
Bilirubin-Induced Transcriptomic Imprinting in Neonatal Hyperbilirubinemia

 ,
,  ,
,  , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Hematoxylin-Eosin Stain and Morphometric Analysis
2.3. GO Analysis, Selection of the Genes, and RT-qPCR
2.4. Correlation and Clustering Studies
2.5. Behavioral Tests
2.6. Statistical Analysis
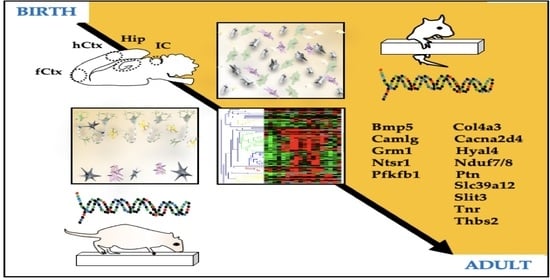
3. Results
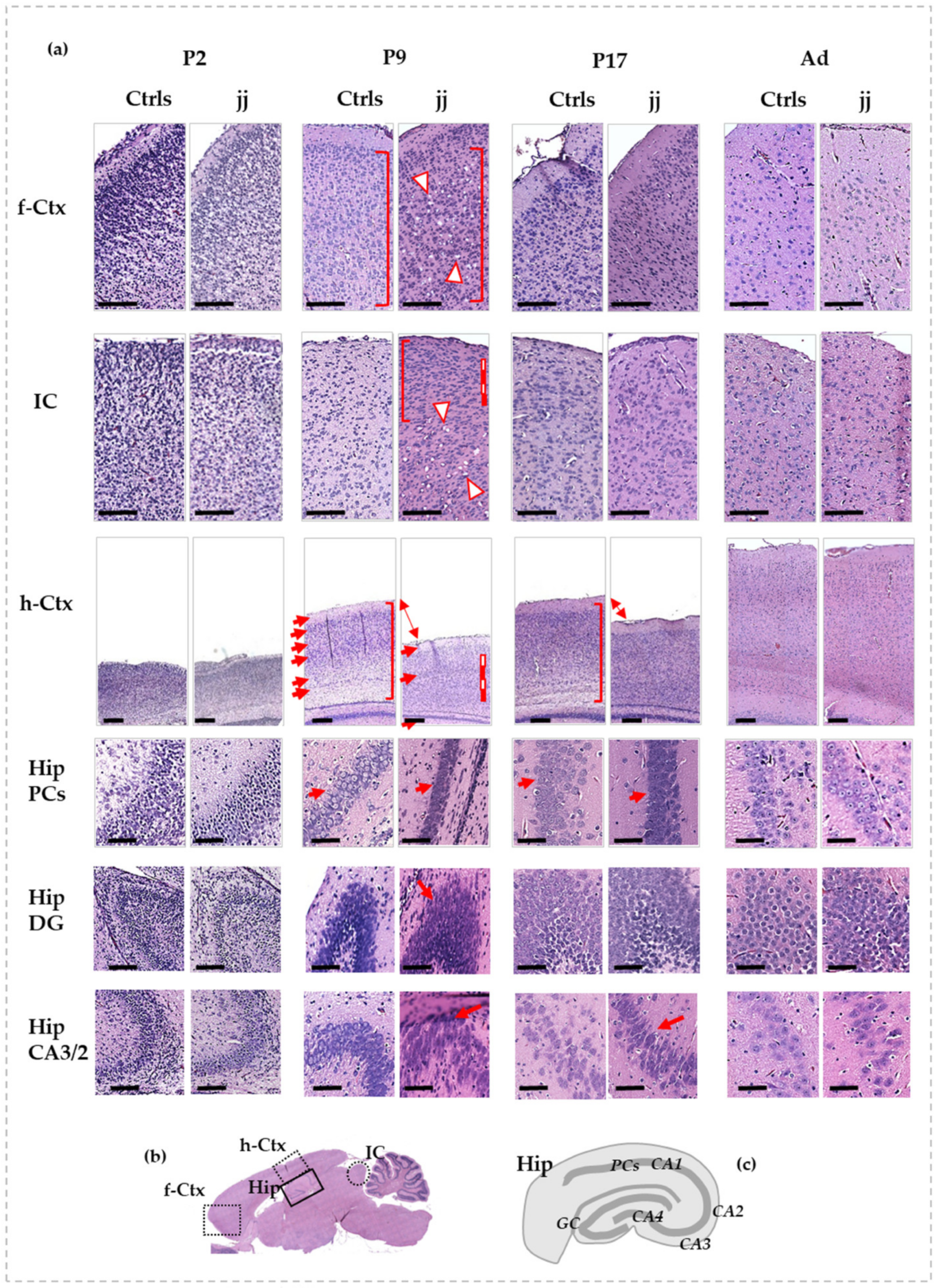
3.1. Histological Features of the Gunn Rat Brain during Postnatal Development
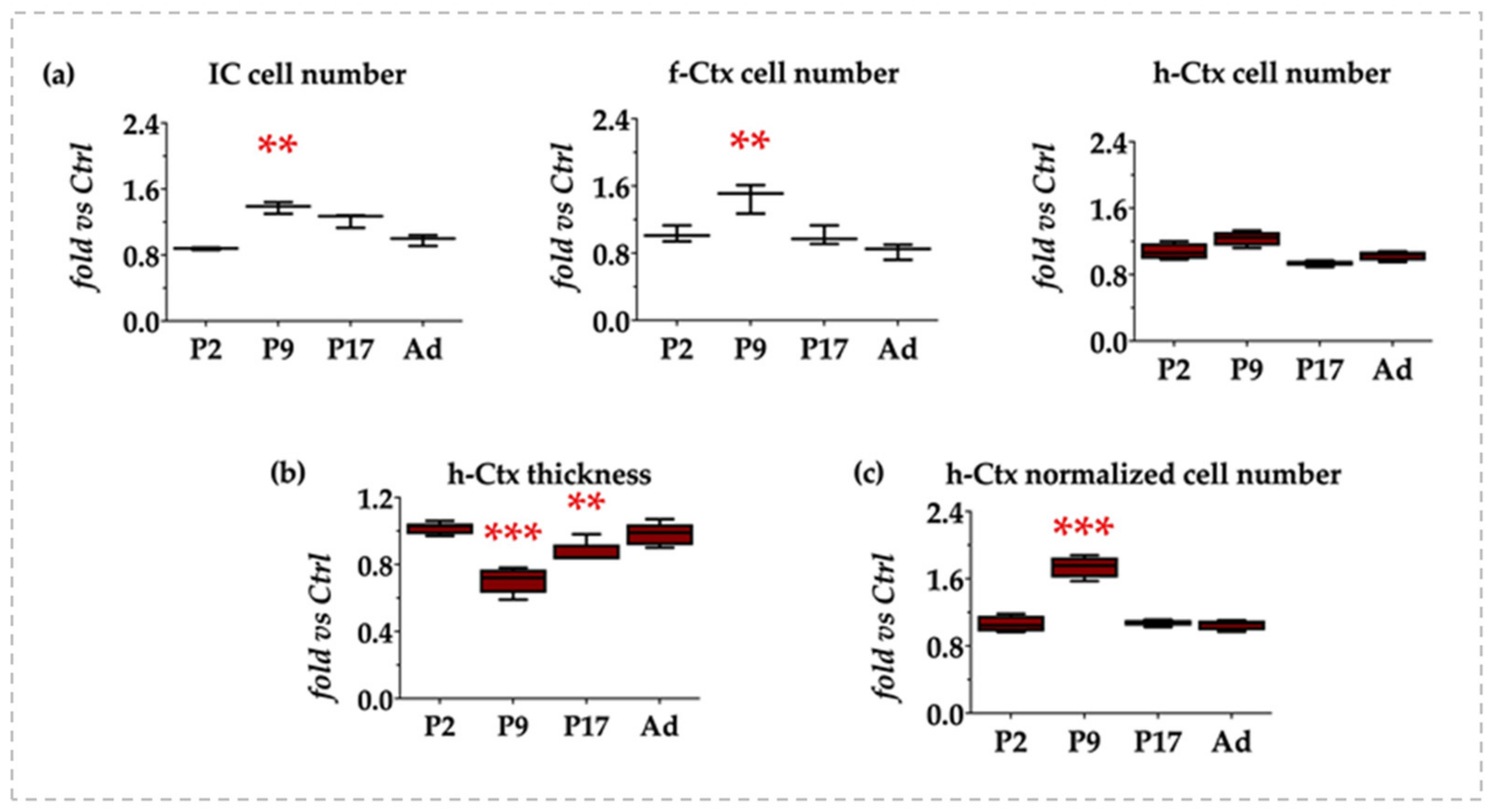
3.2. Morphometric Analysis: Cell Counting and h-Ctx Thickness Quantification
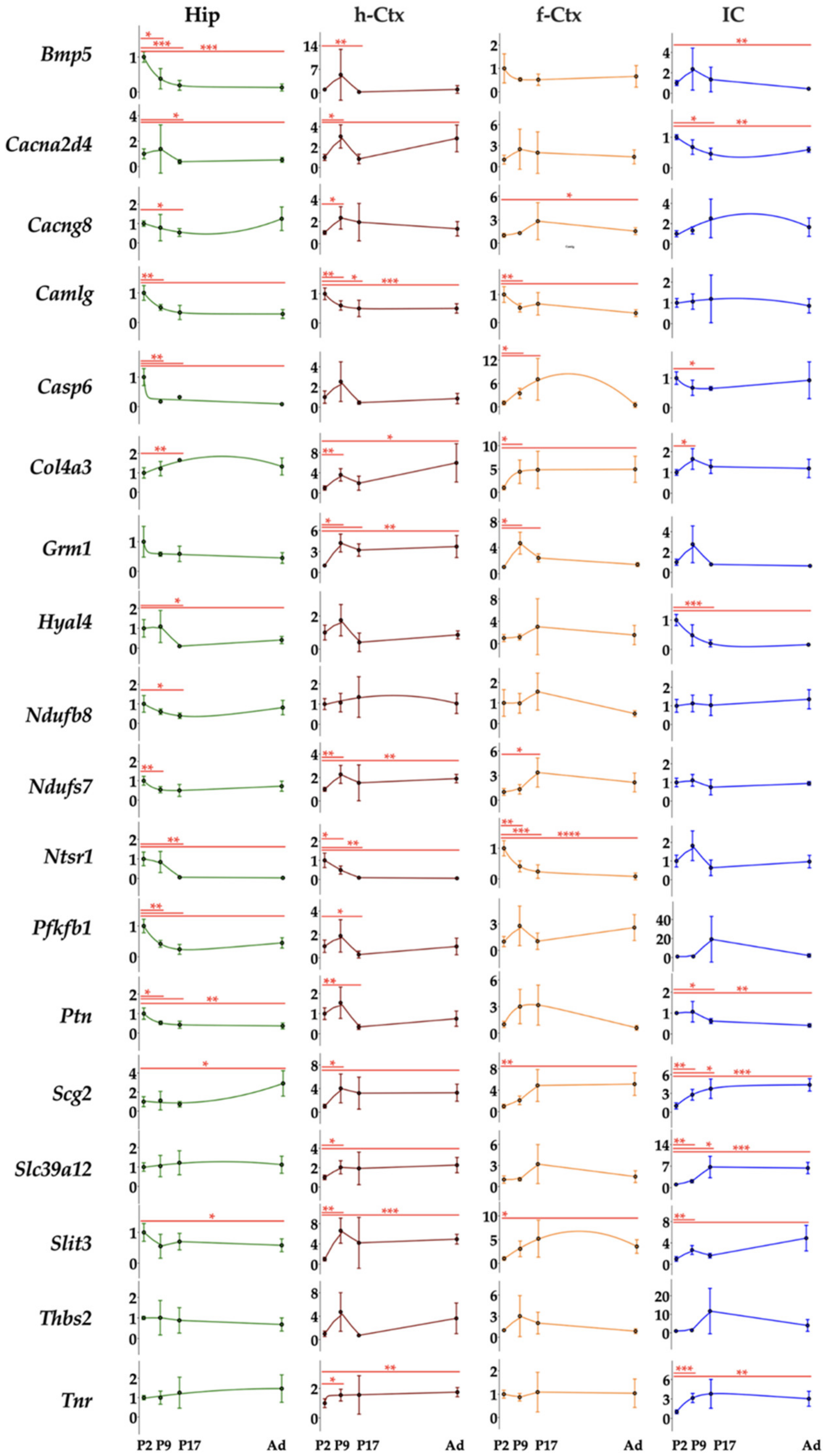
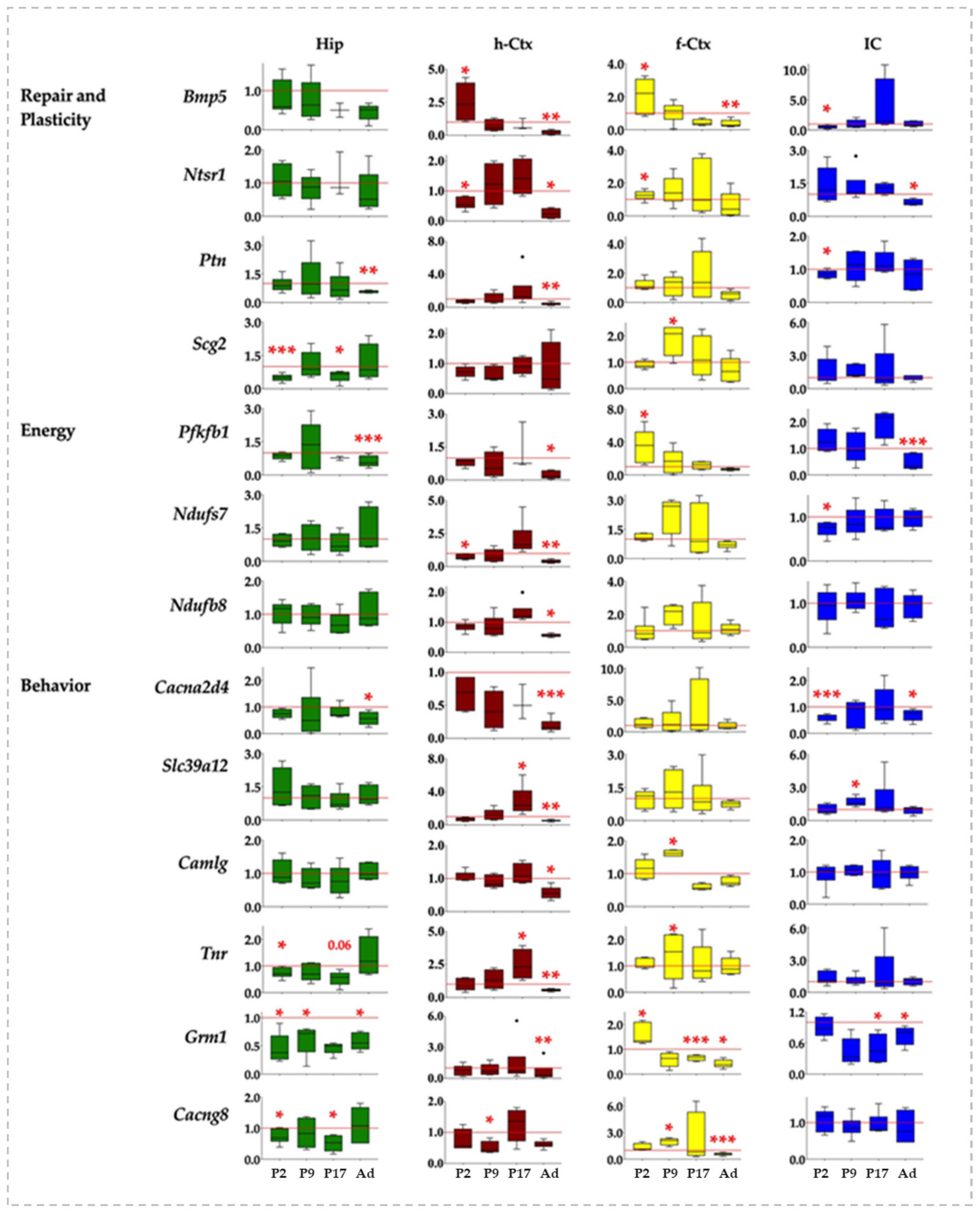
3.3. Gene Ontology and RT-qPCR of Selected Genes
3.4. RTqPCR Results in Ctrls (Physiologic Expression)
3.5. Heatmap Analysis of the Gene Expression in Ctrls along the Postnatal Brain Development
3.6. RTqPCR Results on Hyperbilirubinemic Animals
3.7. Correlation Analysis of Bilirubin Impact on Genes Expression
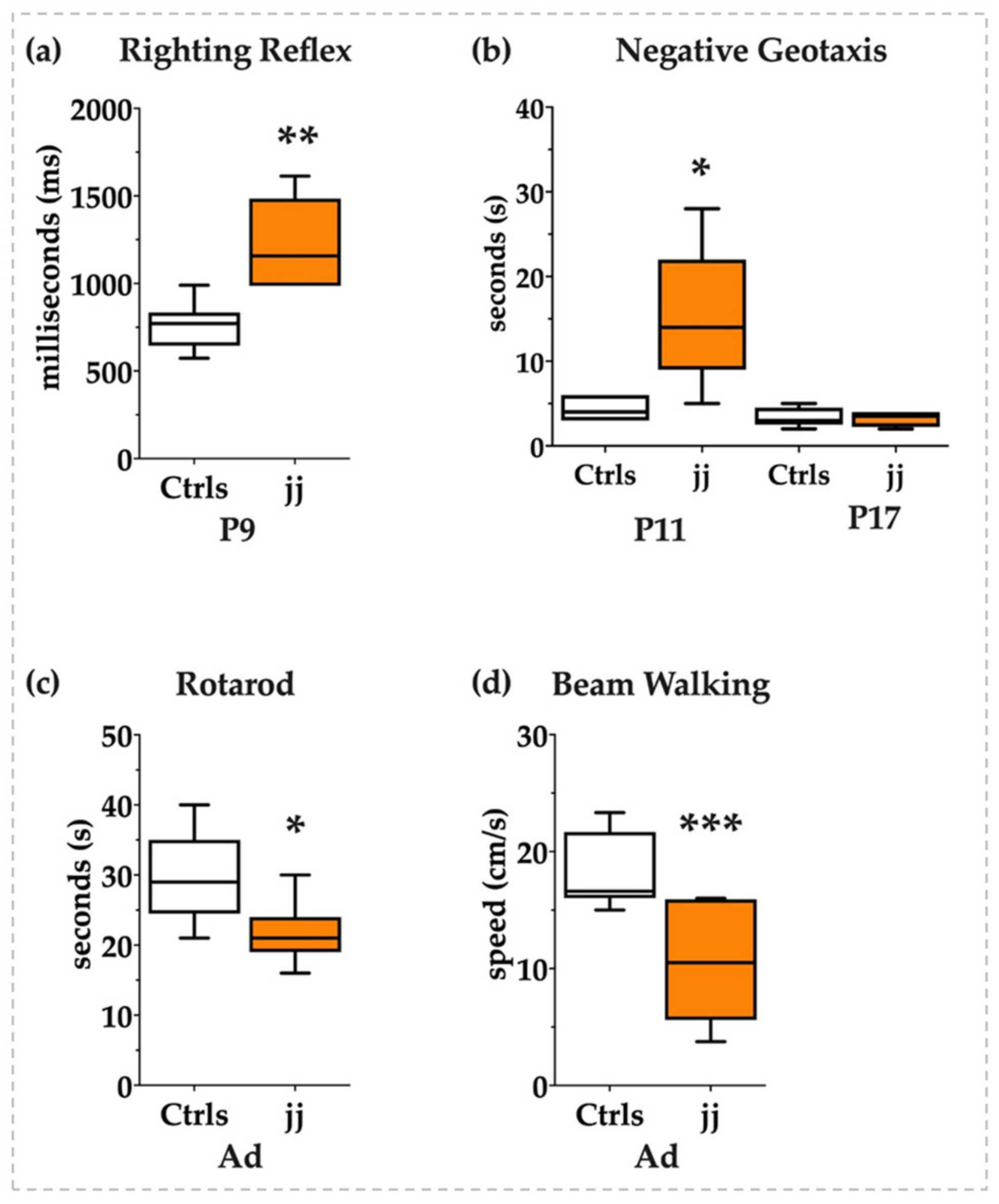
3.8. Behavioral Tests
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Slusher, T.M.; Zamora, T.G. Burden of Severe Neonatal Jaundice: A Systematic Review and Meta-Analysis. BMJ Paediatr. Open 2017, 1, e000105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayanti, S.; Ghersi-Egea, J.-F. Severe Neonatal Hyperbilirubinemia and the Brain: The Old but Still Evolving Story. Pediatr. Med. 2021, 4, 37. [Google Scholar] [CrossRef]
- Le Pichon, J.-B.; Riordan, S.M. The Neurological Sequelae of Neonatal Hyperbilirubinemia: Definitions, Diagnosis and Treatment of the Kernicterus Spectrum Disorders (KSDs). Curr. Pediatr. Rev. 2017, 13, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.M. Definition of the Clinical Spectrum of Kernicterus and Bilirubin-Induced Neurologic Dysfunction (BIND). J. Perinatol. 2005, 25, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Olusanya, B.O.; Teeple, S.; Kassebaum, N.J. The Contribution of Neonatal Jaundice to Global Child Mortality: Findings from the GBD 2016 Study. Pediatrics 2018, 141, e20171471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AAP. Management of Hyperbilirubinemia in the Newborn Infant 35 or More Weeks of Gestation. Pediatrics 2004, 114, 297–316. [Google Scholar] [CrossRef] [Green Version]
- Ip, S.; Chung, M. Subcommittee on Hyperbilirubinemia. An Evidence-Based Review of Important Issues Concerning Neonatal Hyperbilirubinemia. Pediatrics 2004, 114, e130–e153. [Google Scholar] [CrossRef]
- Bhutani, V.K.; Zipursky, A. Neonatal Hyperbilirubinemia and Rhesus Disease of the Newborn: Incidence and Impairment Estimates for 2010 at Regional and Global Levels. Pediatr. Res. 2013, 74, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Gunn, C.H. Hereditary acholuric jaundice in a New Mutant Strain of Rats. J. Hered. 1938, 29, 137–139. [Google Scholar] [CrossRef]
- Chowdhury, J.R.; Kondapalli, R. Gunn Rat: A Model for Inherited Deficiency of Bilirubin Glucuronidation. Adv. Vet. Sci. Comp. Med. 1993, 37, 149–173. [Google Scholar]
- Vianello, E.; Zampieri, S. Histone Acetylation as a New Mechanism for Bilirubin-Induced Encephalopathy in the Gunn Rat. Sci. Rep. 2018, 8, 13690. