

3D Chromatin Architecture Re-Wiring at the CDH3/CDH1 Loci Contributes to E-Cadherin to P-Cadherin Expression Switch in Gastric Cancer
 , , , and
, , , and 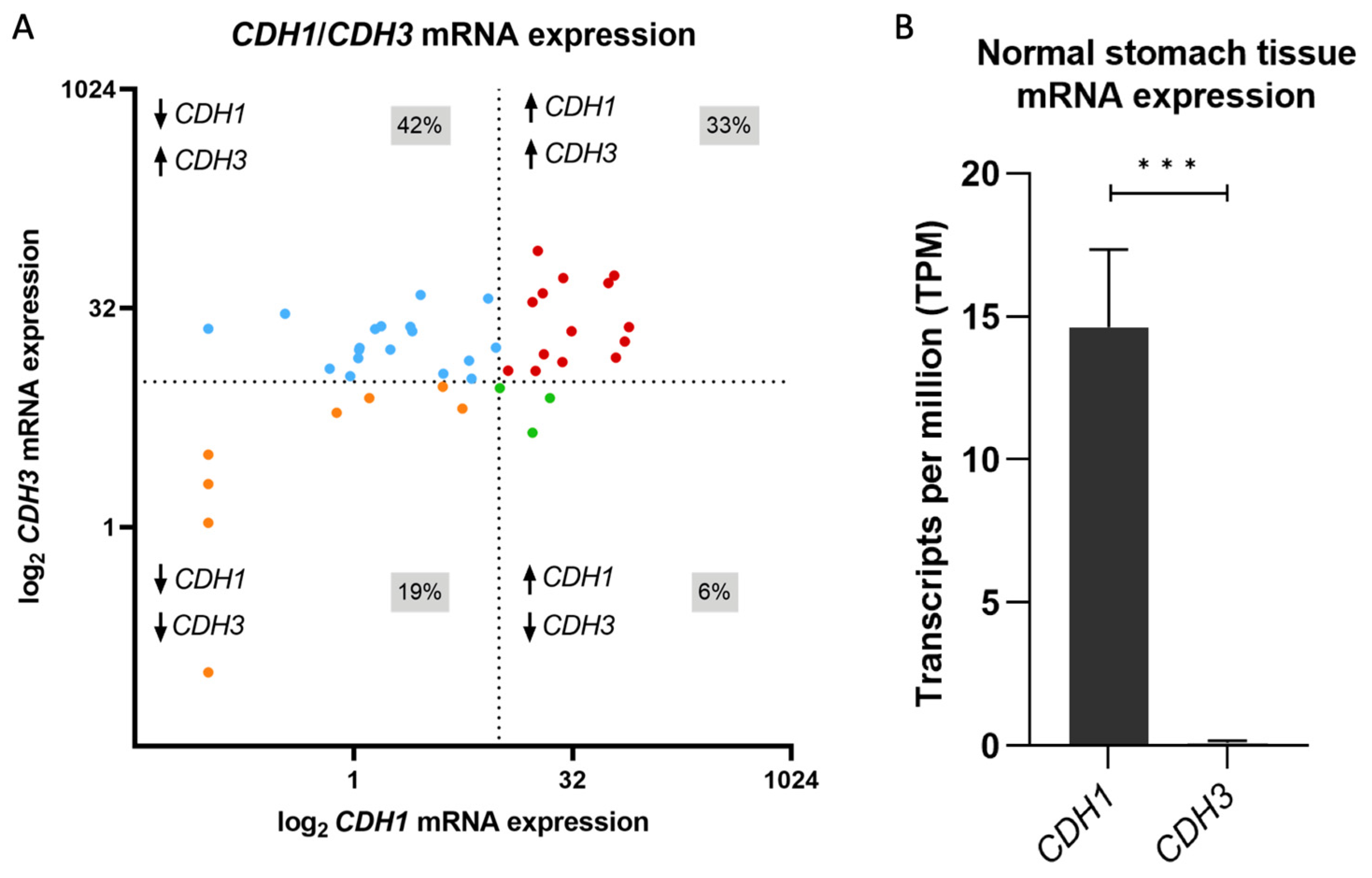{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. RNA Extraction
2.3. RNA-Seq and Whole Transcriptome Sequencing Bioinformatics Analysis
2.4. Cell Lines and Culture
2.5. Generation of CRISPR-Cas9 E-Cadherin Deletion
2.6. Genotyping of Edited Clones
2.7. mRNA and Protein Expression Analysis
2.8. Statistical Analysis
2.9. Immunofluorescence
2.10. Protein Extraction
2.11. Proteomics Sample Processing
2.12. Protein Identification and Quantification, and Enrichment GO Terms Analysis
2.13. ATAC-Seq
2.14. C-Seq
3. Results
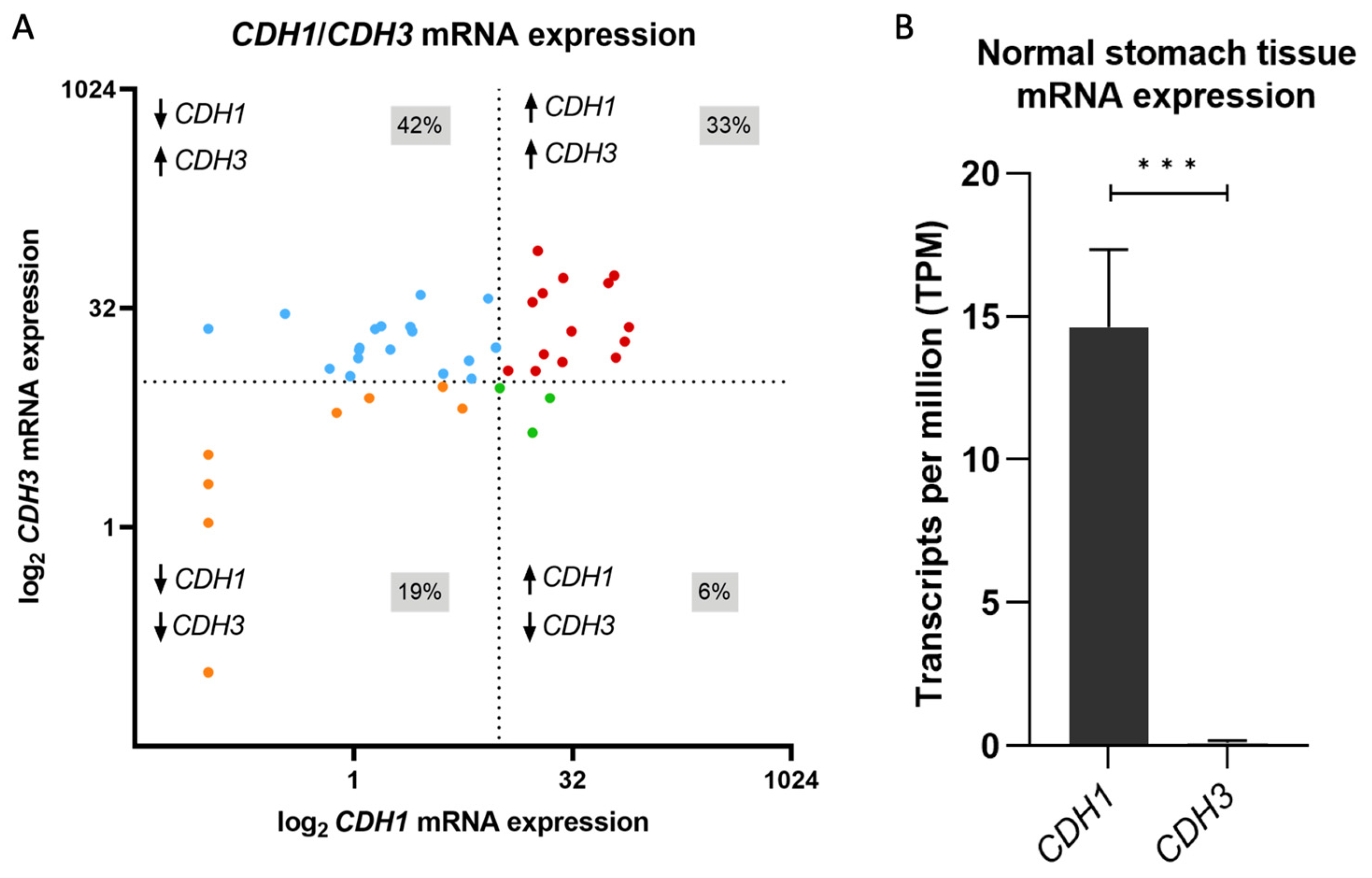
3.1. CDH1 to CDH3 mRNA Expression Switch Is a Frequent Event in Gastric Cancer
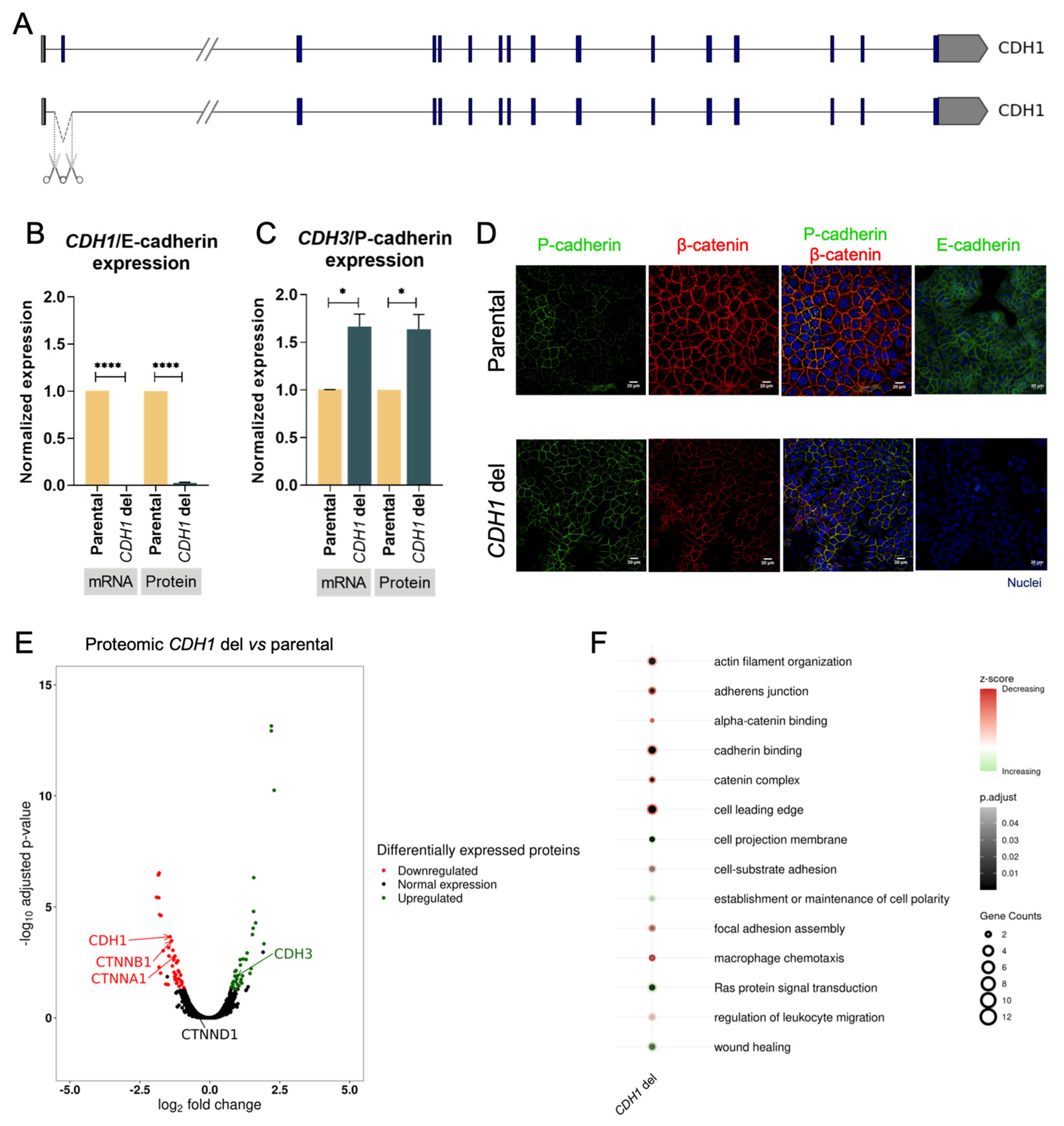
3.2. E-Cadherin Loss of Function Triggers E-Cadherin to P-Cadherin Switch in Gastric Cancer
3.3. E-Cadherin Expression Loss Triggers Downregulation of Adhesion Complex Partners and Adhesion-Related Pathways
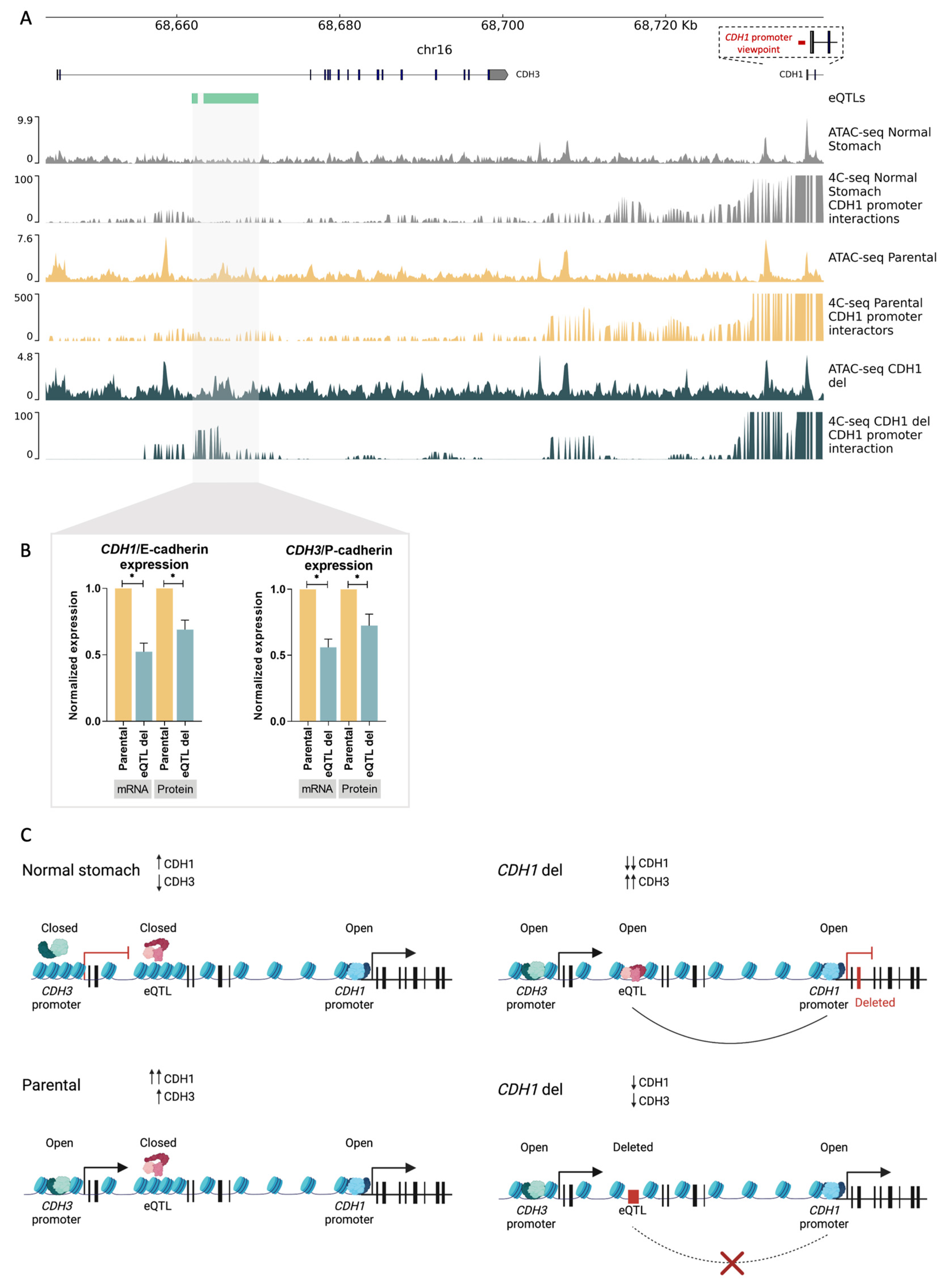
3.4. D Chromatin Architecture Re-Wiring at the CDH3–CDH1 Loci Contributes to E-Cadherin to P-Cadherin Switch in Gastric Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ivanov, D.B.; Philippova, M.P.; Tkachuk, V.A. Structure and Functions of Classical Cadherins. Biochemistry 2001, 66, 1174–1186. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, T.; Rizvi, A.; Batta, S.P.R.; Kataria, S.; Jamora, C. Signaling and Mechanical Roles of E-cadherin. Cell Commun. Adhes. 2013, 20, 189–199. [Google Scholar] [CrossRef]
