
Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation
 ,
,  , and
, and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Animals and TBI Model
2.3. MTT Assay
2.4. Western Blotting (WB)
2.5. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.6. DCF-DA ROS Staining
2.7. Immunohistochemistry
2.8. Drug Treatments/Administration
2.9. Statistical Analysis
3. Results
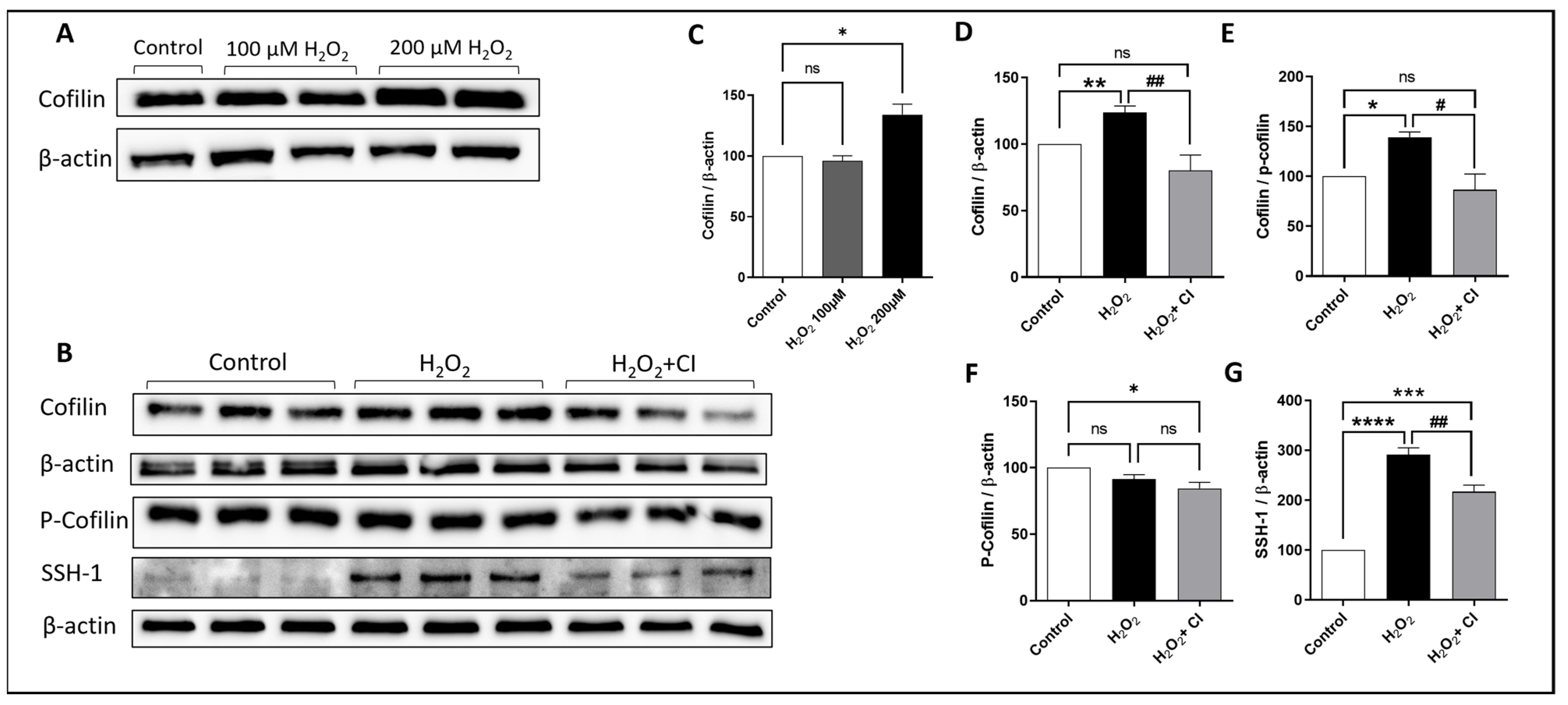
3.1. Cofilin and SSH1 Are Activated in Microglial Cells after H2O2 Treatment
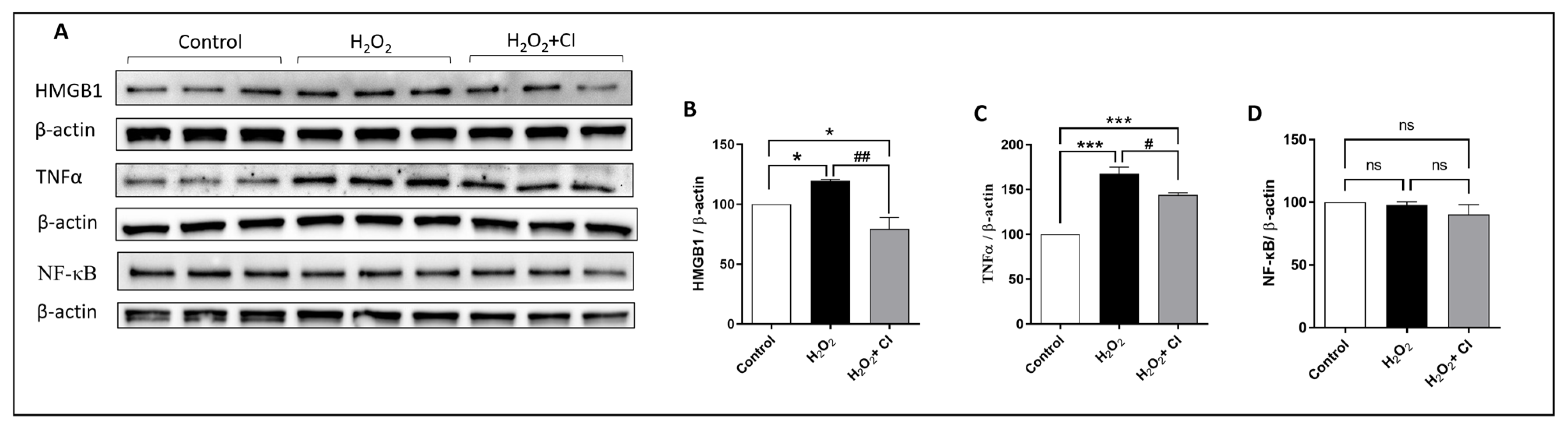
3.2. CI Treatment Reduced the H2O2-Induced Microglial Activation
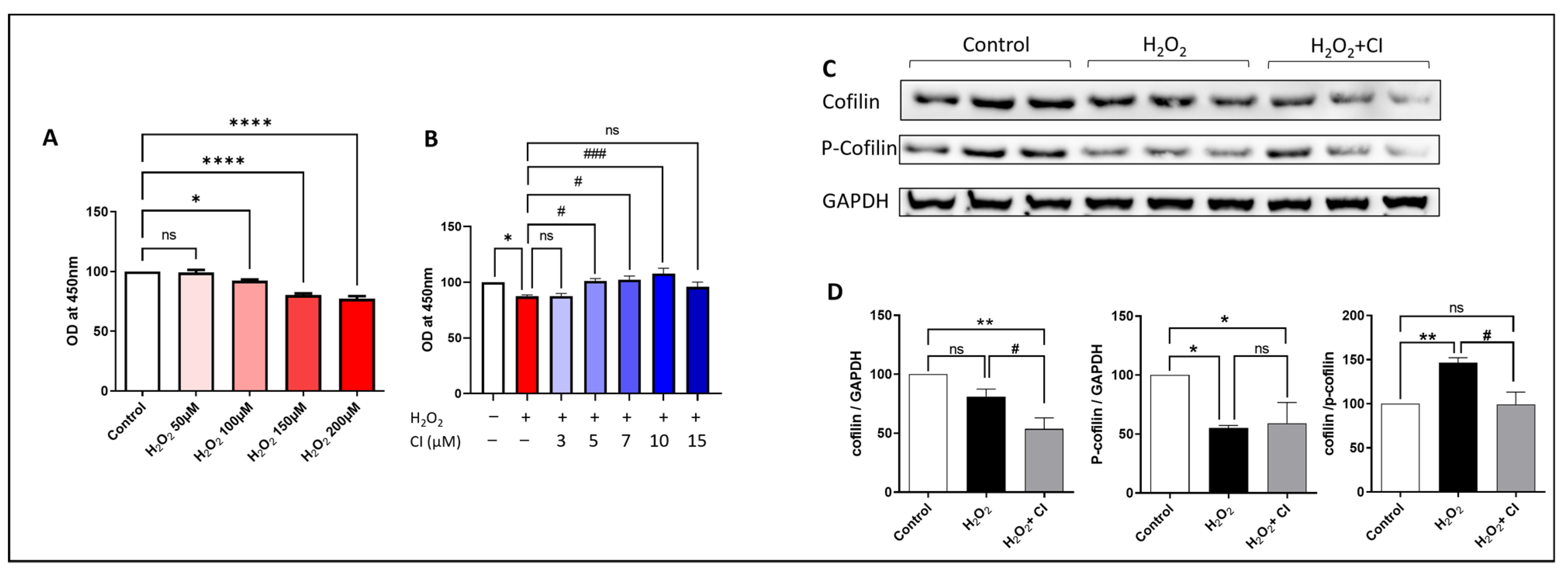
3.3. H2O2 Activates Cofilin Signaling, and CI Treatment Protects against H2O2-Induced Neurotoxicity in SH-SH5Y Cells
3.4. CI Treatment Reduces the H2O2-Induced ROS Production in SH-SY5Y Cells
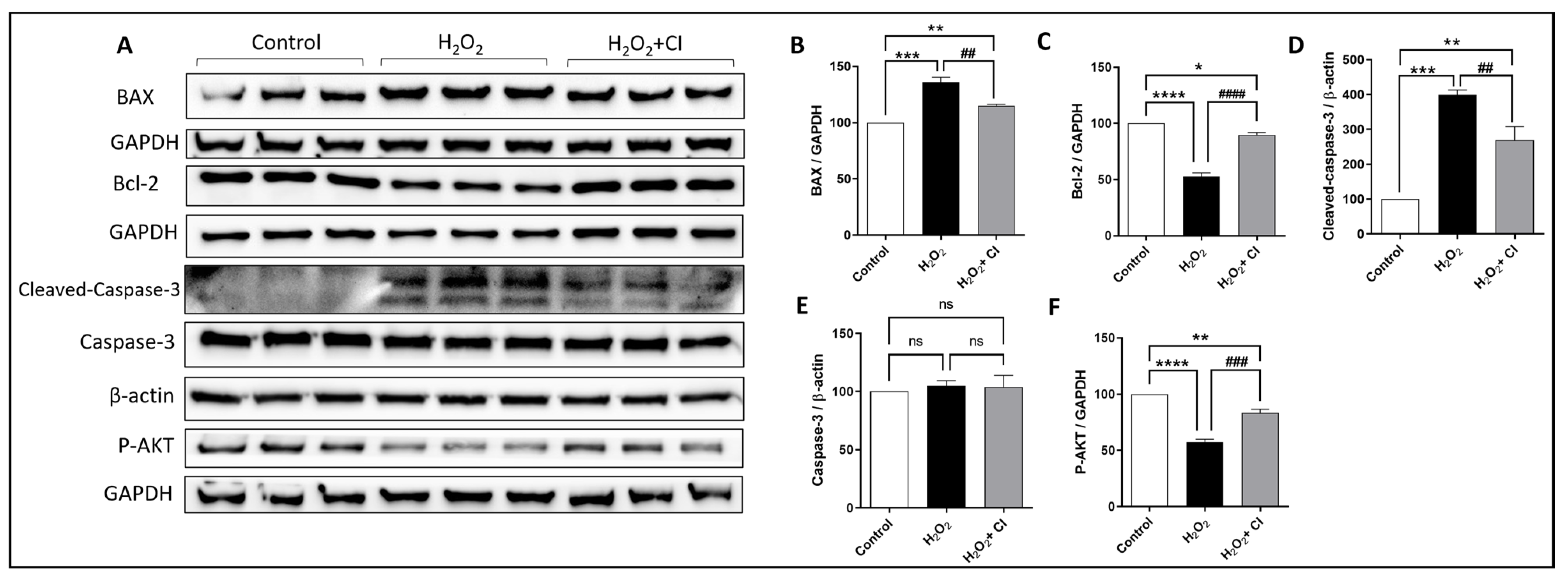
3.5. CI Treatment Enhances the Expression of Anti-Apoptotic and Survival Proteins after H2O2 Stimulation in SH-SY5Y Cells
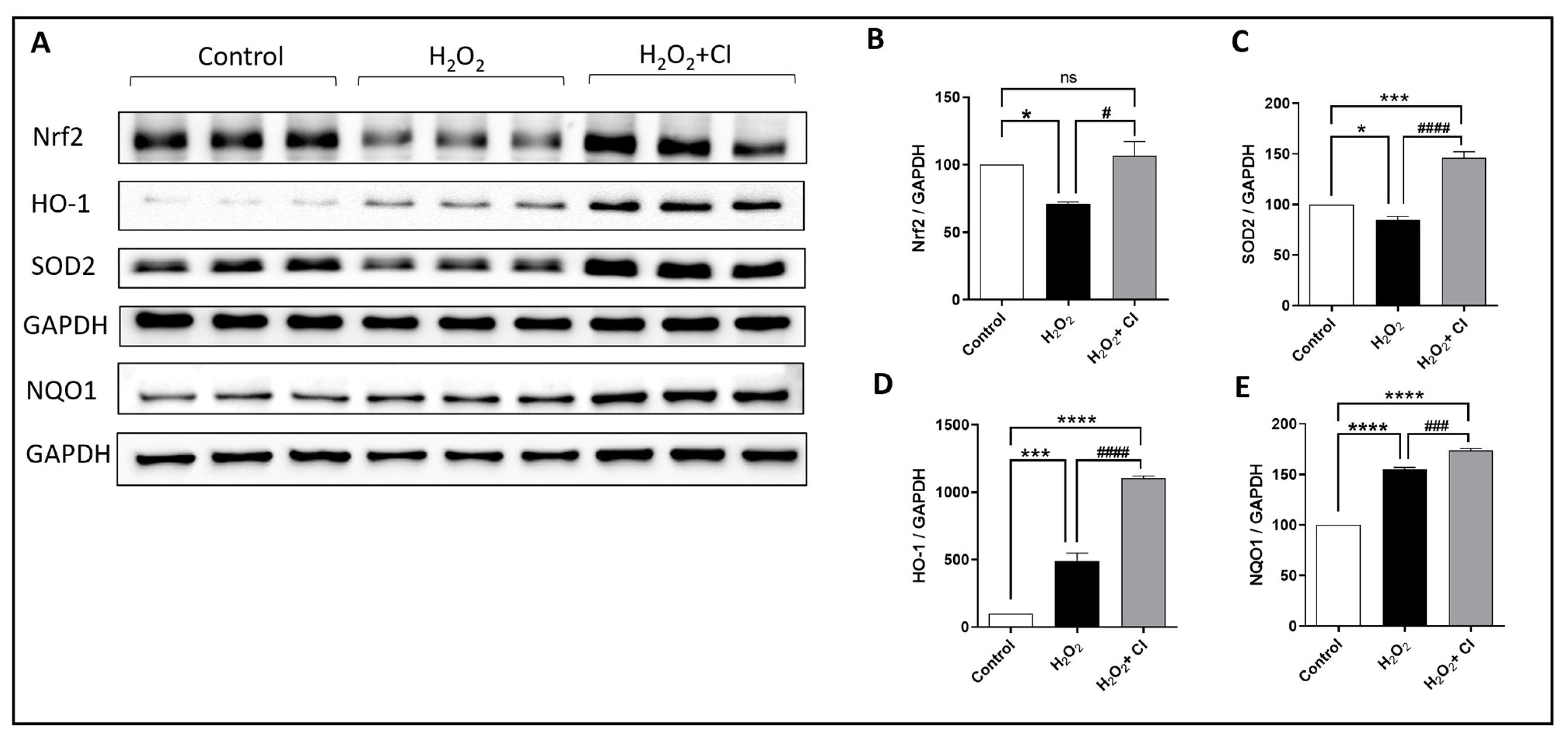
3.6. CI Treatment Induces the Expression of Nrf2 and Antioxidant Enzymes in SY-SY5Y Cells
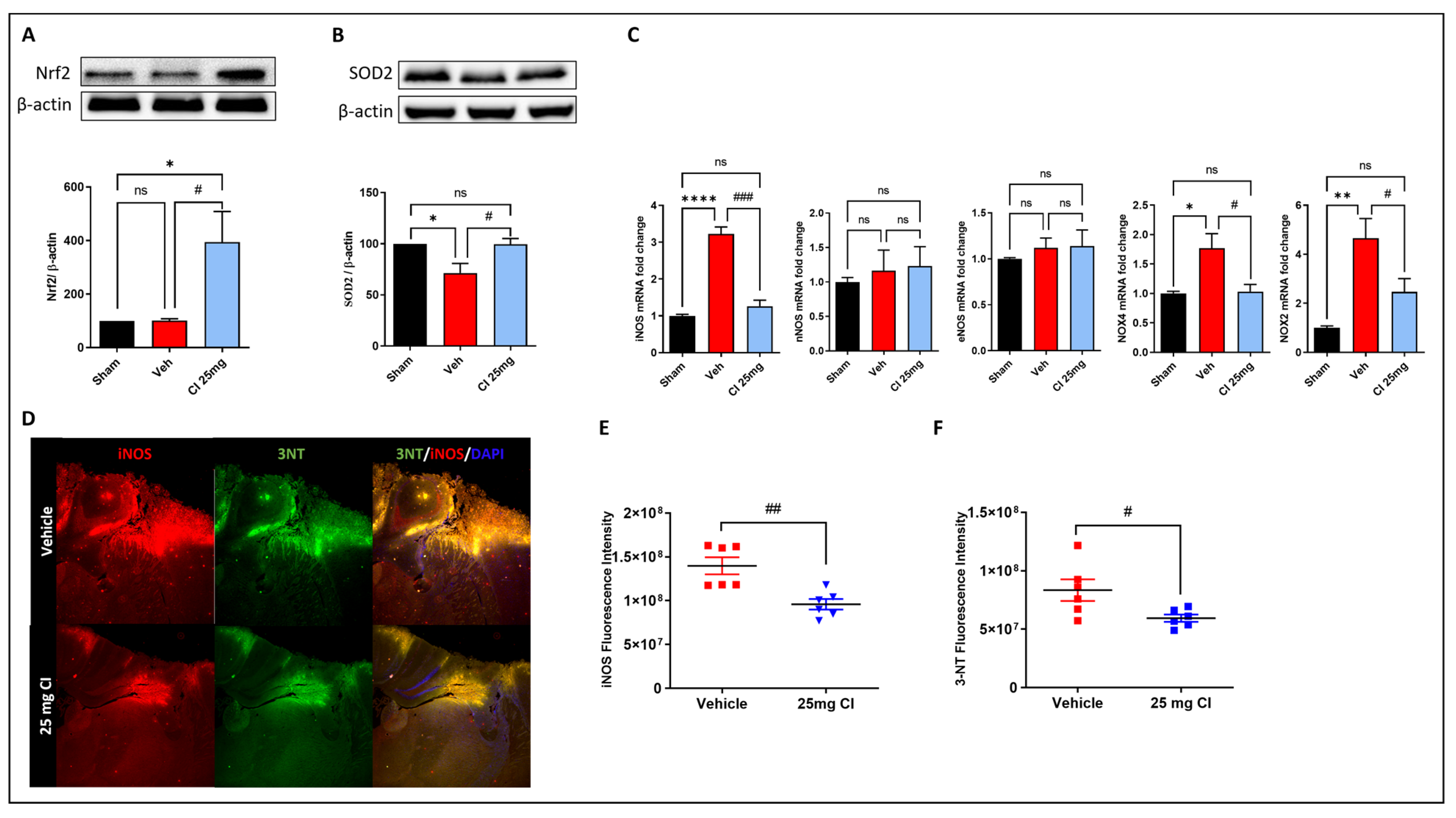
3.7. CI treatment Attenuates TBI-Induced Oxidative and Nitrosative Stress in Mice
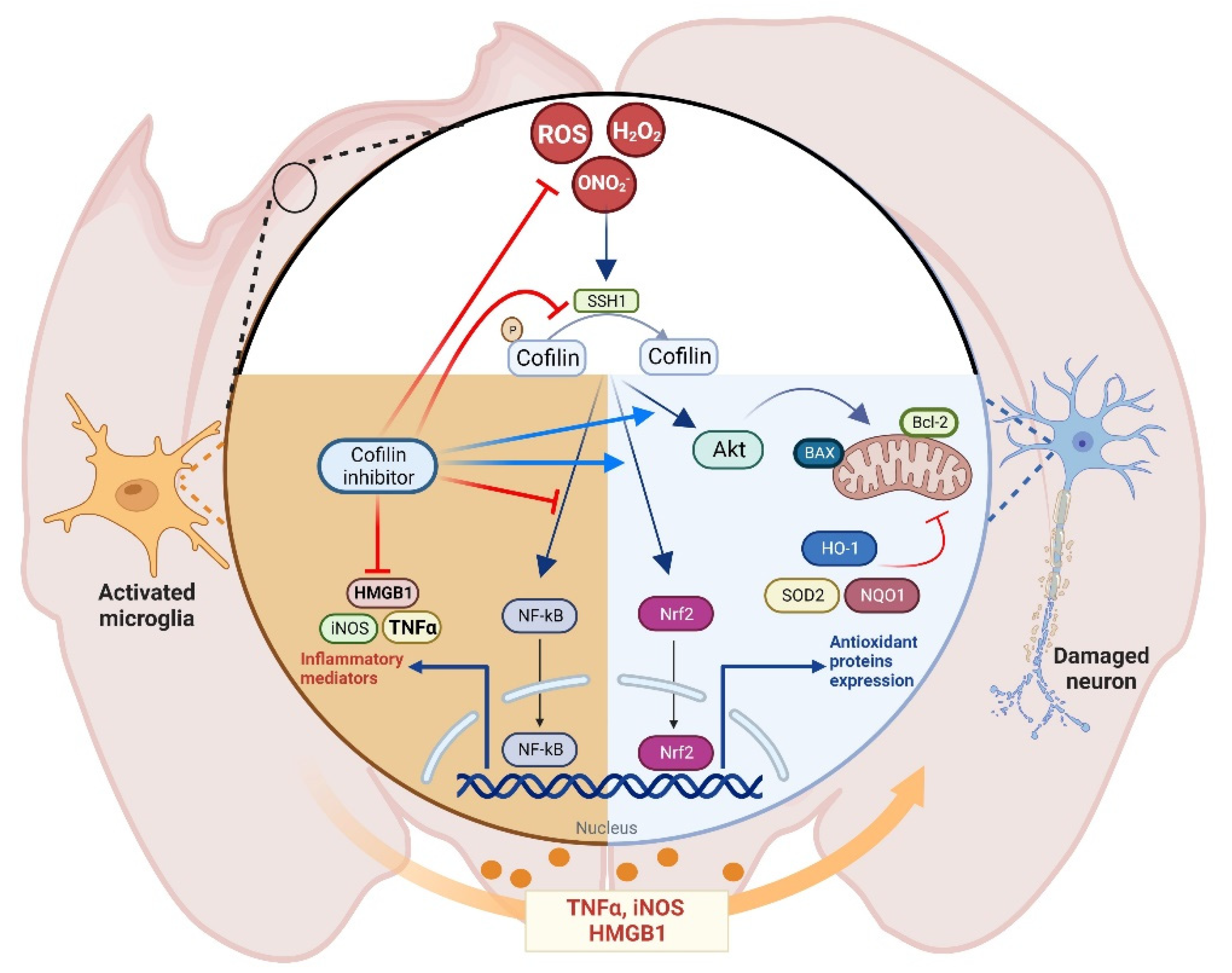