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Zelenka, J. Bilirubin Accumulation and Cyp MRNA Expression in Selected Brain Regions of Jaundiced Gunn Rat Pups. Pediatr. Res. 2012, 71, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Robert, M.C.; Furlan, G. Alterations in the Cell Cycle in the Cerebellum of Hyperbilirubinemic Gunn Rat: A Possible Link with Apoptosis? PLoS ONE 2013, 8, e79073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolussi, G.; Zentilin, L. Rescue of Bilirubin-Induced Neonatal Lethality in a Mouse Model of Crigler-Najjar Syndrome Type I by AAV9-Mediated Gene Transfer. FASEB J. 2012, 26, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, R.; Nguyen, N. Developmental Hyperbilirubinemia and CNS Toxicity in Mice Humanized with the UDP Glucuronosyltransferase 1 (UGT1) Locus. Proc. Natl. Acad. Sci. USA 2010, 107, 5024–5029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riordan, S.M.; Shapiro, S.M. Review of Bilirubin Neurotoxicity I: Molecular Biology and Neuropathology of Disease. Pediatr. Res. 2019, 87, 327–331. [Google Scholar] [CrossRef]
- Shapiro, S.M. Chronic Bilirubin Encephalopathy: Diagnosis and Outcome. Semin. Fetal. Neonatal Med. 2010, 15, 157–163. [Google Scholar] [CrossRef]
- Shapiro, S.M.; Nakamura, H. Bilirubin and the Auditory System. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2001, 21 (Suppl. S1), S52–S55; discussion S59–S62. [Google Scholar] [CrossRef] [Green Version]
- Watchko, J.F.; Maisels, M.J. The Enigma of Low Bilirubin Kernicterus in Premature Infants: Why Does It Still Occur, and Is It Preventable? Semin. Perinatol. 2014, 38, 397–406. [Google Scholar] [CrossRef]
- Watchko, J.F.; Painter, M.J. Are the Neuromotor Disabilities of Bilirubin-Induced Neurologic Dysfunction Disorders Related to the Cerebellum and Its Connections? Semin. Fetal. Neonatal Med. 2015, 20, 47–51. [Google Scholar] [CrossRef]
- Wisnowski, J.L.; Panigrahy, A. Magnetic Resonance Imaging Abnormalities in Advanced Acute Bilirubin Encephalopathy Highlight Dentato-Thalamo-Cortical Pathways. J. Pediatr. 2016, 174, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, M.; Bottin, C. Evaluation of Region Selective Bilirubin-Induced Brain Damage as a Basis for a Pharmacological Treatment. Sci. Rep. 2017, 7, 41032. [Google Scholar] [CrossRef] [Green Version]
- Schutta, H.S.; Johnson, L. Bilirubin Encephalopathy in the Gunn Rat: A Fine Structure Study of the Cerebellar Cortex. J. Neuropathol. Exp. Neurol. 1967, 26, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Schutta, H.S.; Johnson, L. Clinical Signs and Morphologic Abnormalities in Gunn Rats Treated with Sulfadimethoxine. J. Pediatr. 1969, 75, 1070–1079. [Google Scholar] [CrossRef]
- Gazzin, S.; Jayanti, S. Models of Bilirubin Neurological Damage: Lessons Learned and New Challenges. Pediatr. Res. 2022, 1–8. [Google Scholar] [CrossRef]
- Keino, H.; Sato, H. Mode of Prevention by Phototherapy of Cerebellar Hypoplasia in a New Sprague-Dawley Strain of Jaundiced Gunn Rats. Pediatr. Neurosurg. 1985, 12, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Berengeno, A.L. Modulation of Mrp1 (ABCc1) and Pgp (ABCb1) by Bilirubin at the Blood-CSF and Blood-Brain Barriers in the Gunn Rat. PLoS ONE 2011, 6, e16165. [Google Scholar] [CrossRef] [Green Version]
- Mody, M.; Cao, Y. Genome-Wide Gene Expression Profiles of the Developing Mouse Hippocampus. Proc. Natl. Acad. Sci. USA 2001, 98, 8862–8867. [Google Scholar] [CrossRef] [Green Version]
- Urbán, N.; Guillemot, F. Neurogenesis in the Embryonic and Adult Brain: Same Regulators, Different Roles. Front. Cell. Neurosci. 2014, 8, 396. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Iwabuchi, T. Identification and Expression of Frizzled-3 Protein in Rat Frontal Cortex After Antidepressant and Electroconvulsive Treatment. J. Pharmacol. Sci. 2005, 99, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Moeendarbary, E.; Weber, I.P. The Soft Mechanical Signature of Glial Scars in the Central Nervous System. Nat. Commun. 2017, 8, 14787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slomianka, L.; Amrein, I. Hippocampal Pyramidal Cells: The Reemergence of Cortical Lamination. Brain Struct. Funct. 2011, 216, 301–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.A.; Nathanson, J. Conserved Molecular Signatures of Neurogenesis in the Hippocampal Subgranular Zone of Rodents and Primates. Dev. Camb. Engl. 2013, 140, 4633–4644. [Google Scholar] [CrossRef] [Green Version]
