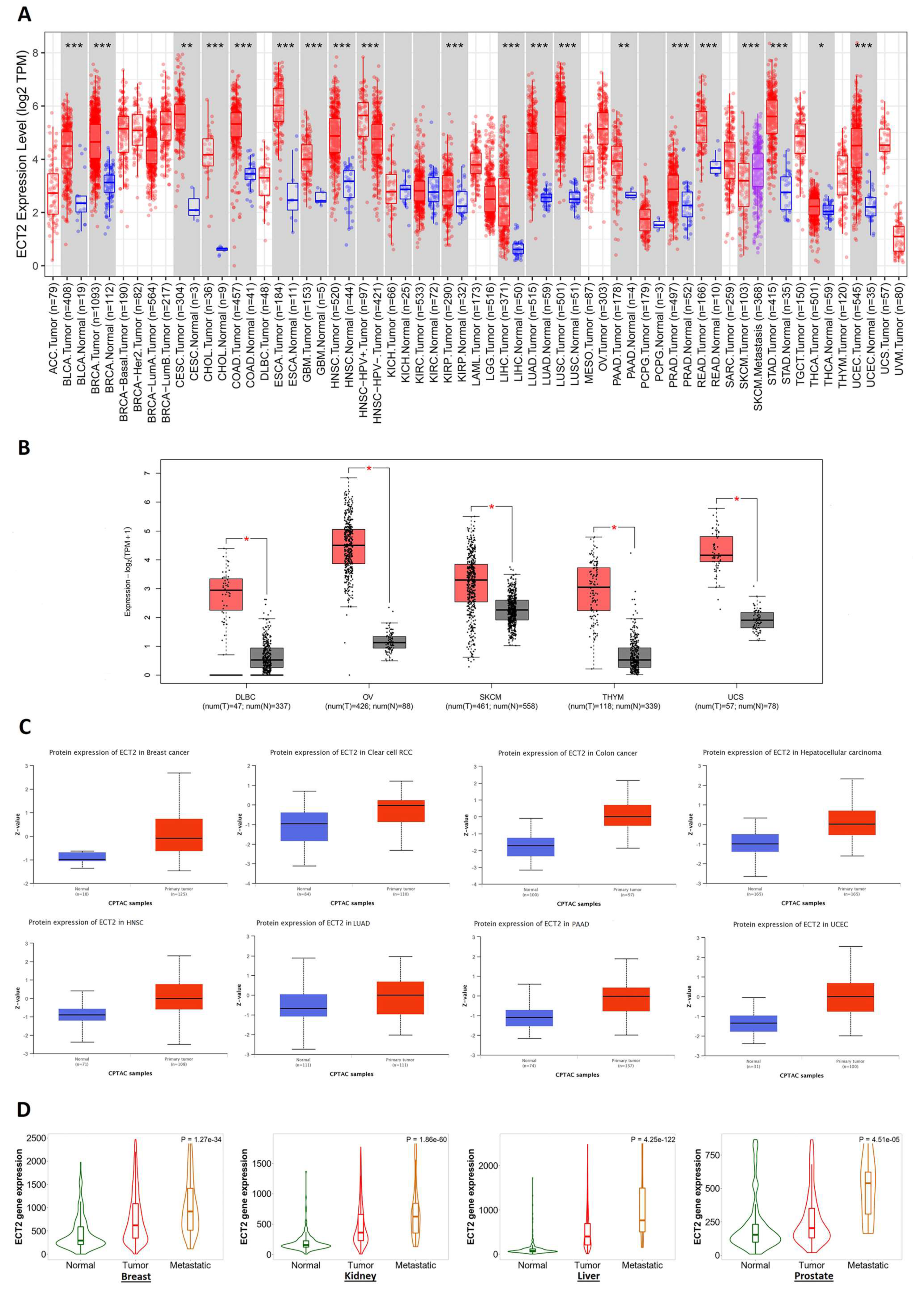
2.1. ECT2 Differential Level in Tumor and Normal Tissues
2.2. Association of ECT2 with Tumor Grade and Stage
2.3. Survival Prognosis Analysis
2.4. ECT2 Genetic Alteration and Its Correlation with Patients’ Survival
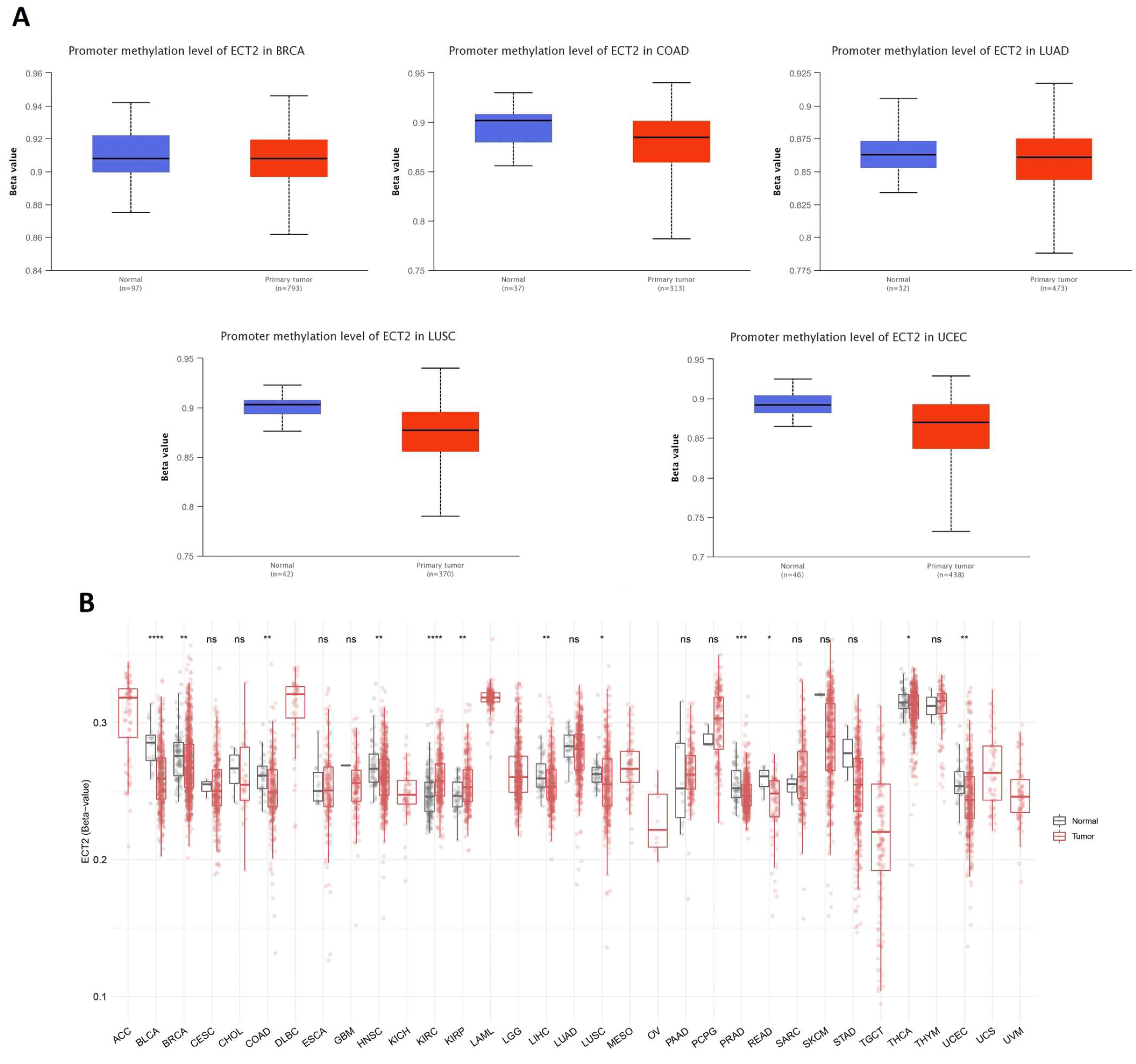
2.5. Methylation and Phosphorylation Analysis of ECT2
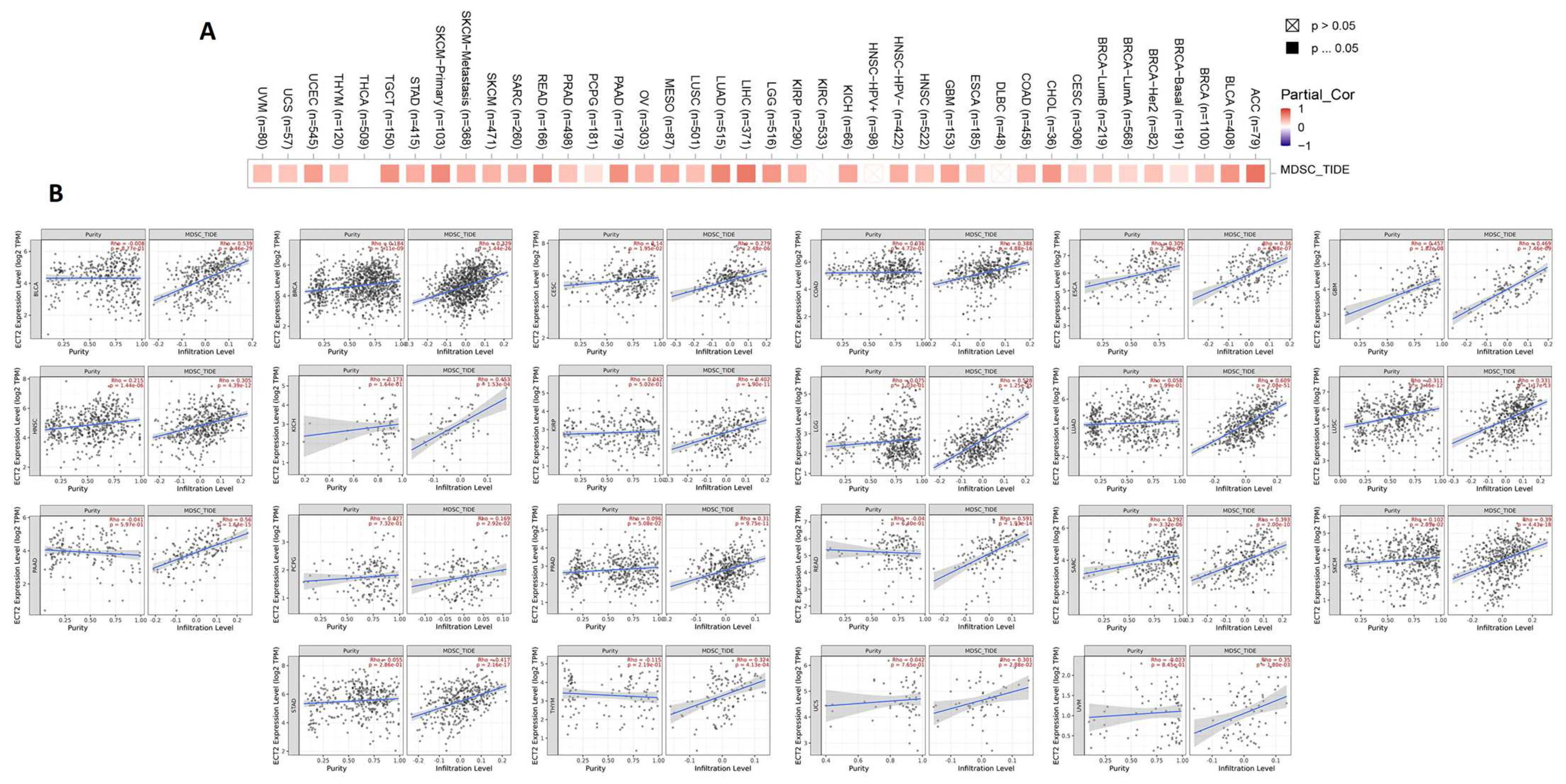
2.6. Assessment of ECT2 Effects on the Tumor Microenvironment
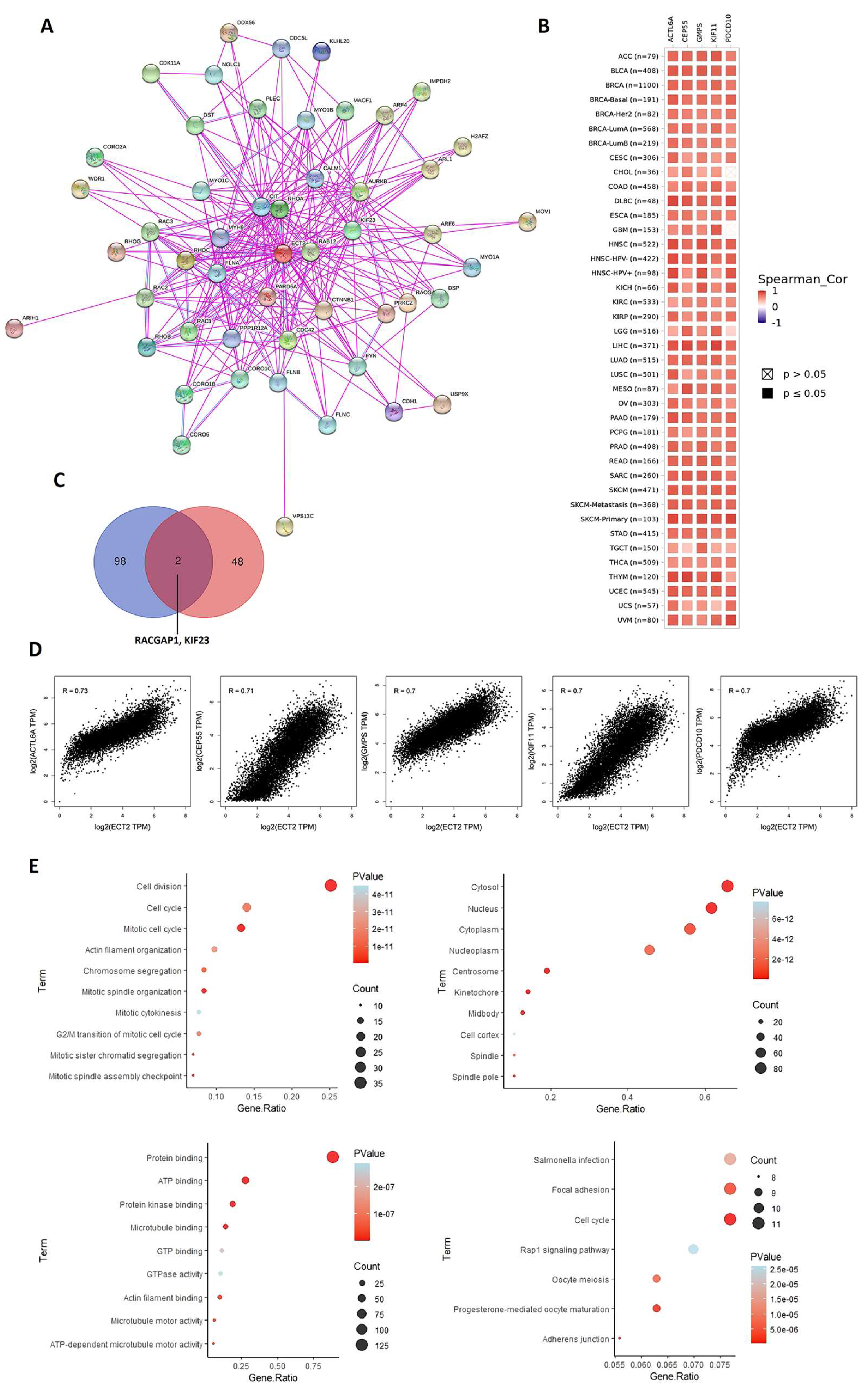
2.7. Analysis of ECT2 Protein–Protein Interactions
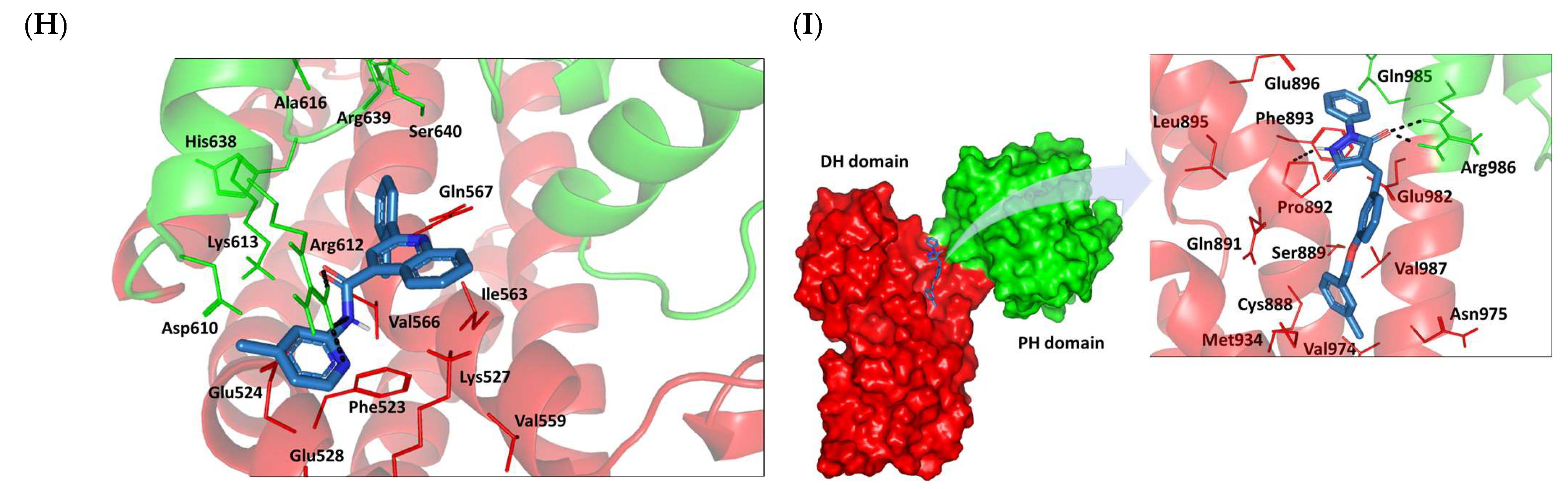
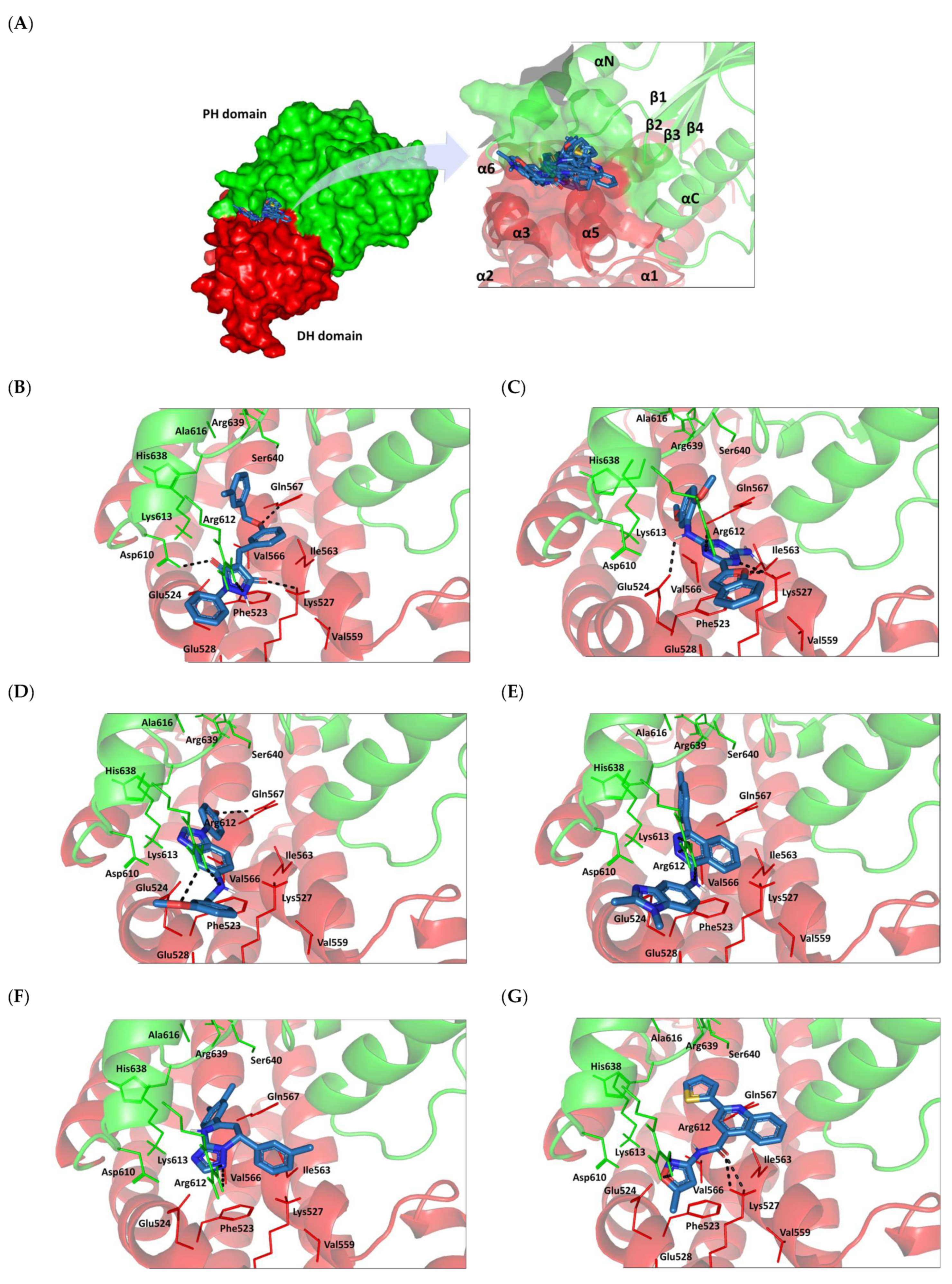
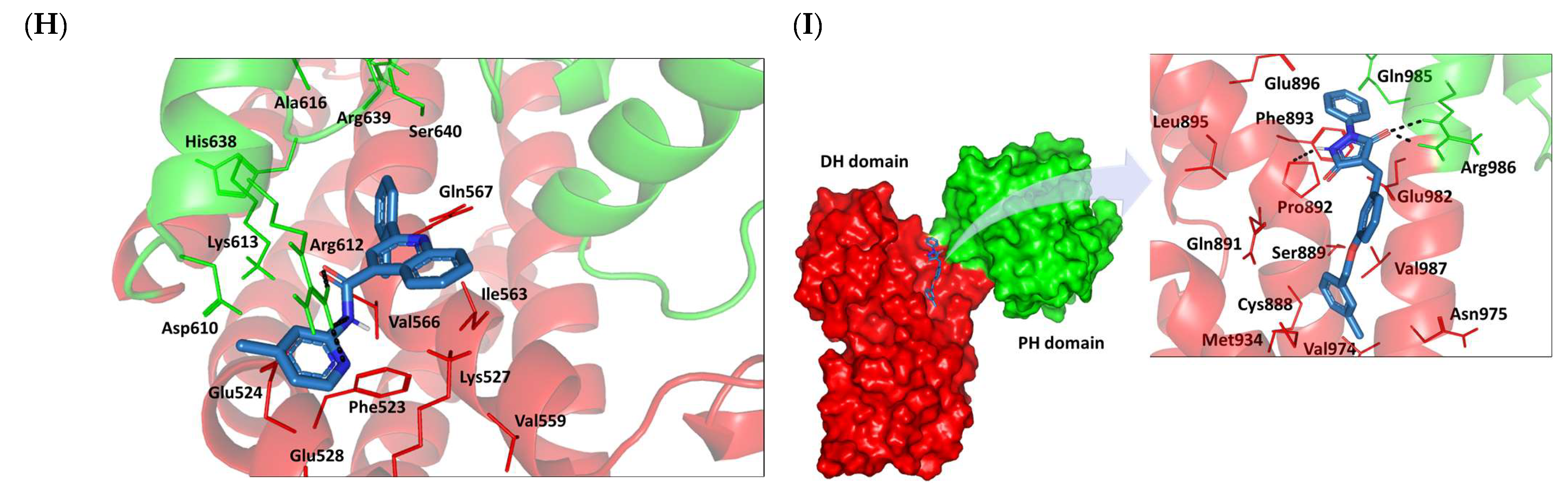
2.8. Small Molecule–ECT2 Binding Investigation
3. Results
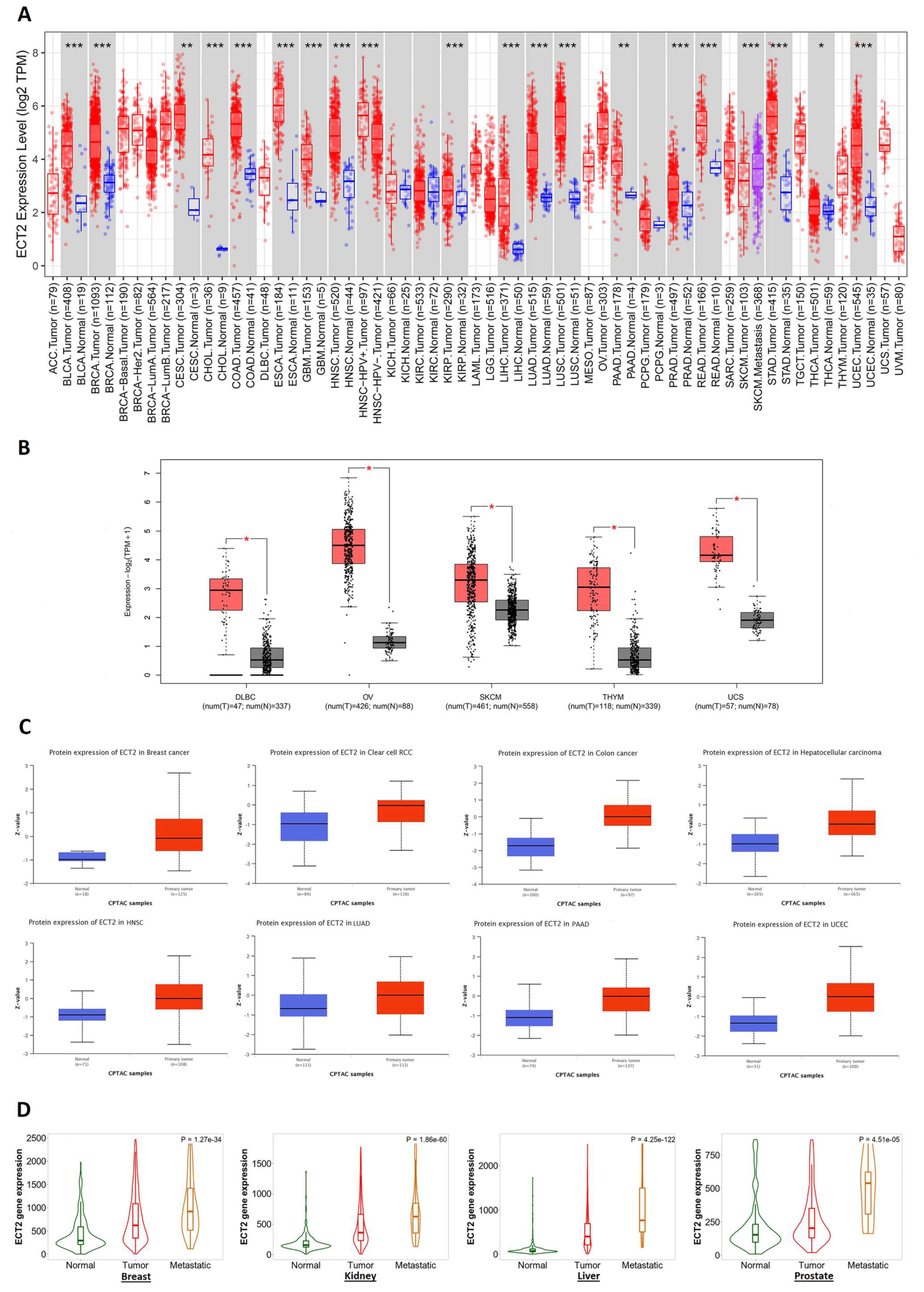
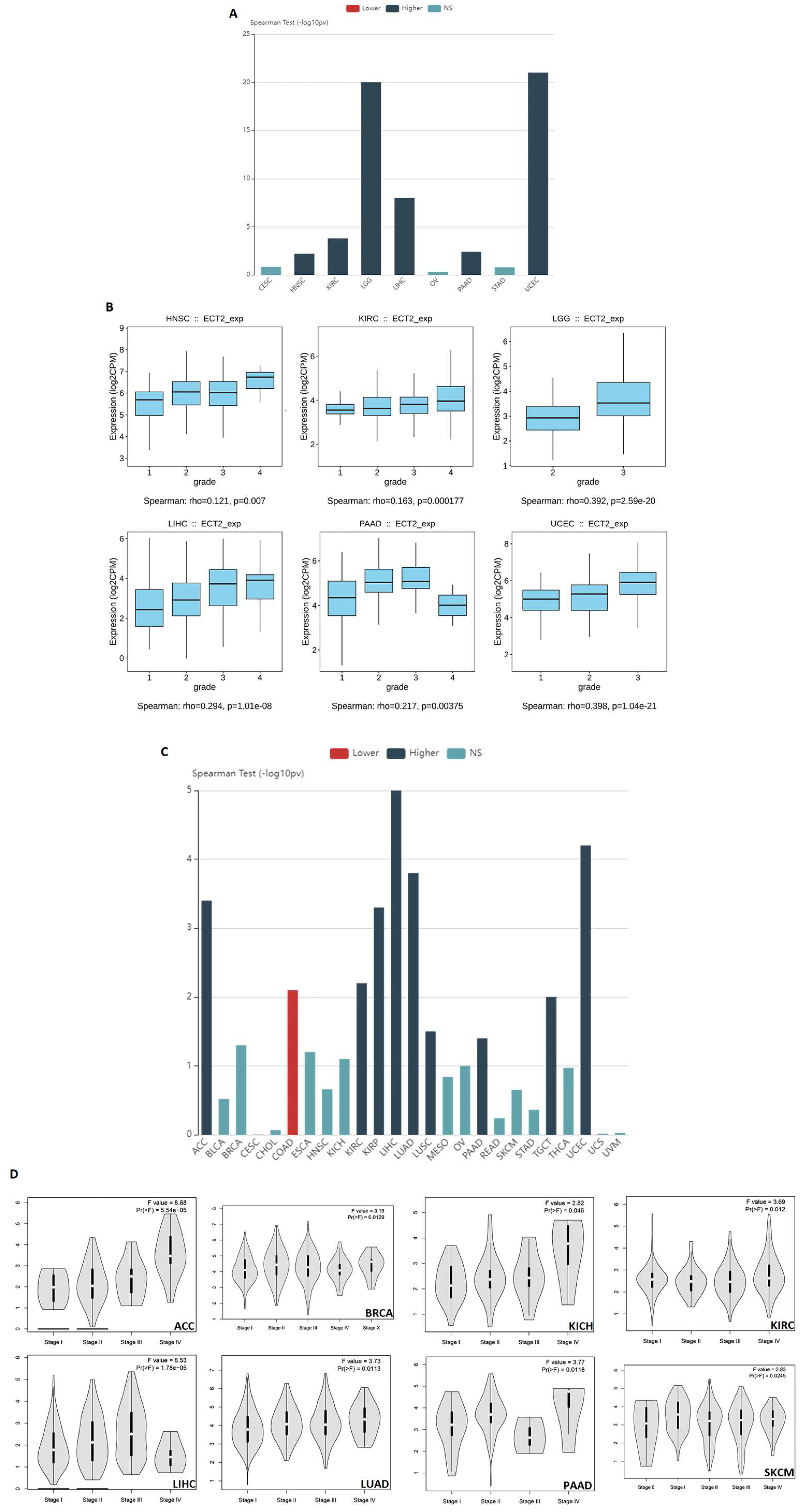
3.1. ECT2 Elevated Expression in Several Human Tumors versus Normal Tissue
3.2. Several Human Cancers Demonstrated a Correlation between ECT2 Expression and Tumor Stage and Grade
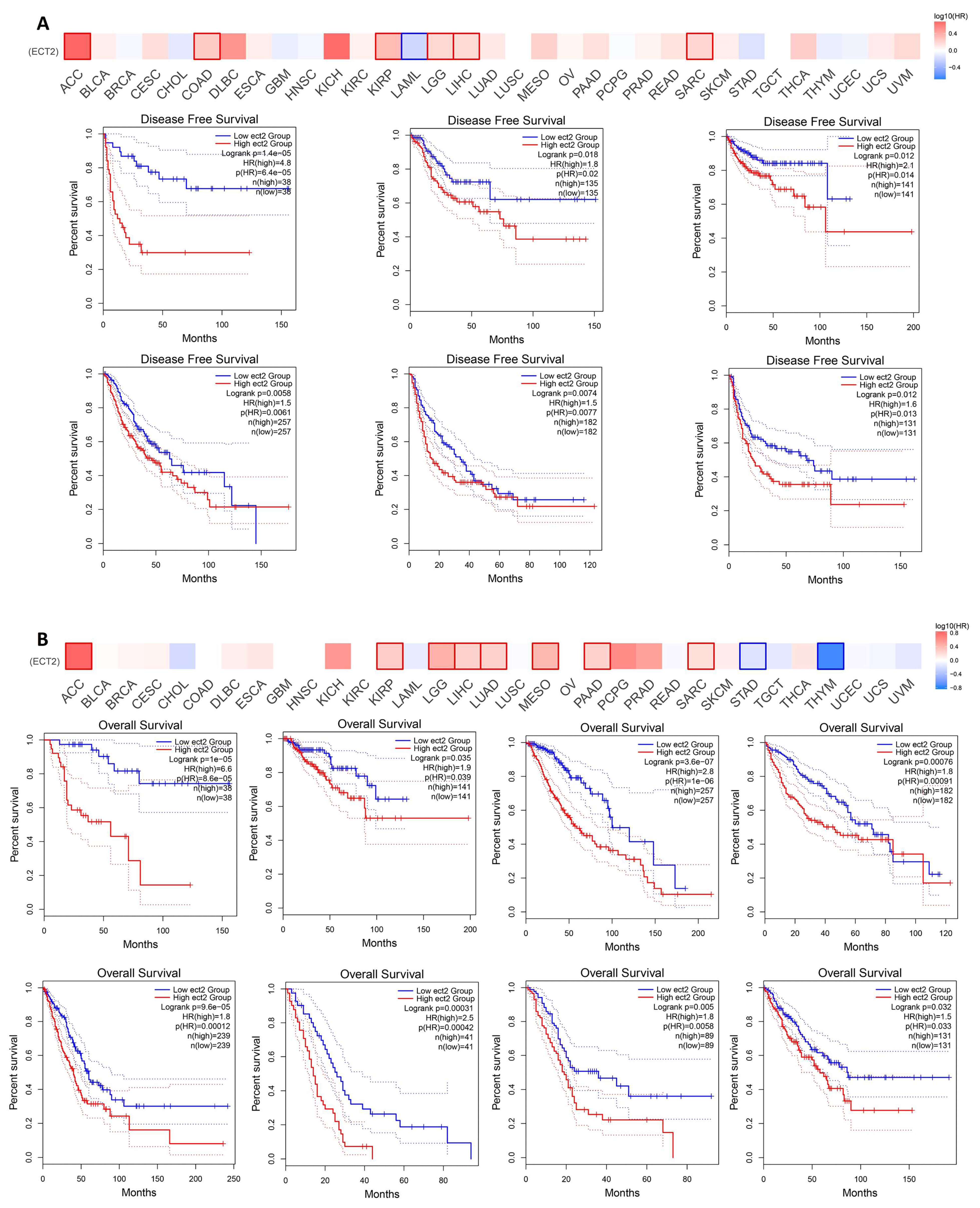
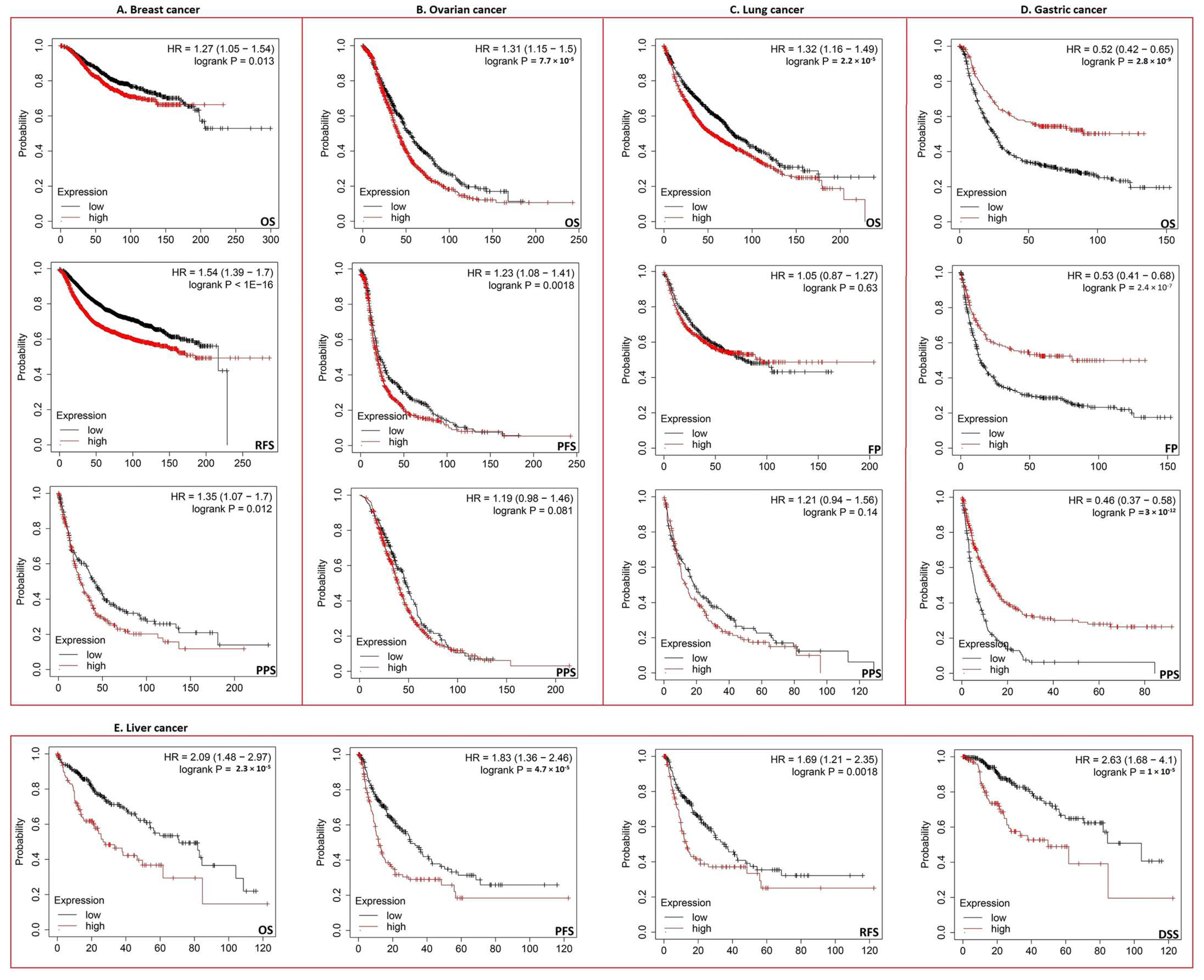
3.3. Increased ECT2 Levels Were Negatively Correlated with the Clinical Outcomes
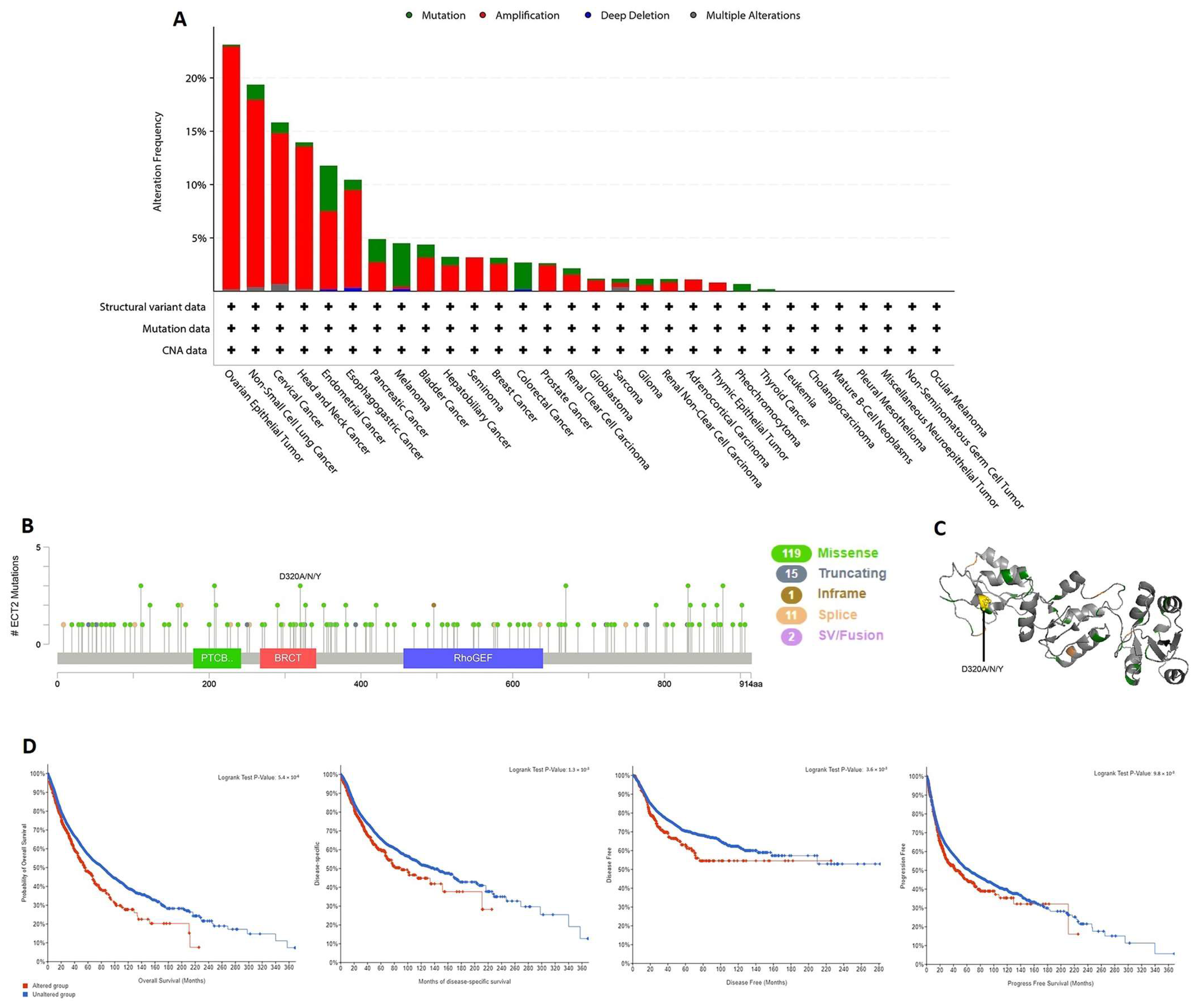
3.4. ECT2 Genetic Alteration Predicts Poor Patient Outcome
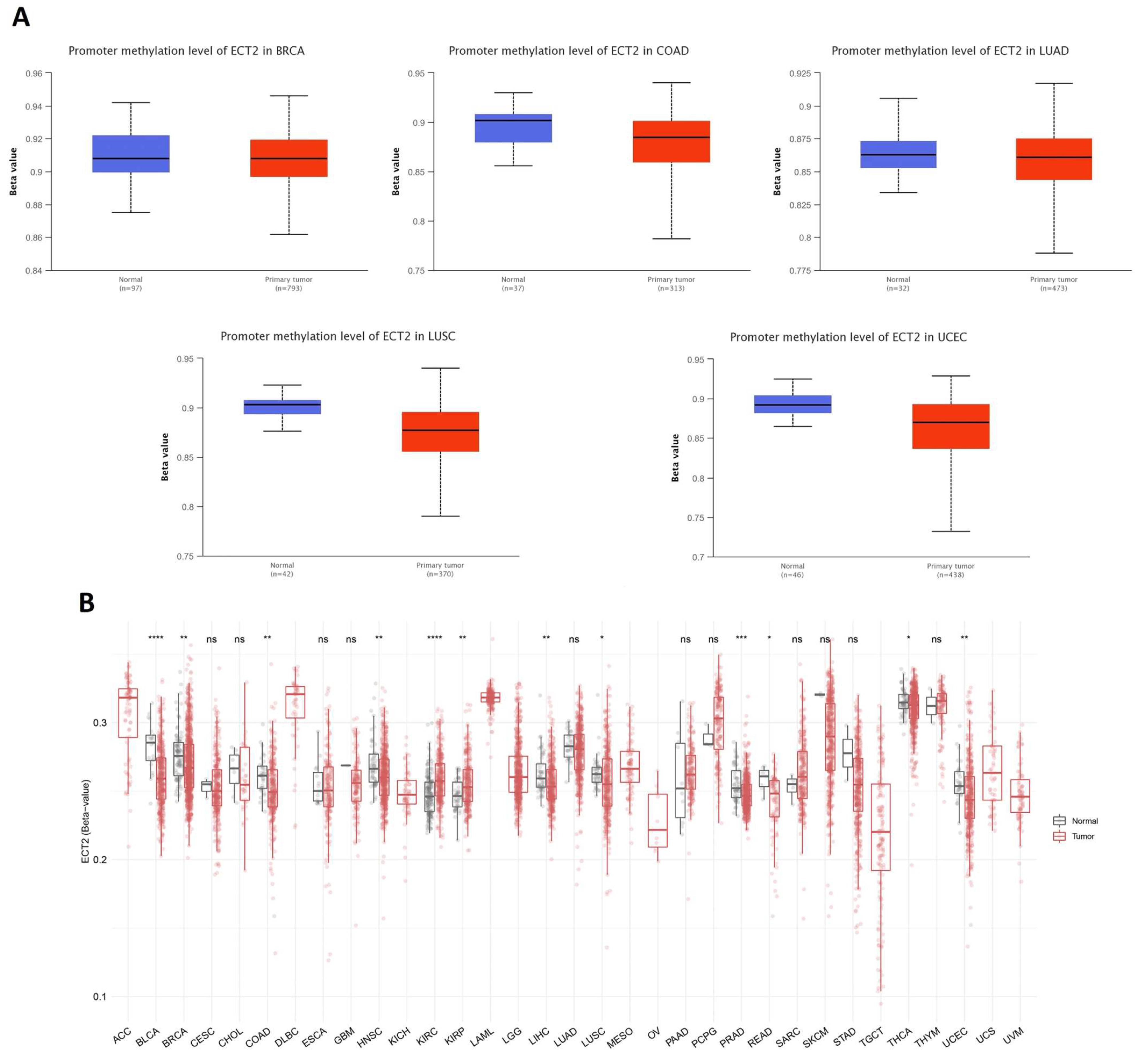
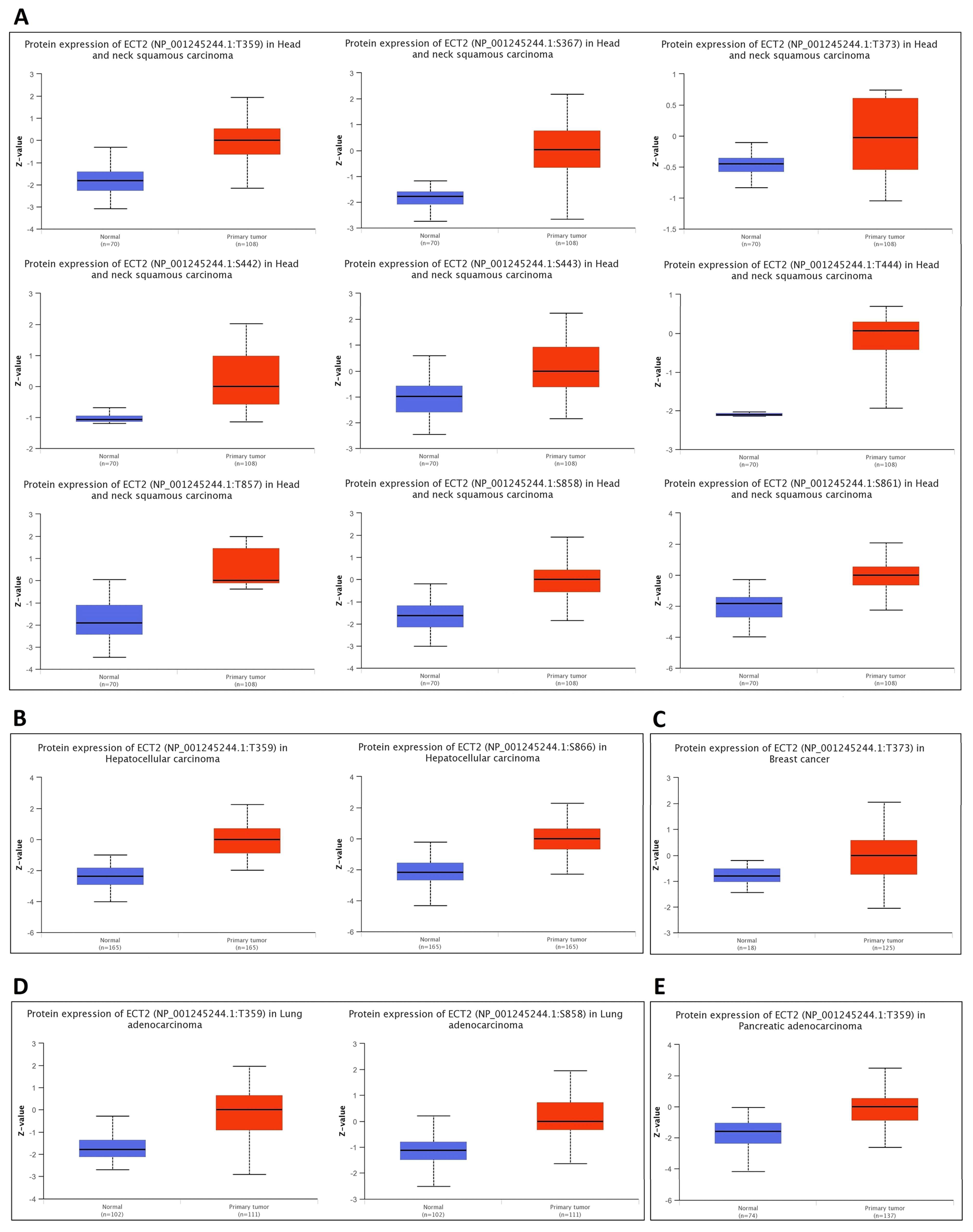
3.5. Opposing Methylation–Phosphorylation Status of ECT2 in Several Human Cancers
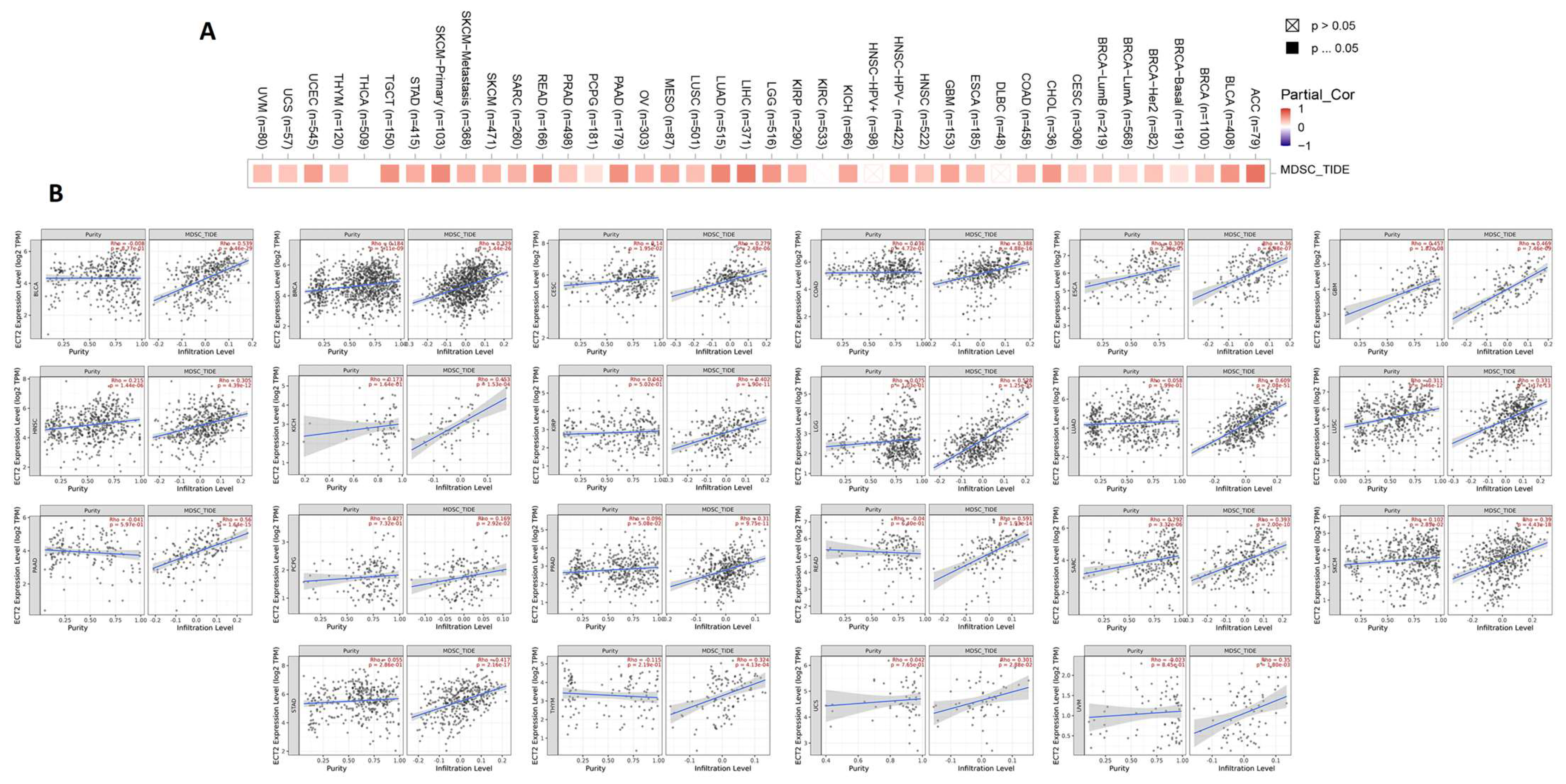
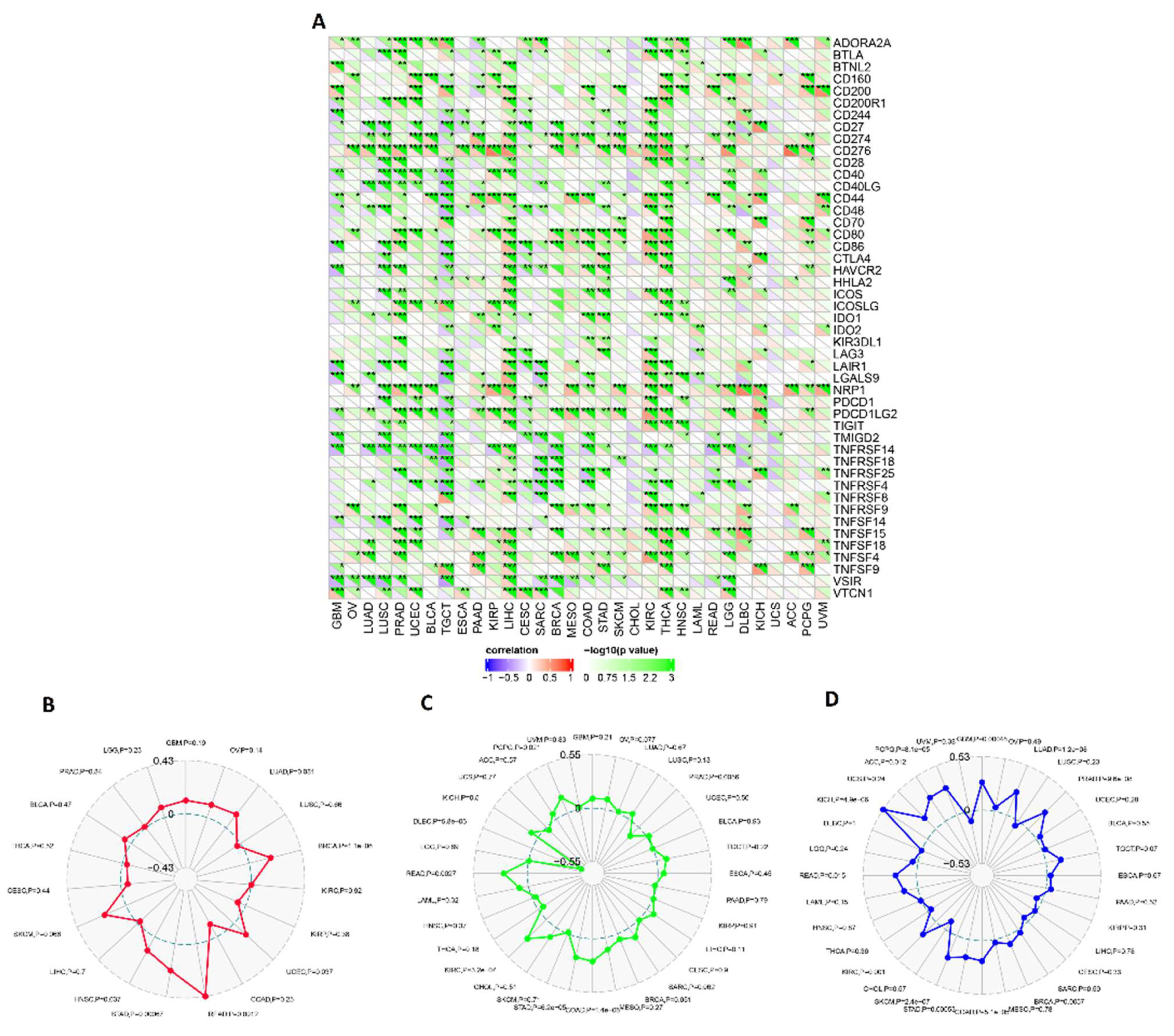
3.6. ECT2 Expression in Cancerous Tissue Was Positively Correlated with the Infiltration of Cells with Immunosuppressive Characteristics
3.7. ECT2-Protein Interactions and Enrichment Analysis
3.8. Docked Small Molecules Showed Promising Binding Affinities at DH-PH Catalytic Interface
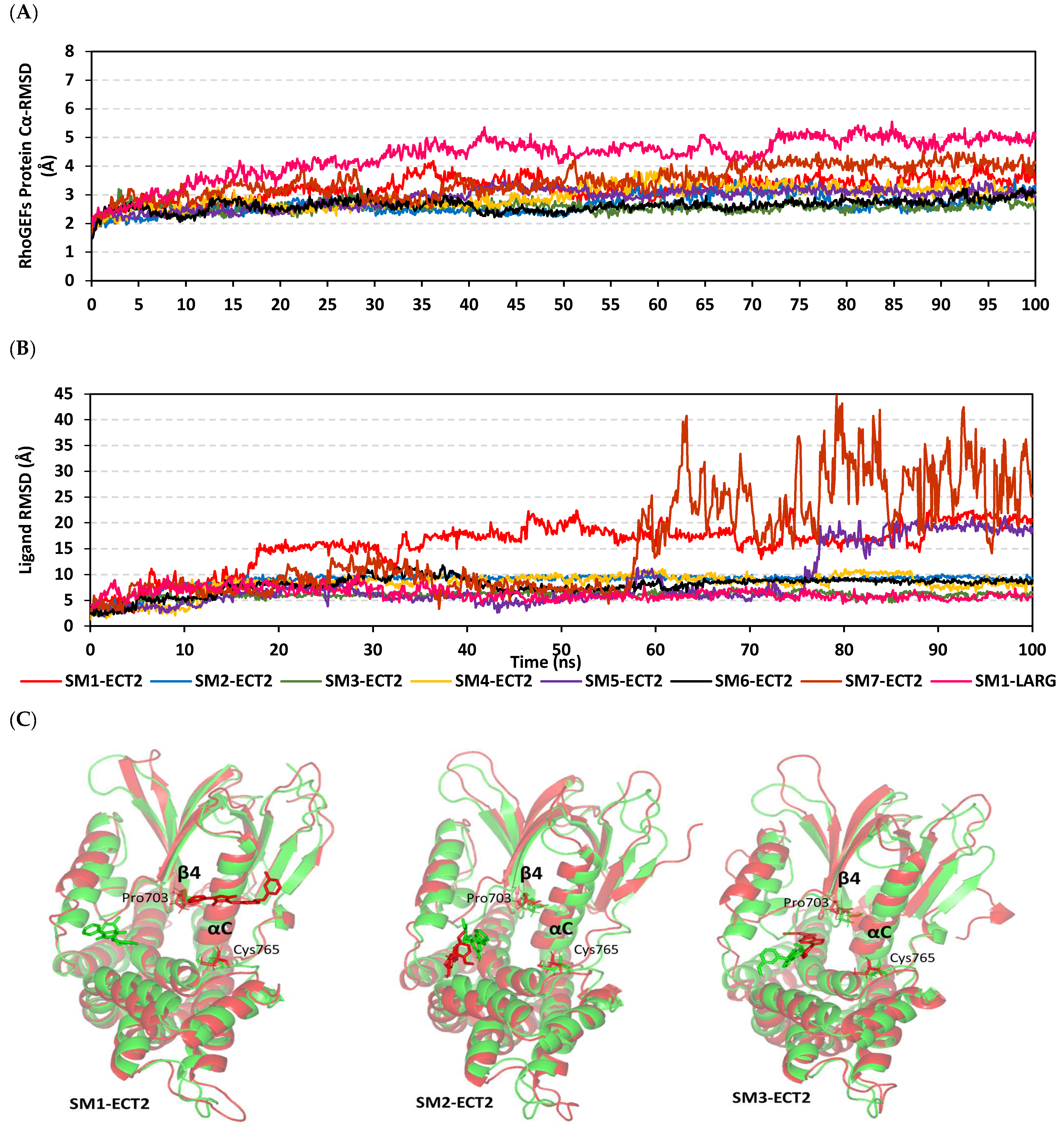
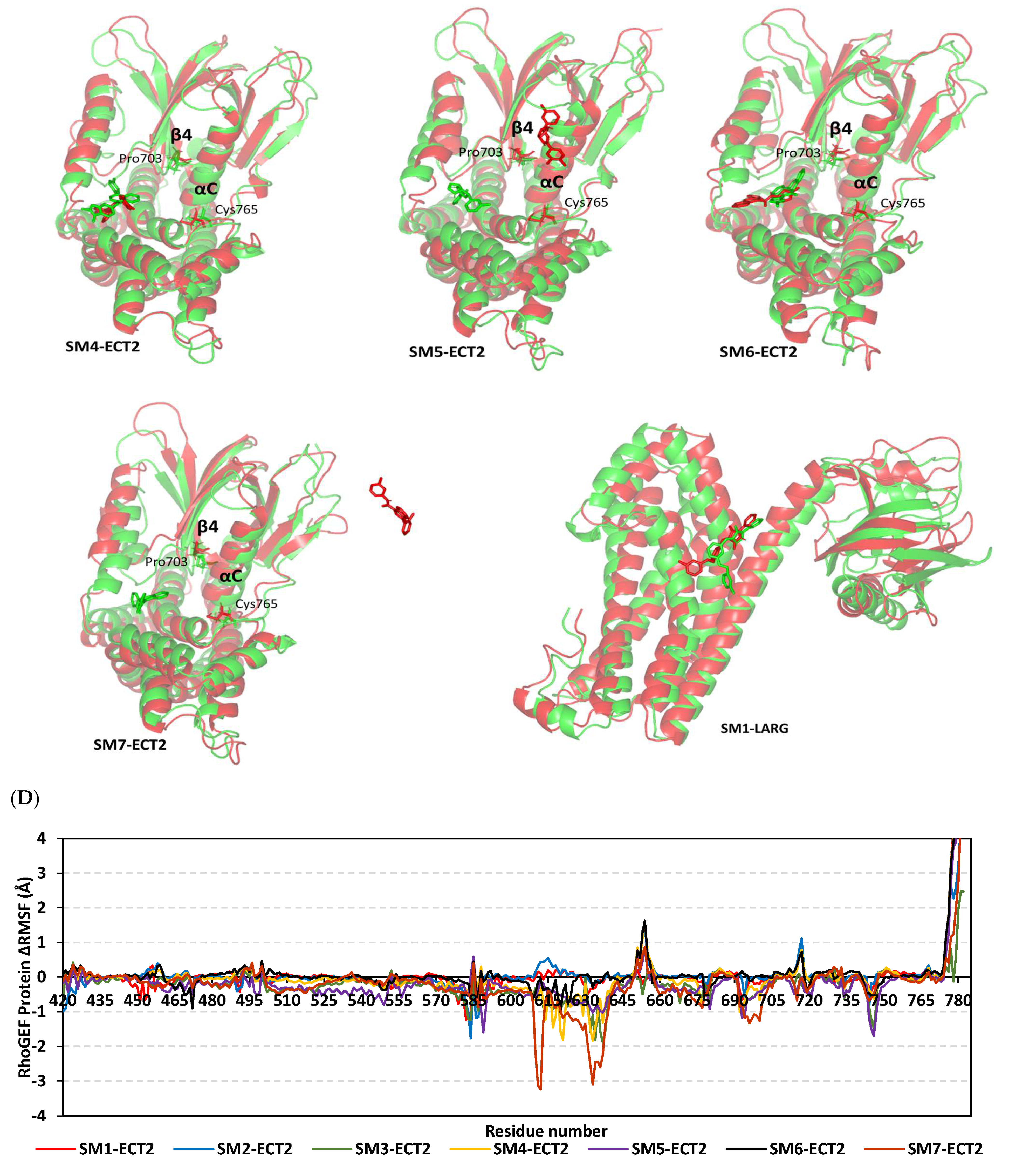
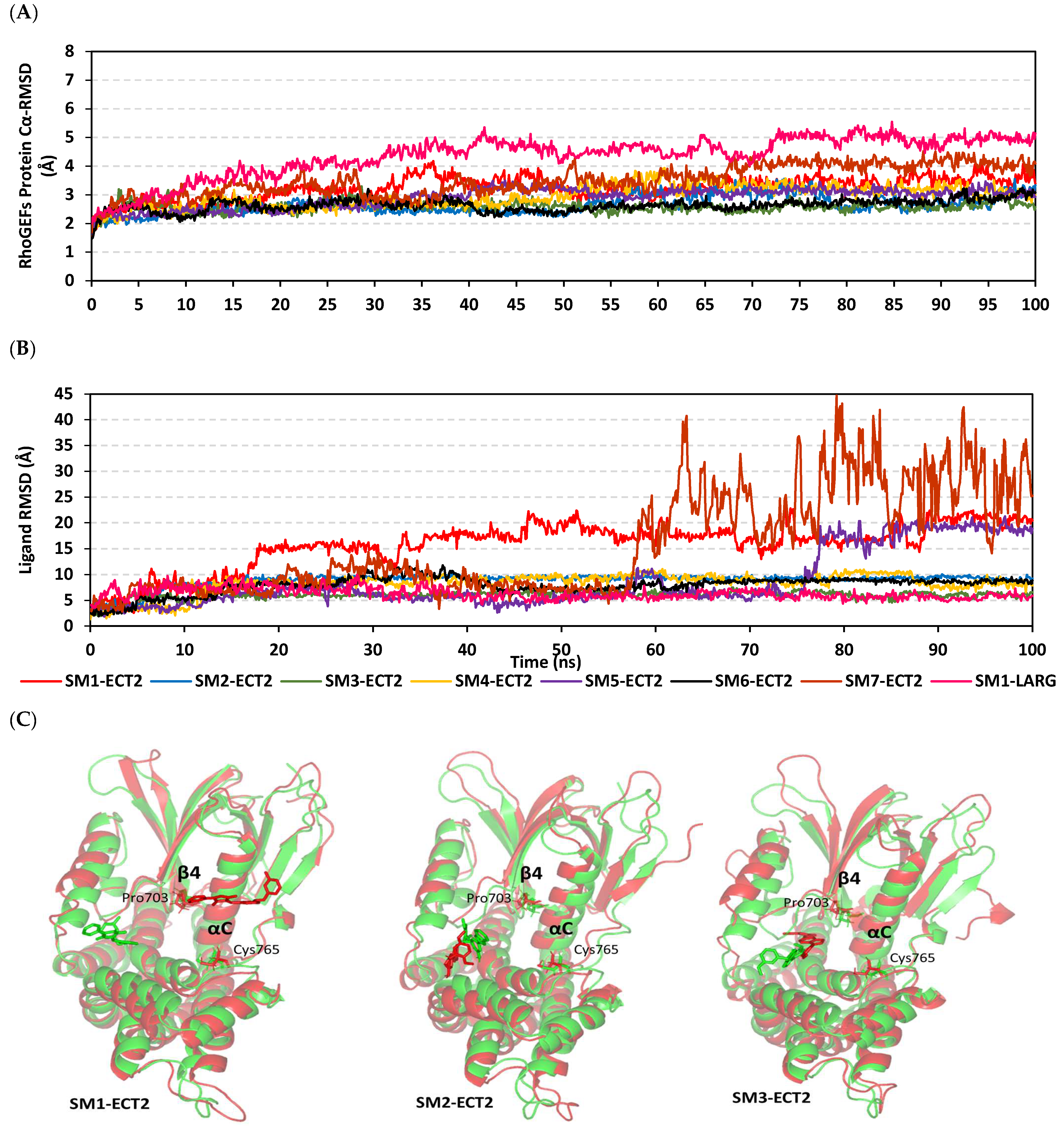
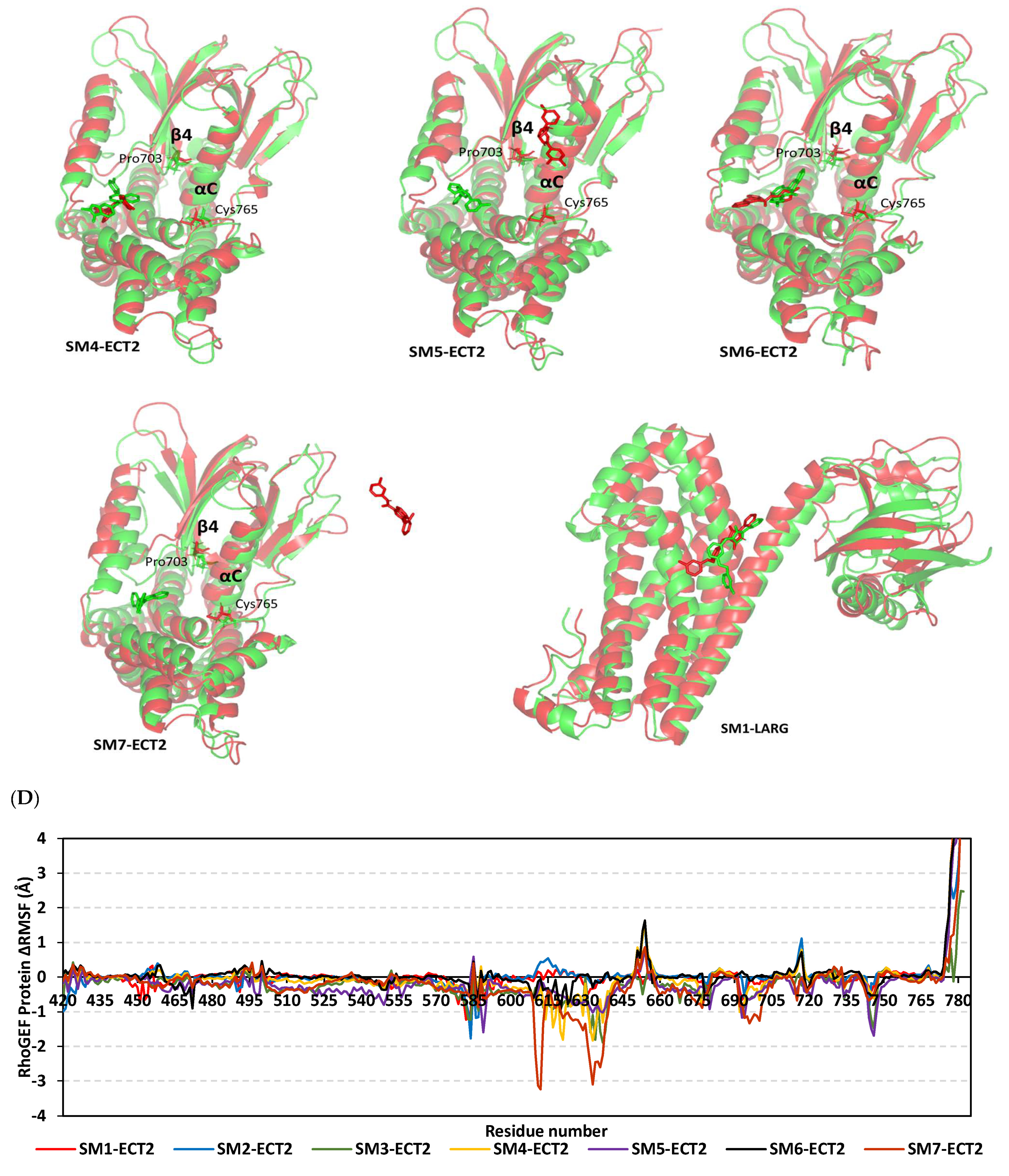
3.9. Several Small Molecules Exhibited Thermodynamic Stability at DH-PH Catalytic Interface
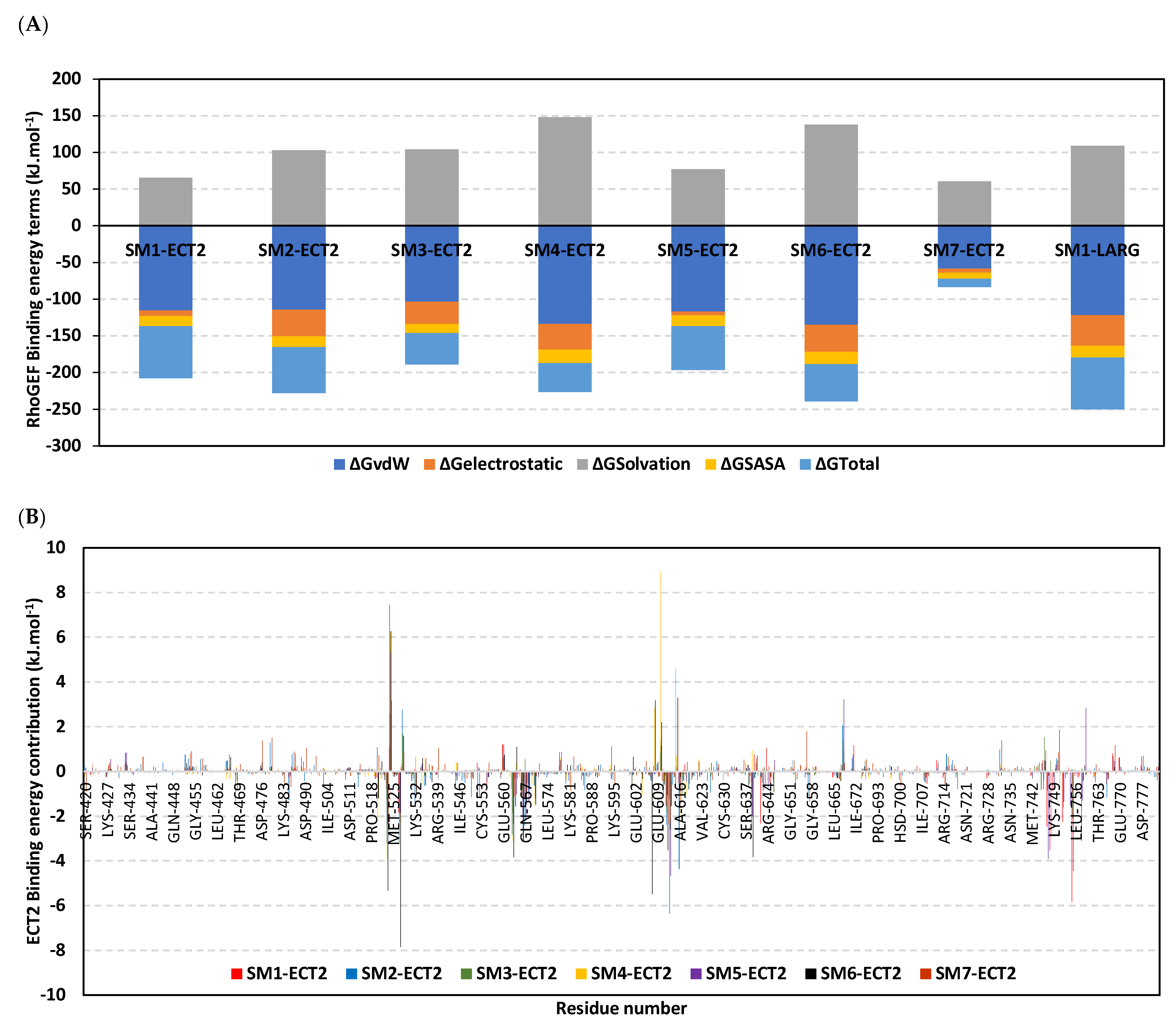
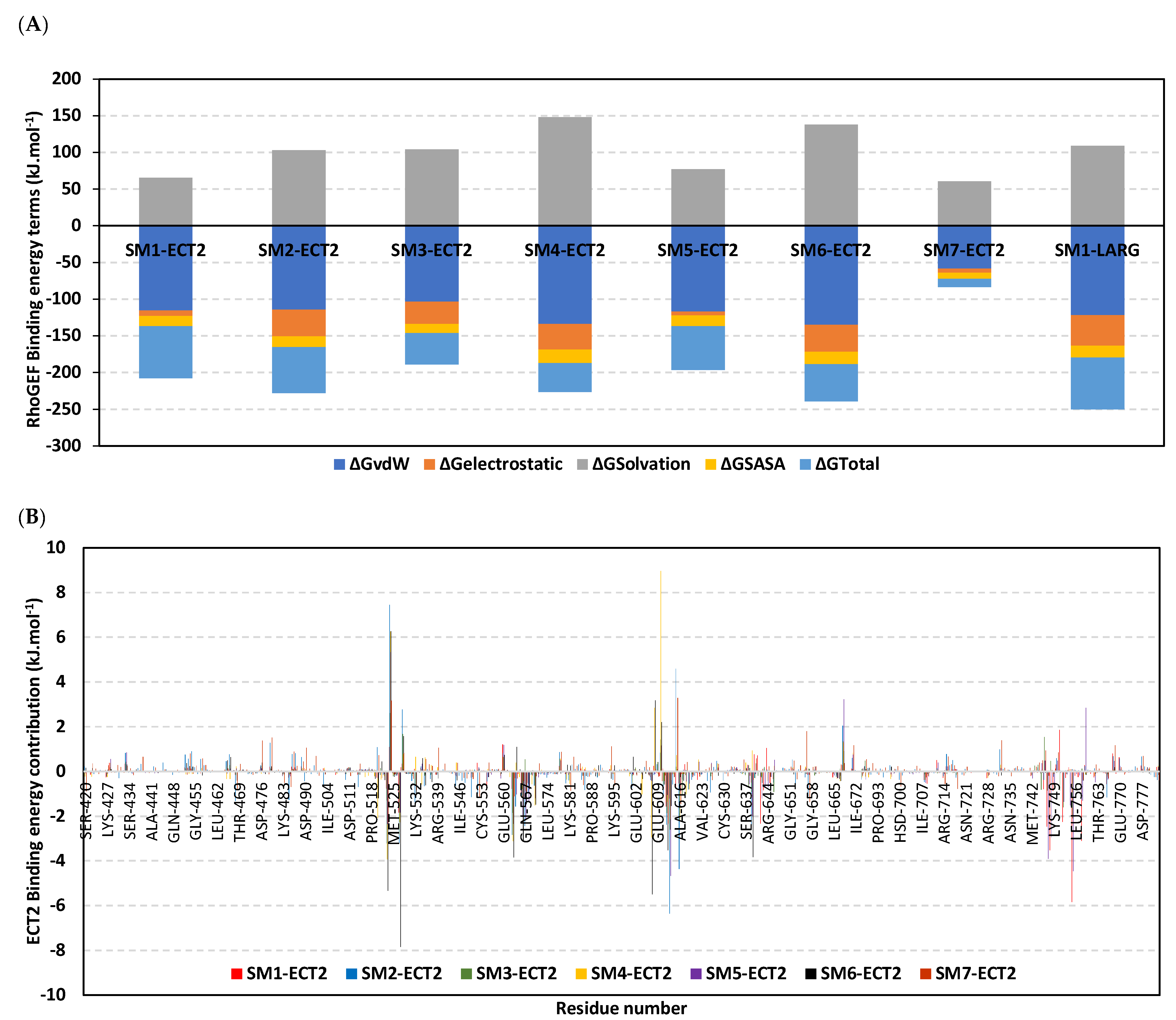
3.10. Hydrophobic Potentials Dominated the Binding-Free Energy Contributions at DH-PH Catalytic Site
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gharib, A.F.; Eldeen, M.A.; Khalifa, A.S.; Elsawy, W.H.; Eed, E.M.; El Askary, A.; Eid, R.A.; Soltan, M.A.; Raafat, N. Assessment of Glutathione Peroxidase-1 ( GPX1 ) Gene Expression as a Specific Diagnostic and Prognostic Biomarker in Malignant Pleural Mesothelioma. Diagnostics 2021, 11, 2285. [Google Scholar] [CrossRef] [PubMed]
- Zabady, S.; Mahran, N.; Soltan, M.A.; Eldeen, M.A.; Eid, R.A.; Albogami, S.; Fayad, E.; Matboli, M.; Habib, E.K.; Hasanin, A.H.; et al. Cyanidin-3-Glucoside Modulates hsa_circ_0001345/miRNA106b/ATG16L1 Axis Expression as a Potential Protective Mechanism against Hepatocellular Carcinoma. Curr. Issues Mol. Biol. 2022, 44, 115. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Abdureyim, M.; Yao, Q.; Li, X. Analysis of Differentially Expressed Genes in Endothelial Cells Following Tumor Cell Adhesion, and the Role of PRKAA2 and miR-124-3p. Front. Cell Dev. Biol. 2021, 9, 604038. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Chen, Y.J.; Liao, W.X.; Huang, S.Z.; Yu, Y.F.; Wen, J.Y.; Chen, J.; Lin, D.G.; Wu, X.Y.; Jiang, N.; Li, X. Prognostic and immunological role of CD36: A pan-cancer analysis. J. Cancer 2021, 12, 4762–4773. [Google Scholar] [CrossRef]
- Eid, R.A.; Soltan, M.A.; Eldeen, M.A.; Shati, A.A.; Dawood, S.A.; Eissa, M.; Zaki, M.S.A.; Algahtani, M.; Theyab, A.; Abdel-Daim, M.M.; et al. Assessment of RACGAP1 as a Prognostic and Immunological Biomarker in Multiple Human Tumors: A Multiomics Analysis. Int. J. Mol. Sci. 2022, 23, 14102. [Google Scholar] [CrossRef]