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Georges, A.; Das, J.M. Traumatic Brain Injury; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Phillips, S.; Woessner, D. Sports-related traumatic brain injury. Prim. Care Clin. Off. Pract. 2015, 42, 243–248. [Google Scholar] [CrossRef]
- Wojcik, B.E.; Stein, C.R.; Bagg, K.; Humphrey, R.J.; Orosco, J. Traumatic brain injury hospitalizations of US army soldiers deployed to Afghanistan and Iraq. Am. J. Prev. Med. 2010, 38, S108–S116. [Google Scholar] [CrossRef]
- Miller, G.F.; DePadilla, L.; Xu, L. Costs of nonfatal traumatic brain injury in the United States, 2016. Med. Care 2021, 59, 451–455. [Google Scholar] [CrossRef]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S. Traumatic brain injury: Oxidative stress and novel anti-oxidants such as mitoquinone and edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef]
- Eastman, C.L.; D’Ambrosio, R.; Ganesh, T. Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology 2020, 172, 107907. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef]
- A Ma, Z. The role of peroxidation of mitochondrial membrane phospholipids in pancreatic β-cell failure. Curr. Diabetes Rev. 2012, 8, 69–75. [Google Scholar] [CrossRef]
- Cheng, W.-Y.; Tong, H.; Miller, E.W.; Chang, C.J.; Remington, J.; Zucker, R.M.; Bromberg, P.A.; Samet, J.M.; Hofer, T.P. An integrated imaging approach to the study of oxidative stress generation by mitochondrial dysfunction in living cells. Environ. Health Perspect. 2010, 118, 902–908. [Google Scholar] [CrossRef]
