
Utilization of Computer Classification Methods for Exposure Prediction and Gene Selection in Daphnia magna Toxicogenomics

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Machine Learning for Gene Expression
2.2. Gene Ranking
2.3. Data Collection
3. Results and Discussion
3.1. Classification and Prediction Results
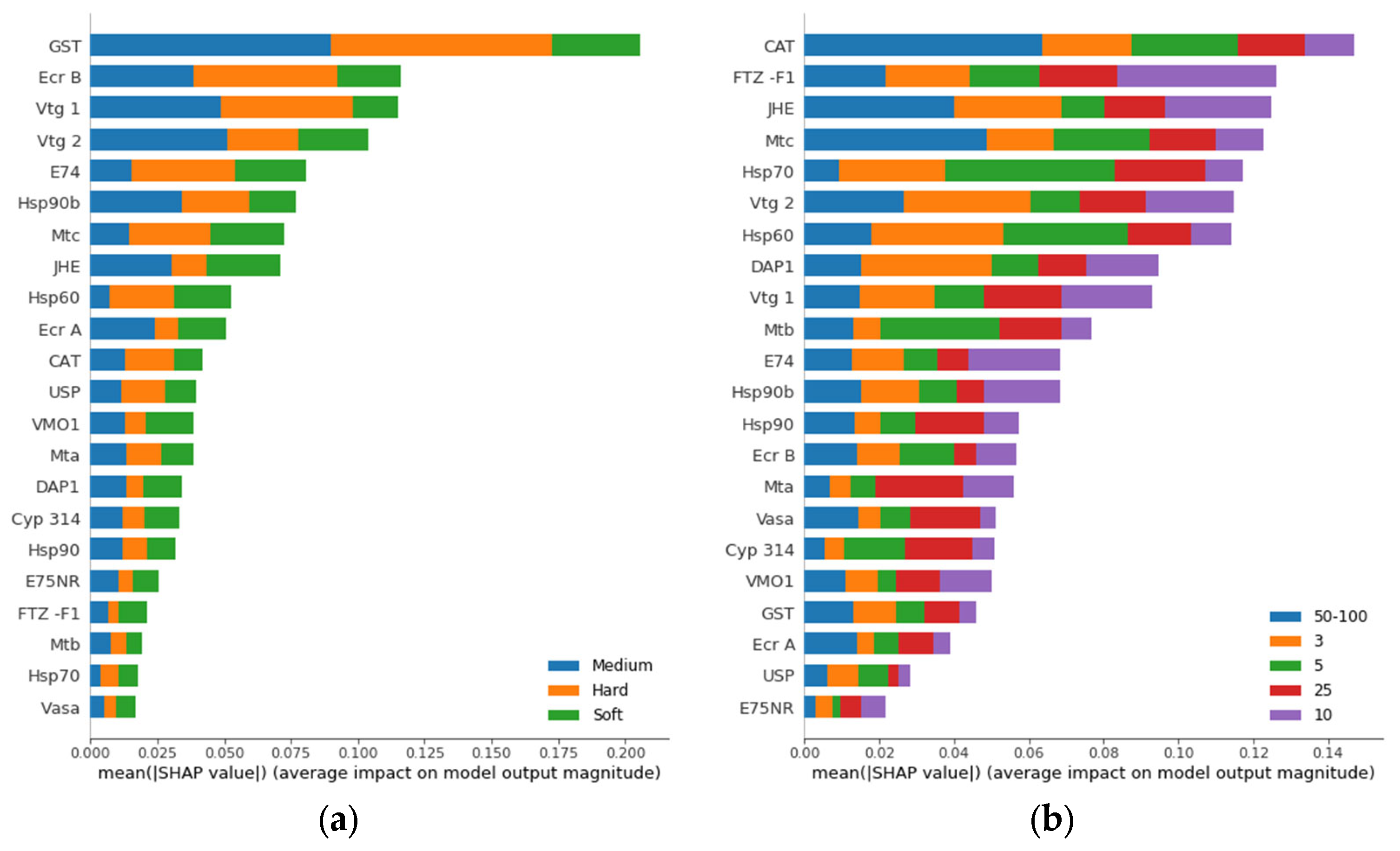
3.2. Gene Ranking
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Scientific Opinion on Dietary Reference Values for Zinc. EFSA J. 2014, 12, 3844. [Google Scholar]
- Fosmire, G.J. Zinc toxicity. Am. J. Clin. Nutr. 1990, 51, 225–227. [Google Scholar] [CrossRef] [PubMed]
- OECD. Test No. 211: Daphnia Magna Reproduction Test; OECD: Paris, France, 2012. [Google Scholar]
- VanGuilder, H.D.; Vrana, K.E.; Freeman, W.M. Twenty-five years of quantitative PCR for gene expression analysis. Biotechniques 2008, 44, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Ma, C.; Ma, J.; Huangfu, X.; He, Q. Machine learning in natural and engineered water systems. Water Res. 2021, 205, 117666. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, M.; Yang, J.Y.; Yang, M.Q.; Deng, Y. A comparative study of different machine learning methods on microarray gene expression data. BMC Genom. 2008, 9, S13. [Google Scholar] [CrossRef]
- Molla, M.; Waddell, M.; Page, D.; Shavlik, J. Using Machine Learning to Design and Interpret Gene-Expression Microarrays. AI Mag. 2004, 25, 23. [Google Scholar]
- Kuo, W.P.; Kim, E.Y.; Trimarchi, J.; Jenssen, T.K.; Vinterbo, S.A.; Ohno-Machado, L. A primer on gene expression and microarrays for machine learning researchers. J. Biomed. Inform. 2004, 37, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Gilbert, D. Ensemble machine learning on gene expression data for cancer classification. Appl. Bioinform. 2003, 2 (Suppl. S3), S75–S83. [Google Scholar]
- Jin, X.; Xu, A.; Bie, R.; Guo, P. Machine Learning Techniques and Chi-Square Feature Selection for Cancer Classification Using SAGE Gene Expression Profiles. In Proceedings of the Data Mining for Biomedical Applications: PAKDD 2006 Workshop, BioDM 2006, Singapore, 9 April 2006. [Google Scholar]
- Spencer, W.C.; Zeller, G.; Watson, J.D.; Henz, S.R.; Watkins, K.L.; McWhirter, R.D.; Miller, D.M. A spatial and temporal map of C. elegans gene expression. Genome Res. 2011, 21, 325–341. [Google Scholar] [CrossRef]
- Gunay, M.; Goceri, E.; Balasubramaniyan, R. Machine learning for optimum CT-prediction for qPCR. In Proceedings of the 15th IEEE International Conference on Machine Learning and Applications, ICMLA 2016, Anaheim, CA, USA, 18–20 December 2016. [Google Scholar]