- Vecchione, R.; Quagliariello, V.; Giustetto, P.; Calabria, D.; Sathya, A.; Marotta, R.; Profeta, M.; Nitti, S.; Silvestri, N.; Pellegrino, T.; et al. Oil/water nano-emulsion loaded with cobalt ferrite oxide nanocubes for photo-acoustic and magnetic resonance dual imaging in cancer: In vitro and preclinical studies. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 275–286. [Google Scholar] [CrossRef]
- Rosso, F.; Quagliariello, V.; Tortora, C.; Di Lazzaro, A.; Barbarisi, A.; Iaffaioli, R.V. Cross-linked hyaluronic acid sub-micron particles: In vitro and in vivo biodistribution study in cancer xenograft model. J. Mater. Sci. Mater. Med. 2013, 24, 1473–1481. [Google Scholar] [CrossRef]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef]
- Ellenbroek, S.I.J.; Collard, J.G. Rho GTPases: Functions and association with cancer. Clin. Exp. Metastasis 2007, 24, 657–672. [Google Scholar] [CrossRef]
- Choudhari, R.; Minero, V.G.; Menotti, M.; Pulito, R.; Brakebusch, C.; Compagno, M.; Voena, C.; Ambrogio, C.; Chiarle, R. Redundant and nonredundant roles for Cdc42 and Rac1 in lymphomas developed in NPM-ALK transgenic mice. Blood 2016, 127, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Clayton, N.S.; Ridley, A.J. Targeting Rho GTPase Signaling Networks in Cancer. Front. Cell Dev. Biol. 2020, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Quilliam, L.A.; Rebhun, J.F.; Castro, A.F. A growing family of guanine nucleotide exchange factors is responsible for activation of ras-family GTPases. Prog. Nucleic Acid Res. Mol. Biol. 2002, 71, 391–444. [Google Scholar] [PubMed]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef]
- Schneid, S.; Wolff, F.; Buchner, K.; Bertram, N.; Baygün, S.; Barbosa, P.; Mangal, S.; Zanin, E. The BRCT domains of ECT2 have distinct functions during cytokinesis. Cell Rep. 2021, 34, 108805. [Google Scholar] [CrossRef]
- Justilien, V.; Fields, A.P. Ect2 links the PKC-Par6α complex to Rac1 activation and cellular transformation. Oncogene 2009, 28, 3597–3607. [Google Scholar] [CrossRef]
- Kosibaty, Z.; Murata, Y.; Minami, Y.; Noguchi, M.; Sakamoto, N. ECT2 promotes lung adenocarcinoma progression through extracellular matrix dynamics and focal adhesion signaling. Cancer Sci. 2021, 112, 703–714. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Y.; Wu, J.; Zhang, Z.; Chen, J.; Xie, M.; Tang, R.; Chen, C.; Chen, L.; Lin, S.; et al. ECT2 overexpression promotes the polarization of tumor-associated macrophages in hepatocellular carcinoma via the ECT2/PLK1/PTEN pathway. Cell Death Dis. 2021, 12, 162. [Google Scholar] [CrossRef]
- Gong, D.Y.; Chen, X.; Yang, T.L.; Wang, Y.; Guo, Y.; Zeng, J.H.; Chen, S.Z. Upregulation of ECT2 is associated with transcriptional program of cancer stem cells and predicts poor clinical outcome in gastric cancer. Oncol. Lett. 2020, 20, 54. [Google Scholar] [CrossRef]
- Cook, D.R.; Kang, M.; Martin, T.D.; Galanko, J.A.; Loeza, G.H.; Trembath, D.G.; Justilien, V.; Pickering, K.A.; Vincent, D.F.; Jarosch, A.; et al. Aberrant Expression and Subcellular Localization of ECT2 Drives Colorectal Cancer Progression and Growth. Cancer Res. 2021, 82, 90–104. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 9, 649–658. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. Tnmplot.Com: A web tool for the comparison of gene expression in normal, tumor and metastatic tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef] [PubMed]
- Almangush, A.; Mäkitie, A.A.; Triantafyllou, A.; de Bree, R.; Strojan, P.; Rinaldo, A.; Hernandez-Prera, J.C.; Suárez, C.; Kowalski, L.P.; Ferlito, A.; et al. Staging and grading of oral squamous cell carcinoma: An update. Oral Oncol. 2020, 107, 104799. [Google Scholar] [CrossRef] [PubMed]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef] [PubMed]
