

Bisphenol-A Neurotoxic Effects on Basal Forebrain Cholinergic Neurons In Vitro and In Vivo

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Treatment Protocol
2.2. Basal Forebrain Preparation for Histopathological Studies
2.3. Immunohistochemistry Studies
2.4. Histopathological Study
2.5. Culture Procedure
2.6. Protein Determination
2.7. GSK-3β Activity Analysis
2.8. Gene Expression Analysis
2.9. Glutamate Content
2.10. Glutaminase Function
2.11. Hdac2 and Ptp1b Silencing
2.12. Analysis of Cell Viability
2.13. Caspase Activation Determination
2.14. Statistical Analysis
3. Results
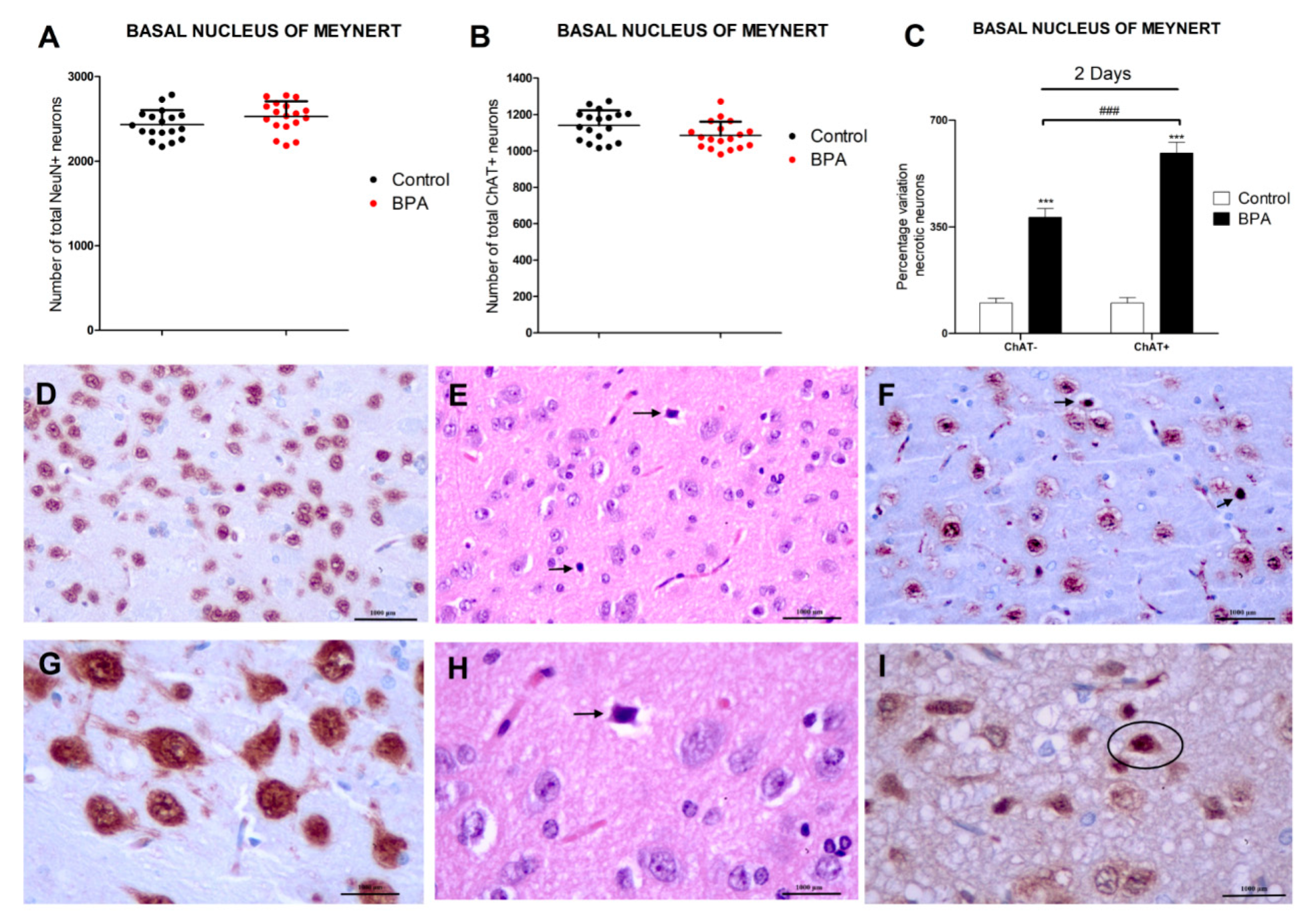
3.1. BPA Acute Exposure Effects on Basal Forebrain Neurons
3.2. Analysis of Glutamatergic Neurotransmission (Glutamate Content, Glutaminase Activity, and VGLUT2 Protein Content and Gene Expression)
3.3. Analysis of Synaptic Proteins (PSD95, SPN, SYP, and NMDAR1 Protein Content and Gene Expression)
3.4. Analysis of WNT/β-Catenin Pathway (GSK3β Activity, and pGSK3β (Ser 9), Cyclin D1, c-Myc and β-Catenin Protein Concentration and Gene Expression)
3.5. Gene Knockdown Evaluation
3.6. Analysis of the Effect of BPA on SN56 Cell Viability and Caspases 3/7 Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eilam-Stock, T.; Serrano, P.; Frankfurt, M.; Luine, V. Bisphenol-A impairs memory and reduces dendritic spine density in adult male rats. Behav. Neurosci. 2012, 126, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, X.; Zhang, Q.; Zhang, G.; Lu, Y.; Ruan, Q.; Dong, F.; Yang, Y. Sex-specific effects of bisphenol-A on memory and synaptic structural modification in hippocampus of adult mice. Horm. Behav. 2013, 63, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo-Solleiro, D.; Castillo-Flores, L.Y.; Solleiro-Villavicencio, H. Impact of BPA on behavior, neurodevelopment and neurodegeneration. Front. Biosci. Landmark Ed. 2021, 26, 363–400. [Google Scholar] [CrossRef] [PubMed]
- Moyano, P.; Flores, A.; García, J.; García, J.M.; Anadon, M.J.; Frejo, M.T.; Sola, E.; Pelayo, A.; Del Pino, J. Bisphenol A single and repeated treatment increases HDAC2, leading to cholinergic neurotransmission dysfunction and SN56 cholinergic apoptotic cell death through AChE variants overexpression and NGF/TrkA/P75NTR signaling disruption. Food. Chem. Toxicol. 2021, 157, 112614. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.; Moyano, P.; García, J.M.; García, J.; Anadon, M.J.; Frejo, M.T.; Sola, E.; Naval, M.V.; De la Cabeza, M.; Del Pino, J. Single and repeated bisphenol A treatment induces ROS, Aβ and hyperphosphorylated-tau accumulation, and insulin pathways disruption, through HDAC2 and PTP1B overexpression, resulting in SN56 cholinergic cell apoptosis. Food. Chem. Toxicol. 2022, 170, 113500. [Google Scholar] [CrossRef]
- Villano, I.; Messina, A.; Valenzano, A.; Moscatelli, F.; Esposito, T.; Monda, V.; Esposito, M.; Precenzano, F.; Carotenuto, M.; Viggiano, A.; et al. Basal Forebrain Cholinergic System and Orexin Neurons: Effects on Attention. Front Behav. Neurosci. 2017, 11, 10. [Google Scholar] [CrossRef]