- Khalaf-Nazzal, R.; Francis, F. Hippocampal Development-Old and New Findings. Neuroscience 2013, 248, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Stead, J.D.H.; Neal, C. Transcriptional Profiling of the Developing Rat Brain Reveals That the Most Dramatic Regional Differentiation in Gene Expression Occurs Postpartum. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Azzarelli, R.; Kerloch, T. Regulation of Cerebral Cortex Development by Rho GTPases: Insights from in Vivo Studies. Front. Cell. Neurosci. 2015, 8, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dityatev, A.; Seidenbecher, C.I. Compartmentalization from the Outside: The Extracellular Matrix and Functional Microdomains in the Brain. Trends Neurosci. 2010, 33, 503–512. [Google Scholar] [CrossRef]
- Cho, Y.; Gong, T.-W.L. Gene Expression Profiles of the Rat Cochlea, Cochlear Nucleus, and Inferior Colliculus. J. Assoc. Res. Otolaryngol. JARO 2002, 3, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Schubert, D.; Martens, G.J.M. Molecular Underpinnings of Prefrontal Cortex Development in Rodents Provide Insights into the Etiology of Neurodevelopmental Disorders. Mol. Psychiatry 2015, 20, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Vallès, A.; Boender, A.J. Genomewide Analysis of Rat Barrel Cortex Reveals Time- and Layer-Specific MRNA Expression Changes Related to Experience-Dependent Plasticity. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 6140–6158. [Google Scholar] [CrossRef] [Green Version]
- Decourt, B.; Bouleau, Y. Identification of Differentially Expressed Genes in the Developing Mouse Inferior Colliculus. Dev. Brain Res. 2005, 159, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [Green Version]
- R Foundation for Statistical Computing, Vienna. R: A Language and Environment for Statistical Computing. R Core Team (2021). Available online: https://www.R-project.org (accessed on 11 March 2023).
- Schemper, M. Predictive Accuracy and Explained Variation. Stat. Med. 2003, 22, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Cai, T. Model Evaluation Based on the Sampling Distribution of Estimated Absolute Prediction Error. Biometrika 2007, 94, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Bacon, C.R. Practical Portfolio Performance Measurement and Attribution, 2nd ed.; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Friendly, M. Corrgrams. Am. Stat. 2002, 56, 316–324. [Google Scholar] [CrossRef]
- Murdoch, D.J.; Chow, E.D. A Graphical Display of Large Correlation Matrices. Am. Stat. 1996, 50, 178–180. [Google Scholar] [CrossRef]
- Howe, E.A.; Sinha, R. RNA-Seq Analysis in MeV. Bioinformatics 2011, 27, 3209–3210. [Google Scholar] [CrossRef] [Green Version]
- Gazzin, S.; Dal Ben, M. Curcumin Prevents Cerebellar Hypoplasia and Restores the Behavior in Hyperbilirubinemic Gunn Rat by a Pleiotropic Effect on the Molecular Effectors of Brain Damage. Int. J. Mol. Sci. 2021, 22, 299. [Google Scholar] [CrossRef]
- Dunham, N.W.; Miya, T.S. A Note on a Simple Apparatus for Detecting Neurological Deficit in Rats and Mice**College of Pharmacy, University of Nebraska, Lincoln 8. J. Am. Pharm. Assoc. Sci. 1957, 46, 208–209. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.B.; Davis, J.N. Beam-Walking in Rats: Studies towards Developing an Animal Model of Functional Recovery after Brain Injury. J. Neurosci. Methods 1990, 31, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Feather-Schussler, D.N.; Ferguson, T.S. A Battery of Motor Tests in a Neonatal Mouse Model of Cerebral Palsy. J. Vis. Exp. JoVE 2016, 117, 53569. [Google Scholar] [CrossRef] [Green Version]