- Lánczky, A.; Győrffy, B. Web-based survival analysis tool tailored for medical research (KMplot): Development and implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Tran, M.T.M.T.; Yeh, K.T.; Chuang, Y.M.; Hsu, P.Y.; Low, J.T.; Kumari, H.; Lee, Y.T.; Chen, Y.C.; Huang, W.H.; Jin, H.; et al. Methylomic analysis identifies C11orf87 as a novel epigenetic biomarker for GI cancers. PLoS ONE 2021, 16, e0250499. [Google Scholar] [CrossRef]
- Søes, S.; Daugaard, I.L.; Sørensen, B.S.; Carus, A.; Mattheisen, M.; Alsner, J.; Overgaard, J.; Hager, H.; Hansen, L.L.; Kristensen, L.S. Hypomethylation and increased expression of the putative oncogene ELMO3 are associated with lung cancer development and metastases formation. Oncoscience 2014, 1, 367–374. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, D.; Lu, C. The SMART App: An interactive web application for comprehensive DNA methylation analysis and visualization. Epigenet. Chromatin 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L. Lo The crucial role of protein phosphorylation in cell signalingand its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cao, X.J.; Kulej, K.; Liu, W.; Ma, T.; MacDonald, M.; Chiang, C.M.; Garcia, B.A.; You, J. Uncovering BRD4 hyperphosphorylation associated with cellular transformation in NUT midline carcinom1. Proc. Natl. Acad. Sci. USA 2017, 114, E5352–E5361. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Shang, X.; Marchioni, F.; Evelyn, C.R.; Sipes, N.; Zhou, X.; Seibel, W.; Wortman, M.; Zheng, Y. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc. Natl. Acad. Sci. USA 2013, 110, 3155–3160. [Google Scholar] [CrossRef]
- Oleg, T.; Arthur, J.O. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Chen, M.; Pan, H.; Sun, L.; Shi, P.; Zhang, Y.; Li, L.; Huang, Y.; Chen, J. Structure and regulation of human epithelial cell transforming 2 protein. Proc. Natl. Acad. Sci. USA 2020, 117, 1027–1035. [Google Scholar] [CrossRef]
- Kristelly, R.; Gao, G.; Tesmer, J.J.G. Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J. Biol. Chem. 2004, 279, 47352–47362. [Google Scholar] [CrossRef] [PubMed]
- Elrayess, R.; Darwish, K.M.; Nafie, M.S.; El-sayyed, G.S.; Said, M.M.; Yassen, A.S.A. Quinoline–hydrazone hybrids as dual mutant EGFR inhibitors with promising metallic nanoparticle loading: Rationalized design, synthesis, biological investigation and computational studies. New J. Chem. 2022, 46, 18207–18232. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, X.; Russell, P.; Li, J.; Pan, W.; Fu, J.; Zhang, A. Evaluation of the binding performance of flavonoids to estrogen receptor alpha by Autodock, Autodock Vina and Surflex-Dock. Ecotoxicol. Environ. Saf. 2022, 233, 113323. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Saleh, A.H.; Abdelwaly, A.; Darwish, K.M.; Eissa, A.A.H.M.; Chittiboyina, A.; Helal, M.A. Deciphering the molecular basis of the kappa opioid receptor selectivity: A Molecular Dynamics study. J. Mol. Graph. Model. 2021, 106, 107940. [Google Scholar] [CrossRef]
- Ross, G.A.; Rustenburg, A.S.; Grinaway, P.B.; Fass, J.; Chodera, J.D. Biomolecular Simulations under Realistic Macroscopic Salt Conditions. J. Phys. Chem. B 2018, 122, 5466–5486. [Google Scholar] [CrossRef]
- Zaki, A.A.; Ashour, A.; Elhady, S.S.; Darwish, K.M.; Al-Karmalawy, A.A. Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies. J. Tradit. Complement. Med. 2022, 12, 16–34. [Google Scholar] [CrossRef]