- Nollet, F.; Koolsa, P.; Van Roy, F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J. Mol. Biol. 2000, 299, 551–572. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; O’day, D.R.; Pliner, H.A.; Kingsley, P.D.; Deng, M.; Daza, R.M.; Zager, M.A.; Aldinger, K.A.; Blecher-Gonen, R.; Zhang, F.; et al. A human cell atlas of fetal gene expression. Science 2020, 370, eaba7721. [Google Scholar] [CrossRef]
- Daniel, C.W.; Strickland, P.; Friedmann, Y. Expression and Functional Role of E- and P-Cadherins in Mouse Mammary Ductal Morphogenesis and Growth. Dev. Biol. 1995, 169, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, E.; Han, S.-W.; Im, S.-A.; Kim, T.-Y.; Kim, W.-H.; Oh, D.-Y.; Bang, Y.-J. Down-regulation of P-cadherin with PF-03732010 inhibits cell migration and tumor growth in gastric cancer. Investig. New Drugs 2012, 30, 1404–1412. [Google Scholar] [CrossRef]
- Kim, M.A.; Jung, E.J.; Lee, H.S.; Lee, H.E.; Yang, H.-K.; Oh, D.-Y.; Bang, Y.-J.; Kim, W.H. P-cadherin expression in gastric carcinoma: Its regulation mechanism and prognostic significance. Hum. Pathol. 2010, 41, 877–885. [Google Scholar] [CrossRef]
- Eom, B.W.; Ryu, K.W.; Yoon, H.M.; Kook, M.-C. Predictive value of E-cadherin and EpCAM for detection of metastatic lymph node in early gastric cancer. Chin. J. Cancer Res. 2020, 32, 614–620. [Google Scholar] [CrossRef]