- Grothe, M.J.; Heinsen, H.; Amaro, E., Jr.; Grinberg, L.T.; Teipel, S.J. Cognitive Correlates of Basal Forebrain Atrophy and Associated Cortical Hypometabolism in Mild Cognitive Impairment. Cereb. Cortex. 2016, 26, 2411–2426. [Google Scholar] [CrossRef] [PubMed]
- Gelfo, F.; Petrosini, L.; Graziano, A.; De Bartolo, P.; Burello, L.; Vitale, E.; Polverino, A.; Iuliano, A.; Sorrentino, G.; Mandolesi, L. Cortical metabolic deficits in a rat model of cholinergic basal forebrain degeneration. Neurochem. Res. 2013, 38, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Lin, L.; Mattsson, A.; Georgievska, B.; Isacson, O. Selective antibody-induced cholinergic cell and synapse loss produce sustained hippocampal and cortical hypometabolism with correlated cognitive deficits. Exp. Neurol. 2021, 170, 36–47. [Google Scholar] [CrossRef]
- Bi, N.; Gu, X.; Fan, A.; Li, D.; Wang, M.; Zhou, R.; Sun, Q.C.; Wang, H.L. Bisphenol-A exposure leads to neurotoxicity through upregulating the expression of histone deacetylase 2 in vivo and in vitro. Toxicology 2022, 465, 153052. [Google Scholar] [CrossRef]
- Wang, C.; Niu, R.; Zhu, Y.; Han, H.; Luo, G.; Zhou, B.; Wang, J. Changes in memory and synaptic plasticity induced in male rats after maternal exposure to bisphenol A. Toxicology 2014, 322, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Moyano, P.; Frejo, M.T.; Anadon, M.J.; García, J.M.; Díaz, M.J.; Lobo, M.; Sola, E.; García, J.; Del Pino, J. SN56 neuronal cell death after 24 h and 14 days chlorpyrifos exposure through glutamate transmission dysfunction, increase of GSK-3β enzyme, β-amyloid and tau protein levels. Toxicology 2018, 402–403, 17–27. [Google Scholar] [CrossRef]
- Khadrawy, Y.A.; Noor, N.A.; Mourad, I.M.; Ezz, H.S. Neurochemical impact of bisphenol A in the hippocampus and cortex of adult male albino rats. Toxicol. Ind. Health. 2016, 32, 1711–1719. [Google Scholar]
- Jardim, N.S.; Sartori, G.; Sari, M.H.M.; Müller, S.G.; Nogueira, C.W. Bisphenol A impairs the memory function and glutamatergic homeostasis in a sex-dependent manner in mice: Beneficial effects of diphenyl diselenide. Toxicol. Appl Pharmacol. 2017, 329, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Li, T.; Gong, H.; Chen, Z.; Jin, Y.; Xu, G.; Wang, M. Bisphenol A Impairs Synaptic Plasticity by Both Pre- and Postsynaptic Mechanisms. Adv. Sci. 2017, 4, 1600493. [Google Scholar] [CrossRef]
- Tiwari, S.K.; Agarwal, S.; Seth, B.; Yadav, A.; Ray, R.S.; Mishra, V.N.; Chaturvedi, R.K. Inhibitory Effects of Bisphenol-A on Neural Stem Cells Proliferation and Differentiation in the Rat Brain Are Dependent on Wnt/β-Catenin Pathway. Mol. Neurobiol. 2015, 52, 1735–1757. [Google Scholar] [CrossRef]
- Narvaes, R.F.; Furini, C.R.G. Role of Wnt signaling in synaptic plasticity and memory. Neurobiol. Learn. Mem. 2022, 187, 107558. [Google Scholar] [CrossRef]
- Scali, C.; Caraci, F.; Gianfriddo, M.; Diodato, E.; Roncarati, R.; Pollio, G.; Gaviraghi, G.; Copani, A.; Nicoletti, F.; Terstappen, G.C.; et al. Inhibition of Wnt signaling, modulation of Tau phosphorylation and induction of neuronal cell death by DKK1. Neurobiol. Dis. 2006, 24, 254–265. [Google Scholar] [CrossRef]