- Tuble, S.C.; Anwar, J.; Gale, J.D. An Approach to Developing a Force Field for Molecular Simulation of Martensitic Phase Transitions between Phases with Subtle Differences in Energy and Structure. J. Am. Chem. Soc. 2004, 126, 396–405. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.S.; Pacheco, B.D.C.; Pinheiro, S.; Muri, E.M.F.; Dias, L.R.S.; Lima, C.H.S.; Garrett, R.; de Moraes, M.B.M.; de Souza, B.E.G.; Puzer, L. 3-Acyltetramic acids as a novel class of inhibitors for human kallikreins 5 and 7. Bioorg. Med. Chem. Lett. 2019, 29, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, A.M.; Hassan, A.M.A.; Elkaeed, E.B.; Alesawy, M.S.; Al-Karmalawy, A.A. Design, synthesis, and SAR studies of novel 4-methoxyphenyl pyrazole and pyrimidine derivatives as potential dual tyrosine kinase inhibitors targeting both EGFR and VEGFR-2. Bioorg. Chem. 2022, 123, 105770. [Google Scholar] [CrossRef] [PubMed]
- Elhady, S.S.; Abdelhameed, R.F.A.; Malatani, R.T.; Alahdal, A.M.; Bogari, H.A.; Almalki, A.J.; Mohammad, K.A.; Ahmed, S.A.; Khedr, A.I.M.; Darwish, K.M. Molecular docking and dynamics simulation study of hyrtios erectus isolated scalarane sesterterpenes as potential sars-cov-2 dual target inhibitors. Biology 2021, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure of Biomolecules Through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Manandhar, A.; Blass, B.E.; Colussi, D.J.; Almi, I.; Abou-Gharbia, M.; Klein, M.L.; Elokely, K.M. Targeting SARS-CoV-2 M3CLpro by HCV NS3/4a Inhibitors: In Silico Modeling and in Vitro Screening. J. Chem. Inf. Model. 2021, 61, 1020–1032. [Google Scholar] [CrossRef]
- Almalki, A.J.; Ibrahim, T.S.; Elhady, S.S.; Hegazy, W.A.H.; Darwish, K.M. Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents. Pharmaceuticals 2022, 15, 110. [Google Scholar] [CrossRef]
- Benson, N.C.; Daggett, V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 8722–8731. [Google Scholar] [CrossRef]
- Zou, Y.; Shao, Z.; Peng, J.; Li, F.; Gong, D.; Wang, C.; Zuo, X.; Zhang, Z.; Wu, J.; Shi, Y.; et al. Crystal structure of triple-BRCT-domain of ECT2 and insights into the binding characteristics to CYK-4. FEBS Lett. 2014, 588, 2911–2920. [Google Scholar] [CrossRef]
- Cavasotto, C.N. Binding Free Energy Calculation Using Quantum Mechanics Aimed for Drug Lead Optimization. In Methods in Molecular Biology; Humana New York: New York, NY, USA, 2020. [Google Scholar]
- Ren, J.; Yuan, X.; Li, J.; Lin, S.; Yang, B.; Chen, C.; Zhao, J.; Zheng, W.; Liao, H.; Yang, Z.; et al. Assessing the performance of the g_mmpbsa tools to simulate the inhibition of oseltamivir to influenza virus neuraminidase by molecular mechanics Poisson–Boltzmann surface area methods. J. Chin. Chem. Soc. 2019, 67, 46–53. [Google Scholar] [CrossRef]
- Hamed, M.I.A.; Darwish, K.M.; Soltane, R.; Chrouda, A.; Mostafa, A.; Abo Shama, N.M.; Elhady, S.S.; Abulkhair, H.S.; Khodir, A.E.; Elmaaty, A.A.; et al. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: Rational based design, in silico, in vitro, and SAR studies for lead optimization. RSC Adv. 2021, 11, 35536–35558. [Google Scholar] [CrossRef] [PubMed]
- Humphries, B.; Wang, Z.; Yang, C. Rho GTPases: Big Players in Breast Cancer Initiation, Metastasis and Therapeutic Responses. Cells 2020, 9, 2167. [Google Scholar] [CrossRef] [PubMed]
- Limzerwala, J.F.; Jeganathan, K.B.; Kloeber, J.A.; Davies, B.A.; Zhang, C.; Sturmlechner, I.; Zhong, J.; Fierro Velasco, R.; Fields, A.P.; Yuan, Y.; et al. FoxM1 insufficiency hyperactivates Ect2–RhoA–mDia1 signaling to drive cancer. Nat. Cancer 2020, 1, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Fields, A.P.; Justilien, V. The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv. Enzyme Regul. 2010, 50, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Huff, L.P.; DeCristo, M.J.; Trembath, D.; Kuan, P.F.; Yim, M.; Liu, J.; Cook, D.R.; Miller, C.R.; Der, C.J.; Cox, A.D. The Role of Ect2 Nuclear RhoGEF Activity in Ovarian Cancer Cell Transformation. Genes Cancer 2013, 4, 460–475. [Google Scholar] [CrossRef]
- Wang, H.K.; Liang, J.F.; Zheng, H.X.; Xiao, H. Expression and prognostic significance of ECT2 in invasive breast cancer. J. Clin. Pathol. 2018, 71, 442–445. [Google Scholar] [CrossRef]