- Liu, N.; Yu, Q.; Liu, T.J.; Gebreamlak, E.P.; Wang, S.L.; Zhang, R.J.; Zhang, J.; Niu, Y. P-cadherin expression and basal-like subtype in breast cancers. Med. Oncol. 2012, 29, 2606–2612. [Google Scholar] [CrossRef]
- Margan, M.M.; Cimpean, A.M.; Ceausu, A.R.; Raica, M. Differential Expression of E-Cadherin and P-Cadherin in Breast Cancer Molecular Subtypes. Anticancer Res. 2020, 40, 5557–5566. [Google Scholar] [CrossRef]
- Turashvili, G.; E McKinney, S.; Goktepe, O.; Leung, S.C.; Huntsman, D.G.; A Gelmon, K.; Los, G.; A Rejto, P.; Aparicio, S.A.J.R. P-cadherin expression as a prognostic biomarker in a 3992 case tissue microarray series of breast cancer. Mod. Pathol. 2011, 24, 64–81. [Google Scholar] [CrossRef]
- Faria, G.; Cardoso, M.J.; Martins, D.; Bettencourt, H.; Amendoeira, I.; Schimitt, F. P-cadherin as prognostic factor for loco-regional relapse in breast cancer. Acta Med. Port. 2012, 25, 97–105. [Google Scholar]
- Sridhar, S.; Rajesh, C.; Jishnu, P.V.; Jayaram, P.; Kabekkodu, S.P. Increased expression of P-cadherin is an indicator of poor prognosis in breast cancer: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2020, 179, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Gravdal, K.; Halvorsen, O.J.; Haukaas, S.A.; Akslen, L.A. A Switch from E-Cadherin to N-Cadherin Expression Indicates Epithelial to Mesenchymal Transition and Is of Strong and Independent Importance for the Progress of Prostate Cancer. Clin. Cancer Res. 2007, 13, 7003–7011. [Google Scholar] [CrossRef]