- Suematsu, N.; Hosoda, M.; Fujimori, K. Protective effects of quercetin against hydrogen peroxide-induced apoptosis in human neuronal SH-SY5Y cells. Neurosci. Lett. 2011, 504, 223–227. [Google Scholar] [CrossRef]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef]
- Park, J.-H.; Choi, J.W.; Ju, E.J.; Pae, A.N.; Park, K.D. Antioxidant and anti-inflammatory activities of a natural compound, shizukahenriol, through Nrf2 activation. Molecules 2015, 20, 15989–16003. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef]
- Alhadidi, Q.; Shah, Z.A. Cofilin mediates LPS-induced microglial cell activation and associated neurotoxicity through activation of NF-κB and JAK–STAT pathway. Mol. Neurobiol. 2018, 55, 1676–1691. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2018, 25, 180–189. [Google Scholar] [CrossRef]
- Simpson, D.S.; Oliver, P.L. ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Bernstein, B.W.; Bamburg, J.R. ADF/cofilin: A functional node in cell biology. Trends Cell Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef]
- Alhadidi, Q.; Bin Sayeed, M.S.; Shah, Z.A. Cofilin as a promising therapeutic target for ischemic and hemorrhagic stroke. Transl. Stroke Res. 2016, 7, 33–41. [Google Scholar] [CrossRef]
- Posadas, I.; Pérez-Martínez, F.C.; Guerra, J.; Sánchez-Verdú, P.; Ceña, V. Cofilin activation mediates Bax translocation to mitochondria during excitotoxic neuronal death. J. Neurochem. 2012, 120, 515–527. [Google Scholar] [CrossRef]