- Yi, M.; Zhang, D.; Song, B.; Zhao, B.; Niu, M.; Wu, Y.; Dai, Z.; Wu, K. Increased expression of ECT2 predicts the poor prognosis of breast cancer patients. Exp. Hematol. Oncol. 2022, 11, 107. [Google Scholar] [CrossRef]
- Jin, Y.; Yu, Y.; Shao, Q.; Ma, Y.; Zhang, R.; Yao, H.; Xu, Y. Up-regulation of ECT2 is associated with poor prognosis in gastric cancer patients. Int. J. Clin. Exp. Pathol. 2014, 7, 8724–8731. [Google Scholar]
- Wang, H.B.; Yan, H.C.; Liu, Y. Clinical significance of ECT2 expression in tissue and serum of gastric cancer patients. Clin. Transl. Oncol. 2016, 18, 735–742. [Google Scholar] [CrossRef]
- Zhang, M.L.; Lu, S.; Zhou, L.; Zheng, S. Sen Correlation between ECT2 gene expression and methylation change of ECT2 promoter region in pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 2008, 7, 533–538. [Google Scholar]
- Luo, Y.; Qin, S.L.; Mu, Y.F.; Wang, Z.S.; Zhong, M.; Bian, Z.Q. Elevated expression of ECT2 predicts unfavorable prognosis in patients with colorectal cancer. Biomed. Pharmacother. 2015, 73, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Sung, W.W.; Chen, H.C.; Chern, Y.J.; Hsu, H.T.; Lin, Y.M.; Lin, S.H.; Peck, K.; Yeh, K.T. Early assessment of colorectal cancer by quantifying circulating tumor cells in peripheral blood: ECT2 in diagnosis of colorectal cancer. Int. J. Mol. Sci. 2017, 18, 743. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xia, H.; Zhang, X.; Karthik, S.; Pratap, S.V.; Ooi, L.L.; Hong, W.; Hui, K.M. ECT2 regulates the Rho/ERK signalling axis to promote early recurrence in human hepatocellular carcinoma. J. Hepatol. 2015, 62, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.X.; Yin, J.Y.; Shen, Y.; Zhang, W.; Zhou, H.H.; Liu, Z.Q. Genome-scale analysis identifies NEK2, DLGAP5 and ECT2 as promising diagnostic and prognostic biomarkers in human lung cancer. Sci. Rep. 2017, 7, 8072. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.A.; Eldeen, M.A.; Soltan, M.A.; Al-Shraim, M.; Aldehri, M.; Alqahtani, L.S.; Alsharif, G.; Albogami, S.; Jafri, I.; Fayad, E.; et al. Integrative analysis of WDR12 as a potential prognostic and immunological biomarker in multiple human tumors. Front. Genet. 2023, 13, 1008502. [Google Scholar] [CrossRef]
- Bell Gorrod, H.; Kearns, B.; Stevens, J.; Thokala, P.; Labeit, A.; Latimer, N.; Tyas, D.; Sowdani, A. A Review of Survival Analysis Methods Used in NICE Technology Appraisals of Cancer Treatments: Consistency, Limitations, and Areas for Improvement. Med. Decis. Mak. 2019, 39, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Shen, H.; Che, K.; Cong, L.; Dong, W.; Zhang, T.; Liu, Q.; Du, J. Diagnostic and prognostic value of blood samples for KRAS mutation identification in lung cancer: A meta-analysis. Oncotarget 2017, 8, 36812–36823. [Google Scholar] [CrossRef]
- Christopoulos, P.; Dietz, S.; Kirchner, M.; Volckmar, A.L.; Endris, V.; Neumann, O.; Ogrodnik, S.; Heussel, C.P.; Herth, F.J.; Eichhorn, M.; et al. Detection of TP53 mutations in tissue or liquid rebiopsies at progression identifies ALK + lung cancer patients with poor survival. Cancers 2019, 11, 124. [Google Scholar] [CrossRef]
- Shire, N.J.; Klein, A.B.; Golozar, A.; Collins, J.M.; Fraeman, K.H.; Nordstrom, B.L.; McEwen, R.; Hembrough, T.; Rizvi, N.A. STK11 (LKB1) mutations in metastatic NSCLC: Prognostic value in the real world. PLoS ONE 2020, 15, e0238358. [Google Scholar] [CrossRef]