- Wang, S.E.; Ko, S.Y.; Kim, Y.S.; Jo, S.; Lee, S.H.; Jung, S.J.; Son, H. Capsaicin upregulates HDAC2 via TRPV1 and impairs neuronal maturation in mice. Exp. Mol. Med. 2018, 2050, e455. [Google Scholar] [CrossRef]
- Wang, X.M.; Gu, P.; Saligan, L.; Iadarola, M.; Wong, S.S.C.; Ti, L.K.; Cheung, C.W. Dysregulation of EAAT2 and VGLUT2 Spinal Glutamate Transports via Histone Deacetylase 2 (HDAC2) Contributes to Paclitaxel-induced Painful Neuropathy. Mol. Cancer. Ther. 2020, 19, 2196–2209. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Li, J.; Wang, Y.; Ren, F.; Zhou, Y.; Wu, Y.; Feng, Y.; Zhou, Y.; Su, F.; et al. p15RS/RPRD1A (p15INK4b-related sequence/regulation of nuclear pre-mRNA domain-containing protein 1A) interacts with HDAC2 in inhibition of the Wnt/β-catenin signaling pathway. J. Biol. Chem. 2015, 290, 9701–9713. [Google Scholar] [CrossRef] [PubMed]
- Bolon, B.; Butt, M.T. Fundamental Neuropathology for Pathologists and Toxicologists: Principles and Techniques; Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Rao, D.B.; Little, P.B.; Sills, R.C. Subsite awareness in neuropathology evaluation of National Toxicology Program (NTP) studies: A review of select neuroanatomical structures with their functional significance in rodents. Toxicol. Pathol. 2014, 42, 487–509. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: New York, NY, USA, 2013. [Google Scholar]
- Defazio, R.; Criado, A.; Zantedeschi, V.; Scanziani, E. Neuroanatomy-based matrix-guided trimming protocol for the rat brain. Toxicol. Pathol. 2015, 43, 249–256. [Google Scholar] [CrossRef]
- Traissard, N.; Herbeaux, K.; Cosquer, B.; Jeltsch, H.; Ferry, B.; Galani, R.; Pernon, A.; Majchrzak, M.; Cassel, J.C. Combined Damage to Entorhinal Cortex and Cholinergic Basal Forebrain Neurons, Two Early Neurodegenerative Features Accompanying Alzheimer′s Disease: Effects on Locomotor Activity and Memory Functions in Rats. Neuropsychopharmacology 2007, 32, 851–871. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Danandeh, A.; Baratta, J.; Lin, C.Y.; Yu, J.; Robertson, R.T. Neurotrophic factors rescue basal forebrain cholinergic neurons and improve performance on a spatial learning test. Exp. Neurol. 2013, 249, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Suzuki, R.; Kitano, Y.; Nishimaki, H.; Kobayashi, H.; Nakanishi, Y.; Yokoi, H. Tissue Thickness Interferes With the Estimation of the Immunohistochemical Intensity: Introduction of a Control System for Managing Tissue Thickness. Appl. Immunohistochem. Mol. Morphol. 2021, 29, 118–126. [Google Scholar] [CrossRef]
- Garman, R.H. Histology of the central nervous system. Toxicol. Pathol. 2011, 39, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Gusel′nikova, V.V.; Korzhevskiy, D.E. NeuN as a neuronal nuclear antigen and neuron differentiation marker. Acta Nat. 2015, 7, 42–47. [Google Scholar] [CrossRef]
- Hammond, D.N.; Lee, H.J.; Tonsgard, J.H.; Wainer, B.H. Development and characterization of clonal cell lines derived from septal cholinergic neurons. Brain Res. 1990, 512, 190–200. [Google Scholar] [CrossRef]