- Bamburg, J.R.; Bernstein, B.W. Actin dynamics and cofilin-actin rods in Alzheimer disease. Cytoskeleton 2016, 73, 477–497. [Google Scholar] [CrossRef]
- Won, S.J.; Minnella, A.M.; Wu, L.; Eun, C.H.; Rome, E.; Herson, P.S.; Shaw, A.E.; Bamburg, J.R.; Swanson, R.A. Cofilin-actin rod formation in neuronal processes after brain ischemia. PLoS ONE 2018, 13, e0198709. [Google Scholar] [CrossRef]
- Sayeed, M.S.B.; Alhadidi, Q.; Shah, Z.A. Cofilin signaling in hemin-induced microglial activation and inflammation. J. Neuroimmunol. 2017, 313, 46–55. [Google Scholar] [CrossRef]
- Madineni, A.; Alhadidi, Q.; Shah, Z.A. Cofilin inhibition restores neuronal cell death in oxygen–glucose deprivation model of ischemia. Mol. Neurobiol. 2016, 53, 867–878. [Google Scholar] [CrossRef]
- Alaqel, S.I.; Dlamini, S.; Almarghalani, D.A.; Shettigar, A.; Alhadidi, Q.; Kodithuwakku, S.H.; Stary, C.; Tillekeratne, L.V.; Shah, Z.A. Synthesis and Development of a Novel First-in-Class Cofilin Inhibitor for Neuroinflammation in Hemorrhagic Brain Injury. ACS Chem. Neurosci. 2022, 13, 1014–1029. [Google Scholar] [CrossRef]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef]
- Lee, H.-S.; Kim, E.-N.; Jeong, G.-S. Lupenone protects neuroblastoma SH-SY5y cells against methamphetamine-induced apoptotic cell death via PI3K/Akt/mTOR signaling pathway. Int. J. Mol. Sci. 2020, 21, 1617. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Cruz-Haces, M.; Tang, J.; Acosta, G.; Fernandez, J.; Shi, R. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 20. [Google Scholar] [CrossRef]