- Rosellini, P.; Amintas, S.; Caumont, C.; Veillon, R.; Galland-Girodet, S.; Cuguillière, A.; Nguyen, L.; Domblides, C.; Gouverneur, A.; Merlio, J.P.; et al. Clinical impact of STK11 mutation in advanced-stage non-small cell lung cancer. Eur. J. Cancer 2022, 172, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, J.; Scholz, C.J.; Kneitz, S.; Weber, D.; Fuchs, J.; Hedrich, R.; Deeken, R. DNA Methylation Mediated Control of Gene Expression Is Critical for Development of Crown Gall Tumors. PLoS Genet. 2013, 9, e1003267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Chen, M.; Sun, L.; Han, J.; Elena, V.K.; Qiao, H. CXCL12 methylation-mediated epigenetic regulation of gene expression in papillary thyroid carcinoma. Sci. Rep. 2017, 7, 44033. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Han, Y.K.; Song, J.M.; Lee, C.H.; Kang, K.; Yi, J.M.; Park, H.R. Aberrantly hypermethylated tumor suppressor genes were identified in oral squamous cell carcinoma (OSCC). Clin. Epigenet. 2019, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Wan, Y.C.; Li, S.; Qiu, H.D.; Jiang, Y.; Ma, X.; Zhou, S.L.; Cheng, W.J. Identification of aberrantly methylated differentially expressed genes and associated pathways in endometrial cancer using integrated bioinformatic analysis. Cancer Med. 2020, 9, 3522–3536. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yang, J.F.; Ho, F.; Robertson, E.S.; You, J. Bromodomain-containing protein BRD4 is hyperphosphorylated in mitosis. Cancers 2020, 12, 1637. [Google Scholar] [CrossRef] [PubMed]
- Ooshima, A.; Park, J.; Kim, S.J. Phosphorylation status at Smad3 linker region modulates transforming growth factor-β-induced epithelial-mesenchymal transition and cancer progression. Cancer Sci. 2019, 110, 481–488. [Google Scholar] [CrossRef]
- Yao, W.; Feng, D.; Bian, W.; Yang, L.; Li, Y.; Yang, Z.; Xiong, Y.; Zheng, J.; Zhai, R.; He, J. EBP50 inhibits EGF-induced breast cancer cell proliferation by blocking EGFR phosphorylation. Amino Acids 2012, 43, 2027–2035. [Google Scholar] [CrossRef]
- Jiang, W.; He, Y.; He, W.; Wu, G.; Zhou, X.; Sheng, Q.; Zhong, W.; Lu, Y.; Ding, Y.; Lu, Q.; et al. Exhausted CD8+T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front. Immunol. 2021, 11, 622509. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Loeuillard, E.; Yang, J.; Buckarma, E.L.N.; Wang, J.; Liu, Y.; Conboy, C.; Pavelko, K.D.; Li, Y.; O’Brien, D.; Wang, C.; et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J. Clin. Investig. 2020, 130, 5380–5396. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Berzofsky, J.A. Tissue-specific roles of NKT cells in tumor immunity. Front. Immunol. 2018, 9, 1838. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Lin, M.; Brouwer-Visser, J.; Heim, J.; Smotkin, D.; Hebert, T.; Gunter, M.J.; Goldberg, G.L.; Zheng, D.; Huang, G.S. RNA-seq identification of RACGAP1 as a metastatic driver in uterine carcinosarcoma. Clin. Cancer Res. 2016, 22, 4676–4686. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, B.; Guo, D.; Pan, L.; Luo, X.; Tang, J.; Yang, W.; Zhang, Y.; Zhang, L.; Huang, J.; et al. Up-Regulation of RACGAP1 Promotes Progressions of Hepatocellular Carcinoma Regulated by GABPA via PI3K/AKT Pathway. Oxid. Med. Cell. Longev. 2022, 2022, 3034150. [Google Scholar] [CrossRef]
- Li, X.L.; Ji, Y.M.; Song, R.; Li, X.N.; Guo, L.S. KIF23 promotes gastric cancer by stimulating cell proliferation. Dis. Markers 2019, 2019, 9751923. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.-T.; Ren, J.; Yu, J.; Li, S.-N.; Guo, X.-F.; Zhou, Y.-Z. KIF23 enhances cell proliferation in pancreatic ductal adenocarcinoma and is a potent therapeutic target. Ann. Transl. Med. 2020, 8, 1394. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef]
- Baker, M.J.; Cooke, M.; Kazanietz, M.G. Nuclear PKCι-ECT2-Rac1 and Ribosome Biogenesis: A Novel Axis in Lung Tumorigenesis. Cancer Cell 2017, 31, 167–169. [Google Scholar] [CrossRef]
- Jatoi, A.; Radecki Breitkopf, C.; Foster, N.R.; Block, M.S.; Grudem, M.; Wahner Hendrickson, A.; Carlson, R.E.; Barrette, B.; Karlin, N.; Fields, A.P. A mixed-methods feasibility trial of protein kinase C iota inhibition with auranofin in asymptomatic ovarian cancer patients. Oncology 2015, 88, 208–213. [Google Scholar] [CrossRef]