- Yuan, F.; Lu, L.; Zou, Q. Analysis of gene expression profiles of lung cancer subtypes with machine learning algorithms. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165822. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2016, 18, 851–869. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.K.K.; Delong, A.; Alipanahi, B.; Frey, B.J. Machine Learning in Genomic Medicine: A Review of Computational Problems and Data Sets. Proc. IEEE 2016, 104, 176–197. [Google Scholar] [CrossRef]
- Mamoshina, P.; Vieira, A.; Putin, E.; Zhavoronkov, A. Applications of Deep Learning in Biomedicine. Mol. Pharm. 2016, 13, 1445–1454. [Google Scholar] [CrossRef]
- Abraham, R.S.A. Comparison of Supervised and Unsupervised Learning Algorithms for Pattern Classification. Int. J. Adv. Res. Artif. Intell. 2013, 2, 34–38. [Google Scholar]
- Ramaswamy, S.; Tamayo, P.; Rifkin, R.; Mukherjee, S.; Yeang, C.-H.; Angelo, M.; Ladd, C.; Reich, M.; Latulippe, E.; Mesirov, J.P.; et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. USA 2001, 98, 15149–15154. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef]
- Nikkilä, J.; Törönen, P.; Kaski, S.; Venna, J.; Castrén, E.; Wong, G. Analysis and visualization of gene expression data using Self-Organizing Maps. Neural Networks 2002, 15, 953–966. [Google Scholar] [CrossRef]
- De Jongh, R.P.; van Dijk, A.D.; Julsing, M.K.; Schaap, P.J.; de Ridder, D. Designing Eukaryotic Gene Expression Regulation Using Machine Learning. Trends Biotechnol. 2020, 38, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Berrar, D. Cross-Validation; Tokyo Institute of Technology: Tokyo, Japan, 2019; pp. 542–545. [Google Scholar]
- Jain, A.K.; Duin, R.P.W.; Mao, J. Statistical pattern recognition: A review. IEEE Trans. Pattern Anal. Mach. Intell. 2000, 22, 4–37. [Google Scholar] [CrossRef]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 14–18 August 2016; Association for Computing Machinery: New York, NY, USA, 2016; pp. 1135–1144. [Google Scholar]
- Bach, S.; Binder, A.; Montavon, G.; Klauschen, F.; Müller, K.-R.; Samek, W. On Pixel-Wise Explanations for Non-Linear Classifier Decisions by Layer-Wise Relevance Propagation. PLoS ONE 2015, 10, e0130140. [Google Scholar] [CrossRef]
- Shrikumar, A.; Greenside, P.; Kundaje, A. Learning Important Features Through Propagating Activation Differences. In Proceedings of the 34th International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; PMLR: Proceedings of Machine Learning Research; Precup, D., Teh, Y.W., Eds.; JMLR: Cambridge, MA, USA, 2017; Volume 70, pp. 3145–3153. [Google Scholar]
- Lundberg, S.M.; Lee, S.I. A unified approach to interpreting model predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; Curran Associates Inc.: Long Beach, CA, USA, 2017; pp. 4768–4777. [Google Scholar]
- Glaab, E.; Bacardit, J.; Garibaldi, J.M.; Krasnogor, N. Using Rule-Based Machine Learning for Candidate Disease Gene Prioritization and Sample Classification of Cancer Gene Expression Data. PLoS ONE 2012, 7, e39932. [Google Scholar] [CrossRef] [PubMed]