- Shao, F.; Wang, X.; Wu, H.; Wu, Q.; Zhang, J. Microglia and Neuroinflammation: Crucial Pathological Mechanisms in Traumatic Brain Injury-Induced Neurodegeneration. Front. Aging Neurosci. 2022, 14, 825086. [Google Scholar] [CrossRef]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of traumatic brain injury: Time for a paradigm shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, N.; Yenari, M.A. Mechanisms and potential therapeutic applications of microglial activation after brain injury. CNS Neurosci. Ther. 2015, 21, 309–319. [Google Scholar] [CrossRef]
- Lapeña-Luzón, T.; Rodríguez, L.R.; Beltran-Beltran, V.; Benetó, N.; Pallardó, F.V.; Gonzalez-Cabo, P. Cofilin and Neurodegeneration: New Functions for an Old but Gold Protein. Brain Sci. 2021, 11, 954. [Google Scholar] [CrossRef]
- Kim, J.-S.; Huang, T.Y.; Bokoch, G.M. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol. Biol. Cell 2009, 20, 2650–2660. [Google Scholar] [CrossRef]
- Huang, T.Y.; Minamide, L.S.; Bamburg, J.R.; Bokoch, G.M. Chronophin mediates an ATP-sensing mechanism for cofilin dephosphorylation and neuronal cofilin-actin rod formation. Dev. Cell 2008, 15, 691–703. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Reichert, F.; Rotshenker, S. Galectin-3 (MAC-2) controls microglia phenotype whether amoeboid and phagocytic or branched and non-phagocytic by regulating the cytoskeleton. Front. Cell. Neurosci. 2019, 13, 90. [Google Scholar] [CrossRef]