- Topkas, E.; Cai, N.; Cumming, A.; Hazar-Rethinam, M.; Gannon, O.M.; Burgess, M.; Saunders, N.A.; Endo-Munoz, L. Auranofin is a potent suppressor of osteosarcoma metastasis. Oncotarget 2016, 7, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Gamberi, T.; Chiappetta, G.; Fiaschi, T.; Modesti, A.; Sorbi, F.; Magherini, F. Upgrade of an old drug: Auranofin in innovative cancer therapies to overcome drug resistance and to increase drug effectiveness. Med. Res. Rev. 2022, 42, 1111–1146. [Google Scholar] [CrossRef] [PubMed]
- Cardama, G.; Comin, M.; Hornos, L.; Gonzalez, N.; Defelipe, L.; Turjanski, A.; Alonso, D.; Gomez, D.; Menna, P. Preclinical Development of Novel Rac1-GEF Signaling Inhibitors using a Rational Design Approach in Highly Aggressive Breast Cancer Cell Lines. Anticancer Agents Med. Chem. 2014, 14, 840–851. [Google Scholar] [CrossRef]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [PubMed]
- Cardama, G.A.; Gonzalez, N.; Ciarlantini, M.; Donadío, L.G.; Comin, M.J.; Alonso, D.F.; Menna, P.L.; Gomez, D.E. Proapoptotic and antiinvasive activity of Rac1 small molecule inhibitors on malignant glioma cells. OncoTargets Ther. 2014, 7, 2021–2033. [Google Scholar] [CrossRef]
- Verdine, G.L.; Walensky, L.D. The challenge of drugging undruggable targets in cancer: Lessons learned from targeting BCL-2 family members. Clin. Cancer Res. 2007, 13, 7264–7270. [Google Scholar] [CrossRef]
- Russ, A.P.; Lampel, S. The druggable genome: An update. Drug Discov. Today 2005, 10, 1607–1610. [Google Scholar] [CrossRef] [PubMed]
- Soisson, S.M.; Nimnual, A.S.; Uy, M.; Bar-Sagi, D.; Kuriyan, J. Crystal structure of the Dbl and pleckstrin homology domains from the human Son of sevenless protein. Cell 1998, 95, 259–268. [Google Scholar] [CrossRef]
- Bandekar, S.J.; Arang, N.; Tully, E.S.; Tang, B.A.; Barton, B.L.; Li, S.; Gutkind, J.S.; Tesmer, J.J.G. Structure of the C-terminal guanine nucleotide exchange factor module of Trio in an autoinhibited conformation reveals its oncogenic potential. Sci. Signal. 2019, 12, eaav2449. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, L.; Sprang, S.R.; Sternweis, P.C. Modulation of a GEF switch: Autoinhibition of the intrinsic guanine nucleotide exchange activity of p115-RhoGEF. Protein Sci. 2011, 20, 107–117. [Google Scholar] [CrossRef]
- He, X.; Kuo, Y.C.; Rosche, T.J.; Zhang, X. Structural basis for autoinhibition of the guanine nucleotide exchange factor FARP2. Structure 2013, 21, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided. Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Singh, W.; Karabencheva-Christova, T.G.; Black, G.W.; Ainsley, J.; Dover, L.; Christov, C.Z. Conformational Dynamics, Ligand Binding and Effects of Mutations in NirE an S-Adenosyl-L-Methionine Dependent Methyltransferase. Sci. Rep. 2016, 6, 20107. [Google Scholar] [CrossRef] [PubMed]
- Fatriansyah, J.F.; Rizqillah, R.K.; Yandi, M.Y.; Fadilah; Sahlan, M. Molecular docking and dynamics studies on propolis sulabiroin-A as a potential inhibitor of SARS-CoV-2. J. King Saud Univ.-Sci. 2022, 34, 101707. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small Molecules | Docking Binding Energy (Kcal.mol−1; RMSD Å) | H-Bond Interaction (Length Å/angle°) | Hydrophobic Interaction | π-Driven Interaction (Length Å) | van der Waal with Side Chain Carbons | |
|---|---|---|---|---|---|---|
| Code-Target | Chemical Structure | |||||
| SM1-ECT2 |  | −5.824 (1.890) | Lys527; 2.5/121 Gln567; 2.4/159 Asp610; 2.9/140 Arg612; 3.0/126 | Val520, Phe523, Val559, Ile563, Val566, Ala616, His638 | Phe523; 5.1 Arg612; 3.2 | Glu524 (Cβ,Cγ) Lys613 (Cγ,Cδ) |