- Hudgens, E.D.; Ji, L.; Carpenter, C.D.; Petersen, S.L. The gad2 promoter is a transcriptional target of estrogen receptor (ER)alpha and ER beta: A unifying hypothesis to explain diverse effects of estradiol. J. Neurosci. 2009, 29, 8790–8797. [Google Scholar] [CrossRef]
- Bielarczyk, H.; Jankowska-Kulawy, A.; Gul, S.; Pawełczyk, T.; Szutowicz, A. Phenotype dependent differential effects of interleukin-1beta and amyloid-beta on viability and cholinergic phenotype of T17 neuroblastoma cells. Neurochem. Int. 2005, 47, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Ronowska, A.; Jankowska-Kulawy, A.; Gul-Hinc, S.; Zyśk, M.; Michno, A.; Szutowicz, A. Effects of Marginal Zn Excess and Thiamine Deficiency on Microglial N9 Cell Metabolism and Their Interactions with Septal SN56 Cholinergic Cells. Int. J. Mol. Sci. 2023, 24, 4465. [Google Scholar] [CrossRef]
- Cao, X.L.; Zhang, J.; Goodyer, C.G.; Hayward, S.; Cooke, G.M.; Curran, I.H. Bisphenol A in human placental and fetal liver tissues collected from Greater Montreal area (Quebec) during 1998–2008. Chemosphere 2012, 89, 505–511. [Google Scholar] [CrossRef]
- González, N.; Cunha, S.C.; Monteiro, C.; Fernandes, J.O.; Marquès, M.; Domingo, J.L.; Nadal, M. Quantification of eight bisphenol analogues in blood and urine samples of workers in a hazardous waste incinerator. Environ. Res. 2019, 176, 108576. [Google Scholar] [CrossRef] [PubMed]
- Ikezuki, Y.; Tsutsumi, O.; Takai, Y.; Kamei, Y.; Taketani, Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum. Reprod. 2002, 17, 2839–2841. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Andrianou, X.D.; Van der Meer, T.P.; Den Dunnen, W.F.A.; Swaab, D.F.; Wolffenbuttel, B.H.R.; Makris, K.C.; Van Vliet-Ostaptchouk, J.V. Possible Obesogenic Effects of Bisphenols Accumulation in the Human Brain. Sci. Rep. 2018, 8, 8186. [Google Scholar] [CrossRef]
- Kim, C.S.; Sapienza, P.P.; Ross, I.A.; Johnson, W.; Luu, H.M.; Hutter, J.C. Distribution of bisphenol A in the neuroendocrine organs of female rats. Toxicol. Ind Health. 2004, 20, 41–50. [Google Scholar] [CrossRef]
- Meng, L.; Liu, J.; Wang, C.; Ouyang, Z.; Kuang, J.; Pang, Q.; Fan, R. Sex-specific oxidative damage effects induced by BPA and its analogs on primary hippocampal neurons attenuated by EGCG. Chemosphere 2021, 264 Pt 1, 128450. [Google Scholar] [CrossRef]
- Pang, Q.; Li, Y.; Meng, L.; Li, G.; Luo, Z.; Fan, R. Neurotoxicity of BPA, BPS, and BPB for the hippocampal cell line (HT-22): An implication for the replacement of BPA in plastics. Chemosphere 2019, 226, 545–552. [Google Scholar] [CrossRef]
- Wang, T.; Xie, C.; Yu, P.; Fang, F.; Zhu, J.; Cheng, J.; Gu, A.; Wang, J.; Xiao, H. Involvement of Insulin Signaling Disturbances in Bisphenol A-Induced Alzheimer’s Disease-like Neurotoxicity. Sci. Rep. 2017, 7, 7497. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta DeltaC(T)). Method. Methods. 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Crooks, D.R.; Welch, N.; Smith, D.R. Low-level manganese exposure alters glutamate metabolism in GABAergic AF5 cells. NeuroToxicology 2007, 28, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Gwiazda, R.H.; Lee, D.; Sheridan, J.; Smith, D.R. Low Cumulative Manganese Exposure Affects Striatal GABA but not Dopamine. NeuroToxicology 2002, 23, 69–76. [Google Scholar] [CrossRef]