- Hoffmann, L.; Waclawczyk, M.S.; Tang, S.; Hanschmann, E.-M.; Gellert, M.; Rust, M.B.; Culmsee, C. Cofilin1 oxidation links oxidative distress to mitochondrial demise and neuronal cell death. Cell Death Dis. 2021, 12, 953. [Google Scholar] [CrossRef]
- Ding, X.; Wang, D.; Li, L.; Ma, H. Dehydroepiandrosterone ameliorates H2O2-induced Leydig cells oxidation damage and apoptosis through inhibition of ROS production and activation of PI3K/Akt pathways. Int. J. Biochem. Cell Biol. 2016, 70, 126–139. [Google Scholar] [CrossRef]
- Klamt, F.; Zdanov, S.; Levine, R.L.; Pariser, A.; Zhang, Y.; Zhang, B.; Yu, L.-R.; Veenstra, T.D.; Shacter, E. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat. Cell Biol. 2009, 11, 1241–1246. [Google Scholar] [CrossRef]
- Woo, J.; Zhao, X.; Khan, H.; Penn, C.; Wang, X.; Joly-Amado, A.; Weeber, E.; Morgan, D.; Kang, D. Slingshot-Cofilin activation mediates mitochondrial and synaptic dysfunction via Aβ ligation to β1-integrin conformers. Cell Death Differ. 2015, 22, 921–934. [Google Scholar] [CrossRef]
- Liu, C.-M.; Ma, J.-Q.; Sun, Y.-Z. Puerarin protects rat kidney from lead-induced apoptosis by modulating the PI3K/Akt/eNOS pathway. Toxicol. Appl. Pharmacol. 2012, 258, 330–342. [Google Scholar] [CrossRef]