- Lotfollahi, M.; Rybakov, S.; Hrovatin, K.; Hediyeh-Zadeh, S.; Talavera-López, C.; Misharin, A.V.; Theis, F.J. Biologically informed deep learning to query gene programs in single-cell atlases. Nat. Cell Biol. 2023, 25, 337–350. [Google Scholar] [CrossRef]
- Seninge, L.; Anastopoulos, I.; Ding, H.; Stuart, J. VEGA is an interpretable generative model for inferring biological network activity in single-cell transcriptomics. Nat. Commun. 2021, 12, 5684. [Google Scholar] [CrossRef]
- Janizek, J.D.; Spiro, A.; Celik, S.; Blue, B.W.; Russell, J.C.; Lee, T.-I.; Kaeberlin, M.; Lee, S.-I. PAUSE: Principled feature attribution for unsupervised gene expression analysis. Genome Biol. 2022, 24, 81. [Google Scholar] [CrossRef]
- Weber, G.; Vinterbo, S.; Ohno-Machado, L. Multivariate selection of genetic markers in diagnostic classification. Artif. Intell. Med. 2004, 31, 155–167. [Google Scholar] [CrossRef]
- Moretti, S.; Patrone, F.; Bonassi, S. The class of microarray games and the relevance index for genes. TOP 2007, 15, 256–280. [Google Scholar] [CrossRef]
- Shapley, L.S. A Value for N-Person Games; RAND Corporation: Santa Monica, CA, USA, 1952. [Google Scholar]
- Lipovetsky, S.; Conklin, M. Analysis of regression in game theory approach. Appl. Stoch. Model. Bus. Ind. 2001, 17, 319–330. [Google Scholar] [CrossRef]
- Moretti, S.; Van Leeuwen, D.; Gmuender, H.; Bonassi, S.; Van Delft, J.; Kleinjans, J.; Patrone, F.; Merlo, D.F. Combining Shapley value and statistics to the analysis of gene expression data in children exposed to air pollution. BMC Bioinform. 2008, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.W.; Moretti, S.; Paskov, K.M.; Stockham, N.T.; Varma, M.; Chrisman, B.S.; Washington, P.Y.; Jung, J.-Y.; Wall, D.P. Game theoretic centrality: A novel approach to prioritize disease candidate genes by combining biological networks with the Shapley value. BMC Bioinform. 2020, 21, 356. [Google Scholar] [CrossRef] [PubMed]
- Fragnelli, V.; Moretti, S. A game theoretical approach to the classification problem in gene expression data analysis. Comput. Math. Appl. 2008, 55, 950–959. [Google Scholar] [CrossRef]
- Fayyad, U.M.I.; Keki, B. Multi-Interval Discretization of Continuous-Valued Attributes for Classification Learning. In Proceedings of the 13th International Joint Conference on Artificial Intelligence, Chambéry, France, 28 August–3 September 1993; Morgan Kaufmann Publishers Inc.: San Francisco, CA, USA, 1993. [Google Scholar]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Lu, X.; Peng, X.; Deng, Y.; Feng, B.; Liu, P.; Liao, B. A Novel Feature Selection Method Based on Correlation-Based Feature Selection in Cancer Recognition. J. Comput. Theor. Nanosci. 2014, 11, 427–433. [Google Scholar] [CrossRef]
- Saeys, Y.; Inza, I.; Larrañaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef]