- Wills, T.J.; Muessig, L. The Development of Spatial Behaviour and the Hippocampal Neural Representation of Space. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130409. [Google Scholar] [CrossRef] [Green Version]
- Brites, D. Bilirubin Injury to Neurons and Glial Cells: New Players, Novel Targets, and Newer Insights. Semin. Perinatol. 2011, 35, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Boules, M.; Li, Z. Diverse Roles of Neurotensin Agonists in the Central Nervous System. Front. Endocrinol. 2013, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- St-Gelais, F.; Jomphe, C. The Role of Neurotensin in Central Nervous System Pathophysiology: What Is the Evidence? J Psychiatry Neurosci. 2006, 31, 229–245. [Google Scholar] [PubMed]
- Xiao, Z.; Cilz, N.I. Activation of Neurotensin Receptor 1 Facilitates Neuronal Excitability and Spatial Learning and Memory in the Entorhinal Cortex: Beneficial Actions in an Alzheimer’s Disease Model. J. Neurosci. 2014, 34, 7027–7042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodworth, H.L.; Batchelor, H.M. Neurotensin Receptor-1 Identifies a Subset of Ventral Tegmental Dopamine Neurons That Coordinates Energy Balance. Cell Rep. 2017, 20, 1881–1892. [Google Scholar] [CrossRef] [Green Version]
- Holt, A.G.; Asako, M. Deafness-Related Plasticity in the Inferior Colliculus: Gene Expression Profiling Following Removal of Peripheral Activity. J. Neurochem. 2005, 93, 1069–1086. [Google Scholar] [CrossRef]
- Hagihara, H.; Ohira, K. Expression of the AMPA Receptor Subunits GluR1 and GluR2 Is Associated with Granule Cell Maturation in the Dentate Gyrus. Front. Neurosci. 2011, 5, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe, V.; Wong, B.K.Y. Rescue from Excitotoxicity and Axonal Degeneration Accompanied by Age-Dependent Behavioral and Neuroanatomical Alterations in Caspase-6-Deficient Mice. Hum. Mol. Genet. 2012, 21, 1954–1967. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, M.; Gardner, H.A.R. Postnatal Neuronal Apoptosis in the Cerebral Cortex: Physiological and Pathophysiological Mechanisms. Neuroscience 2013, 254, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, U.; Khodosevich, K. Neuronal Survival in the Brain: Neuron Type-Specific Mechanisms. Cell Death Dis. 2017, 8, e2643. [Google Scholar] [CrossRef] [Green Version]
- Bandeira, F.; Lent, R. Changing Numbers of Neuronal and Non-Neuronal Cells Underlie Postnatal Brain Growth in the Rat. Proc. Natl. Acad. Sci. USA 2009, 106, 14108–14113. [Google Scholar] [CrossRef] [Green Version]
- Okuda, H.; Tatsumi, K. Chondroitin Sulfate Proteoglycan Tenascin-R Regulates Glutamate Uptake by Adult Brain Astrocytes*. J. Biol. Chem. 2014, 289, 2620–2631. [Google Scholar] [CrossRef] [Green Version]
- Gottschling, C.; Wegrzyn, D. Elimination of the Four Extracellular Matrix Molecules Tenascin-C, Tenascin-R, Brevican and Neurocan Alters the Ratio of Excitatory and Inhibitory Synapses. Sci. Rep. 2019, 9, 13939. [Google Scholar] [CrossRef] [Green Version]
- Eckert, A.; Schmitt, K. Mitochondrial Dysfunction–the Beginning of the End in Alzheimer’s Disease? Separate and Synergistic Modes of Tau and Amyloid-β Toxicity. Alzheimers Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef]
- Foti, S.C.; Hargreaves, I. Cerebral Mitochondrial Electron Transport Chain Dysfunction in Multiple System Atrophy and Parkinson’s Disease. Sci. Rep. 2019, 9, 6559. [Google Scholar] [CrossRef] [Green Version]