- Heo, S.R.; Han, A.M.; Kwon, Y.K.; Joung, I. p62 protects SH-SY5Y neuroblastoma cells against H2O2-induced injury through the PDK1/Akt pathway. Neurosci. Lett. 2009, 450, 45–50. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Huang, B.; Liu, J.; Fu, S.; Zhang, Y.; Li, Y.; He, D.; Ran, X.; Yan, X.; Du, J.; Meng, T. α-Cyperone attenuates H2O2-induced oxidative stress and apoptosis in SH-SY5Y cells via activation of Nrf2. Front. Pharmacol. 2020, 11, 281. [Google Scholar] [CrossRef]
- Liu, J.; Wu, P.; Xu, Z.; Zhang, J.; Liu, J.; Yang, Z. Ginkgolide B inhibits hydrogen peroxide-induced apoptosis and attenuates cytotoxicity via activating the PI3K/Akt/mTOR signaling pathway in H9c2 cells. Mol. Med. Rep. 2020, 22, 310–316. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Chandran, R.; Kim, T.; Mehta, S.L.; Udho, E.; Chanana, V.; Cengiz, P.; Kim, H.; Kim, C.; Vemuganti, R. A combination antioxidant therapy to inhibit NOX2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1818–1827. [Google Scholar] [CrossRef]
- Wang, J.; Ma, M.W.; Dhandapani, K.M.; Brann, D.W. Regulatory role of NADPH oxidase 2 in the polarization dynamics and neurotoxicity of microglia/macrophages after traumatic brain injury. Free Radic. Biol. Med. 2017, 113, 119–131. [Google Scholar] [CrossRef]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.-M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef]