- OECD. Test No. 202: Daphnia sp. Acute Immobilisation Test; OECD: Paris, France, 2004. [Google Scholar]
- Paylar, B.; Asnake, S.; Sjöberg, V.; Ragnvaldsson, D.; Jass, J.; Olsson, P. Influence of water hardness on zinc toxicity in Daphnia magna. J. Appl. Toxicol. 2022, 42, 1510–1523. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, J.; Grek, C.; Ye, Z.-W.; Manevich, Y.; Tew, K.D.; Townsend, D.M. Pleiotropic Functions of Glutathione S-Transferase P. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2014; Volume 122, pp. 143–175. [Google Scholar] [CrossRef]
- Copatti, C.E.; Baldisserotto, B.; Souza, C.D.F.; Monserrat, J.M.; Garcia, L. Water pH and hardness alter ATPases and oxidative stress in the gills and kidney of pacu (Piaractus mesopotamicus). Neotropical Ichthyol. 2019, 17, e190032. [Google Scholar] [CrossRef]
- Saliu, J.K.; Bawa-Allah, K.A. Toxicological Effects of Lead and Zinc on the Antioxidant Enzyme Activities of Post Juvenile Clarias gariepinus. Resour. Environ. 2012, 2, 21–26. [Google Scholar] [CrossRef]
- Chupani, L.; Sjöberg, V.; Jass, J.; Olsson, P.-E. Water Hardness Alters the Gene Expression Response and Copper Toxicity in Daphnia magna. Fishes 2022, 7, 248. [Google Scholar] [CrossRef]
- Matozzo, V.; Gagné, F.; Marin, M.G.; Ricciardi, F.; Blaise, C. Vitellogenin as a biomarker of exposure to estrogenic compounds in aquatic invertebrates: A review. Environ. Int. 2008, 34, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.A.; Maki, A.W. Effects of water hardness and diet on productivity of Daphnia magna Straus. in laboratory culture. Hydrobiologia 1981, 85, 175–179. [Google Scholar] [CrossRef]
- Paulauskis, J.D.; Winner, R.W. Effects of water hardness and humic acid on zinc toxicity to Daphnia magna Straus. Aquat. Toxicol. 1988, 12, 273–290. [Google Scholar] [CrossRef]
- Cowgill, U.M.; Milazzo, D.P. The sensitivity of two cladocerans to water quality variables: Alkalinity. Arch. Environ. Contam. Toxicol. 1991, 21, 224–232. [Google Scholar] [CrossRef]
- Cowgill, U.M.; Milazzo, D.P. Demographic effects of salinity, water hardness and carbonate alkalinity on Daphnia magna and Ceriodaphnia dubia. Arch. Für Hydrobiol. 1991, 122, 33–56. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifier | Water Hardness | Zn Level | Combined |
|---|---|---|---|
| Support Vector Machine (SVM) | 0.85 ± 0.12 | 0.82 ± 0.12 | 0.69 ± 0.06 |
| K-nearest neighbor (KNN) | 0.84 ± 0.08 | 0.78 ± 0.08 | 0.74 ± 0.08 |
| Random Forest (RF) | 0.86 ± 0.12 | 0.87 ± 0.08 | 0.82 ± 0.04 |
| Neural Network (NN) | 0.85 ± 0.12 | 0.91 ± 0.09 | 0.81 ± 0.10 |
| Gaussian Process (GP) | 0.81 ± 0.13 | 0.25 ± 0.03 | 0.07 ± 0.03 |
| AdaBoost | 0.73 ± 0.12 | 0.37 ± 0.12 | 0.12 ± 0.00 |
| Naive Bayes (NB) | 0.79 ± 0.10 | 0.60 ± 0.10 | 0.74 ± 0.08 |
| Quadratic Discriminant Analysis (QDA) | 0.88 ± 0.10 | 0.25 ± 0.03 | 0.17 ± 0.07 |
| Predicted | ||||