- Knowlton, W.M.; Hubert, T. A Select Subset of Electron Transport Chain Genes Associated with Optic Atrophy Link Mitochondria to Axon Regeneration in Caenorhabditis Elegans. Front. Neurosci. 2017, 11, 263. [Google Scholar] [CrossRef] [Green Version]
- Stansberg, C.; Ersland, K.M. Gene Expression in the Rat Brain: High Similarity but Unique Differences between Frontomedial-, Temporal- and Occipital Cortex. BMC Neurosci. 2011, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Scarr, E.; Udawela, M. Increased Cortical Expression of the Zinc Transporter SLC39A12 Suggests a Breakdown in Zinc Cellular Homeostasis as Part of the Pathophysiology of Schizophrenia. Npj Schizophr. 2016, 2, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, D.N.; Strong, M.D. A Role for Zinc Transporter Gene SLC39A12 in the Nervous System and Beyond. Gene 2021, 799, 145824. [Google Scholar] [CrossRef] [PubMed]
- Song, M.D.; Hart, M.D. Role of Zinc Transporter ZIP12 in Susceptibility-Weighted Brain Magnetic Resonance Imaging (MRI) Phenotypes and Mitochondrial Function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 10702–12725. [Google Scholar] [CrossRef]
- Elliott, L.T.; Sharp, K. Genome-Wide Association Studies of Brain Imaging Phenotypes in UK Biobank. Nature 2018, 562, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Bly, M. Examination of the Zinc Transporter Gene, SLC39A12. Schizophr. Res. 2006, 81, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Gazzellone, M.J.; Zhou, X. Copy Number Variation in Han Chinese Individuals with Autism Spectrum Disorder. J. Neurodev. Disord. 2014, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Chowanadisai, W.; Graham, D.M. Neurulation and Neurite Extension Require the Zinc Transporter ZIP12 (Slc39a12). Proc. Natl. Acad. Sci. USA 2013, 110, 9903–9908. [Google Scholar] [CrossRef] [Green Version]
- González-Castillo, C.; Ortuño-Sahagún, D. Pleiotrophin as a Central Nervous System Neuromodulator, Evidences from the Hippocampus. Front. Cell. Neurosci. 2015, 8, 443. [Google Scholar] [CrossRef] [Green Version]
- Wanaka, A.; Carroll, S.L. Developmentally Regulated Expression of Pleiotrophin, a Novel Heparin Binding Growth Factor, in the Nervous System of the Rat. Dev. Brain Res. 1993, 72, 133–144. [Google Scholar] [CrossRef]
- Fernández-Calle, R.; Vicente-Rodríguez, M. Pleiotrophin Regulates Microglia-Mediated Neuroinflammation. J. Neuroinflammation 2017, 14, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.-J.; Zhang, H.-N. Alteration of Thrombospondin-1 and -2 in Rat Brains Following Experimental Intracerebral Hemorrhage: Laboratory Investigation. J. Neurosurg. 2010, 113, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Iruela-Arispe, M.L.; Liska, D.J. Differential Expression of Thrombospondin 1, 2, and 3 during Murine Development. Dev. Dyn. 1993, 197, 40–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Hu, T. Macroglia-Derived Thrombospondin 2 Regulates Alterations of Presynaptic Proteins of Retinal Neurons Following Elevated Hydrostatic Pressure. PLoS ONE 2017, 12, e0185388. [Google Scholar] [CrossRef] [Green Version]
- Wisnowski, J.L.; Panigrahy, A. Magnetic Resonance Imaging of Bilirubin Encephalopathy: Current Limitations and Future Promise. Semin. Perinatol. 2014, 38, 422–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, S.B.; Smith, T. Developmental Influence of Unconjugated Hyperbilirubinemia and Neurobehavioral Disorders. Pediatr. Res. 2019, 85, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Uziel, A.; Marot, M. The Gunn Rat: An Experimental Model for Central Deafness. Acta Otolaryngol. 1983, 95, 651–656. [Google Scholar] [CrossRef]