| SM2-ECT2 |  | −6.369 (1.370) | Glu524; 2.6/124 Lys527; 1.9/158 Lys527; 2.4/129 Arg612; 2.7/125 | Val520, Phe523, Ile563, Val566, Pro570, Ile607, Ala616, His638 | Phe523; 5.0 Arg612; 3.1 Arg612; 2.0 | Glu524 (Cβ,Cγ) Lys613 (Cβ,Cγ,Cδ) |
| SM3-ECT2 |  | −6.061 (1.207) | Gln567; 3.2/133 Arg612; 2.6/130 Arg612; 2.4/128 | Val520, Phe523, Ile563, Val566, Pro570, Ile607, Ala616, His638 | Phe523; 4.9 Arg612; 3.1 | Glu524 (Cβ,Cγ) Lys613 (Cγ,Cδ) |
| SM4-ECT2 |  | −5.855 (1.078) | Gln567; 3.2/133 Arg612; 2.7/121 Arg612; 2.3/131 | Phe523, Val559, Ile563, Val566, Ala616, His638 | Phe523; 5.0 Gln567; 5.1 Arg612; 3.1 Arg612; 3.0 | Glu524 (Cβ,Cγ) Lys613 (Cβ,Cγ,Cδ) |
| SM5-ECT2 |  | −4.972 (1.623) | Arg612; 1.3/143 Arg612; 3.2/126 | Val520, Phe523, Ile563, Val566, Pro570, Ile607, Ala616, His638 | Gln567; 3.7 Lys527; 3.0 Arg612; 2.7 | Glu524 (Cγ) Lys613 (Cγ,Cδ) |
| SM6-ECT2 |  | −4.853 (1.569) | Arg612; 2.5/131 Arg612; 2.7/129 | Phe523, Val559, Ile563, Ala616, His638, His759 | Phe523; 4.8 Glu567; 3.9 Arg612; 2.3 | Glu524 (Cβ,Cγ) Lys613 (Cγ,Cδ) |
| SM7-ECT2 |  | −5.469 (1.012) | Gln567; 2.4/159 Asp610; 2.9/140 Arg612; 3.0/120 Lys527; 2.5/126 | Val520, Phe523, Val559, Ile563, Val566, Ala616, His638 | Phe523; 4.8 Lys527; 4.7 Arg612; 3.2 | Glu524 (Cβ,Cγ) Lys613 (Cβ,Cγ) |
| SM1-LARG |  | −5.534 (1.065) | Pro892; 3.2/146 Arg986; 2.5/143 Arg986; 2.1/159 | Cys888, Phe892, Leu895, Met934, Leu937, Leu971, Val974, Val978 | Phe893; 4.9 | Glu896 (Cβ,Cγ) Asn975 (Cβ) Gln985 (Cβ,Cγ) |
| Ligand/RhoGEF | Energy (kJ/mol ± SE) | ||||
|---|---|---|---|---|---|
| ΔGvdW | ΔGelectrostatic | ΔGSolvation | ΔGSASA | ΔGTotal | |
| SM1-ECT2 | −115.21 ± 8.66 | −7.83 ± 14.77 | 65.68 ± 7.55 | −13.75 ± 2.15 | −71.11 ± 9.84 |
| SM2-ECT2 | −114.20 ± 32.03 | −36.58 ± 37.89 | 103.21 ± 30.75 | −14.69 ± 3.95 | −62.26 ± 21.79 |
| SM3-ECT2 | −103.52 ± 9.21 | −30.26 ± 15.56 | 103.99 ± 32.83 | −12.70 ± 2.44 | −42.49 ± 14.68 |
| SM4-ECT2 | −133.56 ± 19.32 | −35.22 ± 23.48 | 147.80 ± 30.33 | −18.34 ± 1.70 | −39.32 ± 24.11 |
| SM5-ECT2 | −116.85 ± 19.44 | −5.20 ± 8.63 | 76.88 ± 31.92 | −14.64 ± 2.23 | −59.81 ± 17.65 |
| SM6-ECT2 | −135.18 ± 5.64 | −36.59 ± 34.42 | 138.11 ± 48.95 | −16.89 ± 0.75 | −50.55 ± 16.45 |
| SM7-ECT2 | −58.45 ± 53.16 | −5.80 ± 14.24 | 60.72 ± 43.58 | −7.91 ± 7.26 | −11.44 ± 69.52 |
| SM1-LARG | −121.79 ± 10.85 | −41.63 ± 16.39 | 109.01 ± 17.33 | −16.09 ± 0.50 | −70.50 ± 10.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soltan, M.A.; Eldeen, M.A.; Sajer, B.H.; Abdelhameed, R.F.A.; Al-Salmi, F.A.; Fayad, E.; Jafri, I.; Ahmed, H.E.M.; Eid, R.A.; Hassan, H.M.; et al. Integration of Chemoinformatics and Multi-Omics Analysis Defines ECT2 as a Potential Target for Cancer Drug Therapy. Biology 2023, 12, 613. https://doi.org/10.3390/biology12040613
Soltan MA, Eldeen MA, Sajer BH, Abdelhameed RFA, Al-Salmi FA, Fayad E, Jafri I, Ahmed HEM, Eid RA, Hassan HM, et al. Integration of Chemoinformatics and Multi-Omics Analysis Defines ECT2 as a Potential Target for Cancer Drug Therapy. Biology. 2023; 12(4):613. https://doi.org/10.3390/biology12040613
Chicago/Turabian StyleSoltan, Mohamed A., Muhammad Alaa Eldeen, Bayan H. Sajer, Reda F. A. Abdelhameed, Fawziah A. Al-Salmi, Eman Fayad, Ibrahim Jafri, Hebatallah Emam Mohammed Ahmed, Refaat A. Eid, Hesham M. Hassan, and et al. 2023. "Integration of Chemoinformatics and Multi-Omics Analysis Defines ECT2 as a Potential Target for Cancer Drug Therapy" Biology 12, no. 4: 613. https://doi.org/10.3390/biology12040613
APA StyleSoltan, M. A., Eldeen, M. A., Sajer, B. H., Abdelhameed, R. F. A., Al-Salmi, F. A., Fayad, E., Jafri, I., Ahmed, H. E. M., Eid, R. A., Hassan, H. M., Al-Shraim, M., Negm, A., Noreldin, A. E., & Darwish, K. M. (2023). Integration of Chemoinformatics and Multi-Omics Analysis Defines ECT2 as a Potential Target for Cancer Drug Therapy. Biology, 12(4), 613. https://doi.org/10.3390/biology12040613