- Appolloni, I.; Barilari, M.; Caviglia, S.; Gambini, E.; Reisoli, E.; Malatesta, P. A cadherin switch underlies malignancy in high-grade gliomas. Oncogene 2015, 34, 1991–2002. [Google Scholar] [CrossRef]
- Araki, K.; Shimura, T.; Suzuki, H.; Tsutsumi, S.; Wada, W.; Yajima, T.; Kobayahi, T.; Kubo, N.; Kuwano, H. E/N-cadherin switch mediates cancer progression via TGF-beta-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br. J. Cancer 2011, 105, 1885–1893. [Google Scholar] [CrossRef]
- Barber, M.; Murrell, A.; Ito, Y.; Maia, A.-T.; Hyland, S.; Oliveira, C.; Save, V.; Carneiro, F.; Paterson, A.; Grehan, N.; et al. Mechanisms and sequelae of E-cadherin silencing in hereditary diffuse gastric cancer. J. Pathol. 2008, 216, 295–306. [Google Scholar] [CrossRef]
- Li, B.; Guo, X.; Li, N.; Chen, Q.; Shen, J.; Huang, X.; Huang, G.; Wang, F. WNT1, a target of miR-34a, promotes cervical squamous cell carcinoma proliferation and invasion by induction of an E-P cadherin switch via the WNT/β-catenin pathway. Cell. Oncol. 2020, 43, 489–503. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, M.; Guerra, R.F.; Cereda, M.; Marchesi, S.; Montani, F.; Nicassio, F.; Di Fiore, P.P.; Ciccarelli, F.D. Recessive Cancer Genes Engage in Negative Genetic Interactions with Their Functional Paralogs. Cell Rep. 2013, 5, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Andrey, G.; Mundlos, S. The three-dimensional genome: Regulating gene expression during pluripotency and development. Development 2017, 144, 3646–3658. [Google Scholar] [CrossRef]