- Del Pino, J.; Martínez, M.A.; Castellano, V.; Ramos, E.; Martínez-Larrañaga, M.R.; Anadón, A. Effects of exposure to amitraz on noradrenaline, serotonin and dopamine levels in brain regions of 30 and 60 days old male rats. Toxicology 2013, 308, 88–95. [Google Scholar] [CrossRef] [PubMed]
- McKenna, J.T.; Yang, C.; Bellio, T.; Anderson-Chernishof, M.B.; Gamble, M.C.; Hulverson, A.; McCoy, J.G.; Winston, S.; Hodges, E.; Katsuki, F.; et al. Characterization of basal forebrain glutamate neurons suggests a role in control of arousal and avoidance behavior. Brain Struct. Funct. 2021, 226, 1755–1778. [Google Scholar] [CrossRef]
- El Tabaa, M.M.; Sokkar, S.S.; Ramadan, E.S.; Abd El Salam, I.Z.; Zaid, A. Neuroprotective role of Ginkgo biloba against cognitive deficits associated with Bisphenol A exposure: An animal model study. Neurochem. Int. 2017, 108, 199–212. [Google Scholar] [CrossRef]
- Elbakry, M.M.M.; Mansour, S.Z.; Helal, H.; Ahmed, E.S.A. Nattokinase attenuates bisphenol A or gamma irradiation-mediated hepatic and neural toxicity by activation of Nrf2 and suppression of inflammatory mediators in rat. Environ. Sci. Pollut. Res. Int. 2022, 29, 75086–75100. [Google Scholar] [CrossRef]
- El Tabaa, M.M.; Sokkar, S.S.; Ramdan, E.S.; El Salam, I.Z.A.; Anis, A. Does (-)-epigallocatechin-3-gallate protect the neurotoxicity induced by bisphenol A in vivo? Environ. Sci. Pollut. Res. Int. 2022, 29, 32190–32203. [Google Scholar] [CrossRef]
- Sevastre-Berghian, A.C.; Casandra, C.; Gheban, D.; Olteanu, D.; Olanescu Vaida Voevod, M.C.; Rogojan, L.; Filip, G.A.; Bâldea, I. Neurotoxicity of Bisphenol A and the Impact of Melatonin Administration on Oxidative Stress, ERK/NF-kB Signaling Pathway, and Behavior in Rats. Neurotox. Res. 2022, 40, 1882–1894. [Google Scholar] [CrossRef]
- Fetterly, T.L.; Oginsky, M.F.; Nieto, A.M.; Alonso-Caraballo, Y.; Santana-Rodriguez, Z.; Ferrario, C.R. Insulin Bidirectionally Alters NAc Glutamatergic Transmission: Interactions between Insulin Receptor Activation, Endogenous Opioids, and Glutamate Release. J. Neurosci. 2021, 41, 2360–2372. [Google Scholar] [CrossRef] [PubMed]
- Lellos, V.; Moraitou, M.; Tselentis, V.; Philippidis, H.; Palaiologos, G. Effect of starvation or streptozotocin-diabetes on phosphate-activated glutaminase of different rat brain regions. Neurochem. Res. 1992, 17, 141–145. [Google Scholar] [CrossRef]