- Stanford, J.A.; Shuler, J.M. Hyperactivity in the Gunn Rat Model of Neonatal Jaundice: Age-Related Attenuation and Emergence of Gait Deficits. Pediatr. Res. 2015, 77, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Daood, M.J.; Hoyson, M. Lipid Peroxidation Is Not the Primary Mechanism of Bilirubin-Induced Neurologic Dysfunction in Jaundiced Gunn Rat Pups. Pediatr. Res. 2012, 72, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Jew, J.Y.; Sandquist, D. CNS Changes in Hyperbilirubinemia: Functional Implications. Arch. Neurol. 1979, 36, 149–154. [Google Scholar] [CrossRef]
- Liaury, K.; Miyaoka, T. Morphological Features of Microglial Cells in the Hippocampal Dentate Gyrus of Gunn Rat: A Possible Schizophrenia Animal Model. J. Neuroinflam. 2012, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeder, S.S.; Burkardt, P. Evidence for Postnatal Neurogenesis in the Human Amygdala. Commun. Biol. 2022, 5, 366. [Google Scholar] [CrossRef]
- Yang, Z.; Ming, G. Postnatal Neurogenesis in the Human Forebrain: From Two Migratory Streams to Dribbles. Cell Stem Cell 2011, 9, 385–386. [Google Scholar] [CrossRef] [Green Version]
- Sanai, N.; Nguyen, T. Corridors of Migrating Neurons in the Human Brain and Their Decline during Infancy. Nature 2011, 478, 382–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierra, A.; Encinas, J. Adult Human Neurogenesis: From Microscopy to Magnetic Resonance Imaging. Front. Neurosci. 2011, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terstege, D.J.; Addo-Osafo, K. New Neurons in Old Brains: Implications of Age in the Analysis of Neurogenesis in Post-Mortem Tissue. Mol. Brain 2022, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jiménez, E.P.; Terreros-Roncal, J. Evidences for Adult Hippocampal Neurogenesis in Humans. J. Neurosci. 2021, 41, 2541–2553. [Google Scholar] [CrossRef]
- Gordo, A.C.; Falcão, A.S. Unconjugated Bilirubin Activates and Damages Microglia. J. Neurosci. Res. 2006, 84, 194–201. [Google Scholar] [CrossRef]
- Brites, D. The Evolving Landscape of Neurotoxicity by Unconjugated Bilirubin: Role of Glial Cells and Inflammation. Front. Pharmacol. 2012, 3, 88. [Google Scholar] [CrossRef] [Green Version]
- Vaz, A.R.; Falcão, A.S.; Scarpa, E.; Semproni, C.; Brites, D. Microglia Susceptibility to Free Bilirubin Is Age-Dependent. Front. Pharmacol. 2020, 11, 1012. [Google Scholar] [CrossRef]
- Ransohoff, R.M. Microgliosis: The Questions Shape the Answers. Nat. Neurosci. 2007, 10, 1507–1509. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulson, A.J.; Squair, J.W. Diversity of Reactive Astrogliosis in CNS Pathology: Heterogeneity or Plasticity? Front. Cell. Neurosci. 2021, 15, 703810. [Google Scholar] [CrossRef] [PubMed]
- Palmela, I. Histological Findings in the Kernicterus-Associated Vulnerable Brain Regions Are Linked to Neurodegeneration, Alterations in Astrocyte and Pericyte Distribution, and Vascular Modifications. Int. J. Pathol. Clin. Res. 2015, 1, 003. [Google Scholar] [CrossRef]
- Hu, W.; Cheng, X. Nuclear Magnetic Resonance Spectroscopy Reveals Systematic Alterations in Cerebral Metabolites as the Key Pathogenetic Mechanism of Bilirubin Encephalopathy. Mol. Brain 2014, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlee, J.W.; Shapiro, S.M. Development of Cerebellar Hypoplasia in Jaundiced Gunn Rats: A Quantitative Light Microscopic Analysis. Acta Neuropathol. 1997, 93, 450–460. [Google Scholar] [CrossRef]
- Watchko, J.F.; Tiribelli, C. Bilirubin-Induced Neurologic Damage—Mechanisms and Management Approaches. N. Engl. J. Med. 2013, 369, 2021–2030. [Google Scholar] [CrossRef]