- Will, A.J.; Cova, G.; Osterwalder, M.; Chan, W.L.; Wittler, L.; Brieske, N.; Heinrich, V.; de Villartay, J.P.; Vingron, M.; Klopocki, E.; et al. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog). Nat Genet. 2017, 49, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Tennekes, M. tmap: Thematic Maps in R. J. Stat. Softw. 2018, 84, 1–39. [Google Scholar] [CrossRef]
- Feingold, E.A.; Good, P.J.; Guyer, M.S.; Kamholz, S.; Liefer, L.; Wetterstrand, K.; Collins, F.S.; Gingeras, T.R.; Kampa, D.; Sekinger, E.A.; et al. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef]
- Zhang, X.O.; Gingeras, T.R.; Weng, Z. Genome-wide analysis of polymerase III–transcribed Alu elements suggests cell-type–specific enhancer function. Genome Res. 2019, 29, 1402–1414. [Google Scholar] [CrossRef]
- Lee, D.; Zhang, J.; Liu, J.; Gerstein, M. Epigenome-based splicing prediction using a recurrent neural network. PLoS Comput. Biol. 2020, 16, e1008006. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef] [PubMed]
- Van de Werken, H.J.; de Vree, P.J.; Splinter, E.; Holwerda, S.J.; Klous, P.; de Wit, E.; de Laat, W. 4C technology: Protocols and data analysis. Methods Enzym. 2012, 513, 89–112. [Google Scholar] [CrossRef]
- Krijger, P.H.; Geeven, G.; Bianchi, V.; Hilvering, C.R.; de Laat, W. 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods 2020, 170, 17–32. [Google Scholar] [CrossRef]