- Viswaprakash, N.; Vaithianathan, T.; Viswaprakash, A.; Judd, R.; Parameshwaran, K.; Suppiramaniam, V. Insulin treatment restores glutamate (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor function in the hippocampus of diabetic rats. J. Neurosci. Res. 2015, 93, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, A.A.; Makboul, R.M.; El-Mokhtar, M.A.; Abdel-Rahman, E.A.; Ahmed, E.A.; Nicola, M.A. Evaluation of the neuroprotective effect of donepezil in type 2 diabetic rats. Fundam. Clin. Pharmacol. 2021, 35, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, B.D.S.; Kanekiyo, T.; Singh, J. Nerve Growth Factor Gene Delivery across the Blood-Brain Barrier to Reduce Beta Amyloid Accumulation in AD Mice. Mol. Pharm. 2020, 17, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Hayashi, M.; Hunt, A. Role of thyroid hormone and nerve growth factor in the development of choline acetyltransferase and other cell-specific marker enzymes in the basal forebrain of the rat. J. Neurochem. 1988, 50, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Kusiak, J.W. Nerve growth factor up-regulates the N-methyl-D-aspartate receptor subunit 1 promoter in PC12 cells. J. Biol. Chem. 1997, 272, 5936–5942. [Google Scholar] [CrossRef]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3β in Regulating Mitochondrial Activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/β-catenin signaling. Biochim Biophys Acta. Mol. Cell. Res. 2021, 1868, 118926. [Google Scholar] [CrossRef]
- Kim, J.G.; Kim, M.J.; Choi, W.J.; Moon, M.Y.; Kim, H.J.; Lee, J.Y.; Kim, J.; Kim, S.C.; Kang, S.G.; Seo, G.Y.; et al. Wnt3A Induces GSK-3β Phosphorylation and β-Catenin Accumulation Through RhoA/ROCK. J. Cell. Physiol. 2017, 232, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Selim, M.A.; Mosaad, S.M.; El-Sayed, N.M. Lycopene protects against Bisphenol A induced toxicity on the submandibular salivary glands via the upregulation of PPAR-γ and modulation of Wnt/β-catenin signaling. Int. Immunopharmacol. 2022, 112, 109293. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Hong, S.H.; Lee, M.K. Insulin receptor-overexpressing β-cells ameliorate hyperglycemia in diabetic rats through Wnt signaling activation. PLoS ONE 2013, 8, e67802. [Google Scholar] [CrossRef]
- Li, B.; Cai, S.; Zhao, Y.; He, Q.; Yu, X.; Cheng, L.; Zhang, Y.; Hu, X.; Ke, M.; Chen, S.; et al. Nerve growth factor modulates the tumor cells migration in ovarian cancer through the WNT/β-catenin pathway. Oncotarget 2016, 7, 81026–81048. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores, A.; Moyano, P.; Sola, E.; García, J.M.; García, J.; Frejo, M.T.; Guerra-Menéndez, L.; Labajo, E.; Lobo, I.; Abascal, L.; et al. Bisphenol-A Neurotoxic Effects on Basal Forebrain Cholinergic Neurons In Vitro and In Vivo. Biology 2023, 12, 782. https://doi.org/10.3390/biology12060782
Flores A, Moyano P, Sola E, García JM, García J, Frejo MT, Guerra-Menéndez L, Labajo E, Lobo I, Abascal L, et al. Bisphenol-A Neurotoxic Effects on Basal Forebrain Cholinergic Neurons In Vitro and In Vivo. Biology. 2023; 12(6):782. https://doi.org/10.3390/biology12060782
Chicago/Turabian StyleFlores, Andrea, Paula Moyano, Emma Sola, José Manuel García, Jimena García, María Teresa Frejo, Lucia Guerra-Menéndez, Elena Labajo, Inés Lobo, Luisa Abascal, and et al. 2023. "Bisphenol-A Neurotoxic Effects on Basal Forebrain Cholinergic Neurons In Vitro and In Vivo" Biology 12, no. 6: 782. https://doi.org/10.3390/biology12060782