- Roger, C.; Koziel, V. Mapping of the Consequences of Bilirubin Exposure in the Immature Rat: Local Cerebral Metabolic Rates for Glucose during Moderate and Severe Hyperbilirubinemia. Early Hum. Dev. 1995, 43, 133–144. [Google Scholar] [CrossRef]
- Das, S.; van Landeghem, F.K.H. Clinicopathological Spectrum of Bilirubin Encephalopathy/Kernicterus. Diagnostics 2019, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Seto-Ohshima, A.; Aoki, E. Appearance of Parvalbumin-Specific Immunoreactivity in the Cerebral Cortex and Hippocampus of the Developing Rat and Gerbil Brain. Histochemistry 1990, 94, 579–589. [Google Scholar] [CrossRef]
- Nahar, L.; Delacroix, B.M. The Role of Parvalbumin Interneurons in Neurotransmitter Balance and Neurological Disease. Front. Psychiatry 2021, 12, 679960. [Google Scholar] [CrossRef]
- Pearce, I.A.; Cambray-Deakin, M.A. Glutamate Acting on NMDA Receptors Stimulates Neurite Outgrowth from Cerebellar Granule Cells. FEBS Lett. 1987, 223, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klomp, A.; Omichi, R. The Voltage-Gated Ca2+ Channel Subunit A2δ-4 Regulates Locomotor Behavior and Sensorimotor Gating in Mice. PLoS ONE 2022, 17, e0263197. [Google Scholar] [CrossRef] [PubMed]
- Grojean, S.; Koziel, V. Bilirubin Induces Apoptosis via Activation of NMDA Receptors in Developing Rat Brain Neurons. Exp. Neurol. 2000, 166, 334–341. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate Receptors, Neurotoxicity and Neurodegeneration. Pflugers Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- McDonald, J.W.; Shapiro, S.M. Role of Glutamate Receptor-Mediated Excitotoxicity in Bilirubin-Induced Brain Injury in the Gunn Rat Model. Exp. Neurol. 1998, 150, 21–29. [Google Scholar] [CrossRef]
- Silva, S.L.; Vaz, A.R. Neuritic Growth Impairment and Cell Death by Unconjugated Bilirubin Is Mediated by NO and Glutamate, Modulated by Microglia, and Prevented by Glycoursodeoxycholic Acid and Interleukin-10. Neuropharmacology 2012, 62, 2398–2408. [Google Scholar] [CrossRef]
- Barateiro, A.; Domingues, H.S. Rat Cerebellar Slice Cultures Exposed to Bilirubin Evidence Reactive Gliosis, Excitotoxicity and Impaired Myelinogenesis That Is Prevented by AMPA and TNF-α Inhibitors. Mol. Neurobiol. 2014, 49, 424–439. [Google Scholar] [CrossRef] [PubMed]
- Falcão, A.S.; Fernandes, A. Bilirubin-Induced Inflammatory Response, Glutamate Release, and Cell Death in Rat Cortical Astrocytes Are Enhanced in Younger Cells. Neurobiol. Dis. 2005, 20, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Ouda, L.; Syka, J. Immunocytochemical Profiles of Inferior Colliculus Neurons in the Rat and Their Changes with Aging. Front. Neural Circuits 2012, 6, 68. [Google Scholar] [CrossRef] [Green Version]
- Gourley, G.R. Bilirubin Metabolism and Kernicterus. Adv. Pediatr. 1997, 44, 173–229. [Google Scholar]
- Hansen, T.W.R. Pioneers in the Scientific Study of Neonatal Jaundice and Kernicterus. Pediatrics 2000, 106, e15. [Google Scholar] [CrossRef] [Green Version]
- Farouk, Z.L.; Muhammed, A. Follow-up of Children with Kernicterus in Kano, Nigeria. J. Trop. Pediatr. 2018, 64, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozmert, E.; Erdem, G. Long-Term Follow-up of Indirect Hyperbilirubinemia in Full-Term Turkish Infants. Acta Paediatr. Oslo Nor. 1996, 85, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, S.T.; Finne, P.H. Males with Neonatal Hyperbilirubinemia Examined at 18 Years of Age. Acta Paediatr. 1984, 73, 176–180. [Google Scholar] [CrossRef]
- Hokkanen, L.; Launes, J. Adult Neurobehavioral Outcome of Hyperbilirubinemia in Full Term Neonates—A 30 Year Prospective Follow-up Study. PeerJ 2014, 2, e294. [Google Scholar] [CrossRef] [Green Version]