|---|---|---|---|---|
| Soft | Medium | Hard | ||
| Soft | 24 | 5 | 3 | |
| True | Medium | 4 | 42 | 2 |
| Hard | 3 | 2 | 43 | |
| Predicted | ||||||
|---|---|---|---|---|---|---|
| 3 | 5 | 10 | 25 | >50 | ||
| 3 | 22 | 0 | 1 | 0 | 1 | |
| 5 | 1 | 22 | 0 | 1 | 0 | |
| True | 10 | 0 | 1 | 21 | 1 | 1 |
| 25 | 2 | 0 | 0 | 22 | 0 | |
| >50 | 0 | 2 | 0 | 1 | 29 | |
| Soft | Medium | Hard | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 5 | 10 | 25 | 3 | 5 | 10 | 25 | 50 | 100 | 3 | 5 | 10 | 25 | 50 | 100 | ||
| Soft | 3 | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| 5 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 10 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 25 | 0 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Medium | 3 | 4 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 10 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Hard | 3 | 3 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | |
| 10 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 1 | 0 | 0 | |
| 25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 7 | 0 | 0 | |
| 50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 7 | 0 | |
| 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 7 | |
| χ2 | RF Ranking | Shapley Values |
|---|---|---|
| gst | gst | gst |
| vtg1 | ecrb | ecrb |
| vtg2 | vtg1 | vtg1 |
| hsp60 | vtg2 | vtg2 |
| jhe | mtc | e74 |
| hsp90b | e74 | hsp90b |
| cat | hsp90b | mtc |
| e74 | jhe | jhe |
| hsp90 | cat | hsp60 |
| vasa | mta | ecra |
| mtc | hsp60 | cat |
| ecrb | cyp314 | usp |
| e75nr | usp | vmo1 |
| mta | ecra | mta |
| vmo1 | dap1 | dap1 |
| dap1 | vmo1 | cyp314 |
| cyp314 | hsp90 | hsp90 |
| ecra | mtb | e75nr |
| usp | e75nr | ftz-f1 |
| ftz-f1 | ftz-f1 | mtb |
| hsp70 | hsp70 | hsp70 |
| mtb | vasa | vasa |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paylar, B.; Längkvist, M.; Jass, J.; Olsson, P.-E. Utilization of Computer Classification Methods for Exposure Prediction and Gene Selection in Daphnia magna Toxicogenomics. Biology 2023, 12, 692. https://doi.org/10.3390/biology12050692
Paylar B, Längkvist M, Jass J, Olsson P-E. Utilization of Computer Classification Methods for Exposure Prediction and Gene Selection in Daphnia magna Toxicogenomics. Biology. 2023; 12(5):692. https://doi.org/10.3390/biology12050692
Chicago/Turabian StylePaylar, Berkay, Martin Längkvist, Jana Jass, and Per-Erik Olsson. 2023. "Utilization of Computer Classification Methods for Exposure Prediction and Gene Selection in Daphnia magna Toxicogenomics" Biology 12, no. 5: 692. https://doi.org/10.3390/biology12050692
APA StylePaylar, B., Längkvist, M., Jass, J., & Olsson, P.-E. (2023). Utilization of Computer Classification Methods for Exposure Prediction and Gene Selection in Daphnia magna Toxicogenomics. Biology, 12(5), 692. https://doi.org/10.3390/biology12050692