- Alhadidi, Q.; Nash, K.M.; Alaqel, S.; Sayeed, M.S.B.; Shah, Z.A. Cofilin knockdown attenuates hemorrhagic brain injury-induced oxidative stress and microglial activation in mice. Neuroscience 2018, 383, 33–45. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | CTGGTGCGAAGTGTGCAAG | TGAGATTAGCGTGGCCCGAA |
| iNOS | CCAAATCCAACGTTCTCCGT | CCAAATCCAACGTTCTCCGT |
| eNOS | CAGATGCCCAACCCAAACCT | ACAGAGAGGTGTCTGGGACT |
| nNOS | CCTTCACAGGGGATGGAACC | AGATCGACAGCTTTGGTGGG |
| NOX2 | CCCTCCCTGTCTAGGTAATGCATGG | GCATTTGCCTTCGGTGATGTGCT |
| NOX4 | CACCAAATGTTGGGCGATTGT | GATGAGGCTGCAGTTGAGGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahader, G.A.; James, A.W.; Almarghalani, D.A.; Shah, Z.A. Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation. Biology 2023, 12, 630. https://doi.org/10.3390/biology12040630
Bahader GA, James AW, Almarghalani DA, Shah ZA. Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation. Biology. 2023; 12(4):630. https://doi.org/10.3390/biology12040630
Chicago/Turabian StyleBahader, Ghaith A., Antonisamy William James, Daniyah A. Almarghalani, and Zahoor A. Shah. 2023. "Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation" Biology 12, no. 4: 630. https://doi.org/10.3390/biology12040630
APA StyleBahader, G. A., James, A. W., Almarghalani, D. A., & Shah, Z. A. (2023). Cofilin Inhibitor Protects against Traumatic Brain Injury-Induced Oxidative Stress and Neuroinflammation. Biology, 12(4), 630. https://doi.org/10.3390/biology12040630