- Geeven, G.; Teunissen, H.; De Laat, W.; De Wit, E. peakC: A flexible, non-parametric peak calling package for 4C and Capture-C data. Nucleic Acids Res. 2018, 46, e91. [Google Scholar] [CrossRef] [PubMed]
- Martins, E.P.; Gonçalves, C.S.; Pojo, M.; Carvalho, R.; Ribeiro, A.S.; Miranda-Gonçalves, V.; Taipa, R.; Pardal, F.; Pinto, A.A.; Custódia, C.; et al. Cadherin-3 is a novel oncogenic biomarker with prognostic value in glioblastoma. Mol. Oncol. 2022, 16, 2611–2631. [Google Scholar] [CrossRef] [PubMed]
- Van Marck, V.; Stove, C.; Jacobs, K.; Eynden, G.V.D.; Bracke, M. P-cadherin in adhesion and invasion: Opposite roles in colon and bladder carcinoma. Int. J. Cancer 2011, 128, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.S.; Sousa, B.; Carreto, L.; Mendes, N.; Nobre, A.R.; Ricardo, S.; Albergaria, A.; Cameselle-Teijeiro, J.F.; Gerhard, R.; Söderberg, O.; et al. P-cadherin functional role is dependent on E-cadherin cellular context: A proof of concept using the breast cancer model. J. Pathol. 2013, 229, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Christgen, M.; Bartels, S.; van Luttikhuizen, J.L.; Bublitz, J.; Rieger, L.U.; Christgen, H.; Stark, H.; Sander, B.; Lehmann, U.; Steinemann, D.; et al. E-cadherin to P-cadherin switching in lobular breast cancer with tubular elements. Mod. Pathol. 2020, 33, 2483–2498. [Google Scholar] [CrossRef]