- Maimburg, R.D.; Væth, M. Neonatal Jaundice: A Risk Factor for Infantile Autism? Paediatr. Perinat. Epidemiol. 2008, 22, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.-J.; Luo, X.-G. Deficiency of Transmembrane AMPA Receptor Regulatory Protein γ-8 Leads to Attention-Deficit Hyperactivity Disorder-like Behavior in Mice. Zool. Res. 2022, 43, 851–870. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Pliszka, S.R. Catecholamine Influences on Prefrontal Cortical Function: Relevance to Treatment of Attention Deficit/Hyperactivity Disorder and Related Disorders. Pharmacol. Biochem. Behav. 2011, 99, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Biederman, J. Attention-Deficit/Hyperactivity Disorder: A Selective Overview. Biol. Psychiatry 2005, 57, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Couture, J. A Review of the Pathophysiology, Etiology, and Treatment of Attention-Deficit Hyperactivity Disorder (ADHD). Ann. Pharmacother. 2014, 48, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bossche, M.J.; Strazisar, M. Identification of a CACNA2D4 Deletion in Late Onset Bipolar Disorder Patients and Implications for the Involvement of Voltage-Dependent Calcium Channels in Psychiatric Disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2012, 159B, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, M.; Miyaoka, T. Hyperbilirubinemia-Related Behavioral and Neuropathological Changes in Rats: A Possible Schizophrenia Animal Model. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 581–588. [Google Scholar] [CrossRef]
- Wilson, M.P.; Durin, Z. CAMLG-CDG: A Novel Congenital Disorder of Glycosylation Linked to Defective Membrane Trafficking. Hum. Mol. Genet. 2022, 31, 2571–2581. [Google Scholar] [CrossRef]
- Blacker, C.J.; Lewis, C.P. Metabotropic Glutamate Receptors as Emerging Research Targets in Bipolar Disorder. Psychiatry Res. 2017, 257, 327–337. [Google Scholar] [CrossRef]
- De Rosa, A.; Fontana, A. Machine Learning Algorithm Unveils Glutamatergic Alterations in the Post-Mortem Schizophrenia Brain. Schizophrenia 2022, 8, 8. [Google Scholar] [CrossRef]
- Wagner, M.; Lévy, J. Loss of TNR Causes a Nonprogressive Neurodevelopmental Disorder with Spasticity and Transient Opisthotonus. Genet. Med. 2020, 22, 1061–1068. [Google Scholar] [CrossRef]
- Zheng, H.; Lin, J. Magnetic Resonance Image of Neonatal Acute Bilirubin Encephalopathy: A Diffusion Kurtosis Imaging Study. Front. Neurol. 2021, 12, 645534. [Google Scholar] [CrossRef]
- Usman, F.; Diala, U. Acute Bilirubin Encephalopathy and Its Progression to Kernicterus: Current Perspectives. Res. Rep. Neonatol. 2018, 8, 33–44. [Google Scholar] [CrossRef] [Green Version]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llido, J.P.; Fioriti, E.; Pascut, D.; Giuffrè, M.; Bottin, C.; Zanconati, F.; Tiribelli, C.; Gazzin, S. Bilirubin-Induced Transcriptomic Imprinting in Neonatal Hyperbilirubinemia. Biology 2023, 12, 834. https://doi.org/10.3390/biology12060834
Llido JP, Fioriti E, Pascut D, Giuffrè M, Bottin C, Zanconati F, Tiribelli C, Gazzin S. Bilirubin-Induced Transcriptomic Imprinting in Neonatal Hyperbilirubinemia. Biology. 2023; 12(6):834. https://doi.org/10.3390/biology12060834
Chicago/Turabian StyleLlido, John Paul, Emanuela Fioriti, Devis Pascut, Mauro Giuffrè, Cristina Bottin, Fabrizio Zanconati, Claudio Tiribelli, and Silvia Gazzin. 2023. "Bilirubin-Induced Transcriptomic Imprinting in Neonatal Hyperbilirubinemia" Biology 12, no. 6: 834. https://doi.org/10.3390/biology12060834
APA StyleLlido, J. P., Fioriti, E., Pascut, D., Giuffrè, M., Bottin, C., Zanconati, F., Tiribelli, C., & Gazzin, S. (2023). Bilirubin-Induced Transcriptomic Imprinting in Neonatal Hyperbilirubinemia. Biology, 12(6), 834. https://doi.org/10.3390/biology12060834