- Paliou, C.; Guckelberger, P.; Schöpflin, R.; Heinrich, V.; Esposito, A.; Chiariello, A.M.; Bianco, S.; Annunziatella, C.; Helmuth, J.; Haas, S.; et al. Preformed chromatin topology assists transcriptional robustness of Shh during limb development. Proc. Natl. Acad. Sci. USA 2019, 116, 12390–12399. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
São José, C.; Pereira, C.; Ferreira, M.; André, A.; Osório, H.; Gullo, I.; Carneiro, F.; Oliveira, C. 3D Chromatin Architecture Re-Wiring at the CDH3/CDH1 Loci Contributes to E-Cadherin to P-Cadherin Expression Switch in Gastric Cancer. Biology 2023, 12, 803. https://doi.org/10.3390/biology12060803
São José C, Pereira C, Ferreira M, André A, Osório H, Gullo I, Carneiro F, Oliveira C. 3D Chromatin Architecture Re-Wiring at the CDH3/CDH1 Loci Contributes to E-Cadherin to P-Cadherin Expression Switch in Gastric Cancer. Biology. 2023; 12(6):803. https://doi.org/10.3390/biology12060803
Chicago/Turabian StyleSão José, Celina, Carla Pereira, Marta Ferreira, Ana André, Hugo Osório, Irene Gullo, Fátima Carneiro, and Carla Oliveira. 2023. "3D Chromatin Architecture Re-Wiring at the CDH3/CDH1 Loci Contributes to E-Cadherin to P-Cadherin Expression Switch in Gastric Cancer" Biology 12, no. 6: 803. https://doi.org/10.3390/biology12060803
APA StyleSão José, C., Pereira, C., Ferreira, M., André, A., Osório, H., Gullo, I., Carneiro, F., & Oliveira, C. (2023). 3D Chromatin Architecture Re-Wiring at the CDH3/CDH1 Loci Contributes to E-Cadherin to P-Cadherin Expression Switch in Gastric Cancer. Biology, 12(6), 803. https://doi.org/10.3390/biology12060803