
Myocarditis: Etiology, Pathogenesis, and Their Implications in Clinical Practice

 , , ,
, , ,  , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Clinical Presentation, Diagnostic Approach and Assessment of Disease Etiology
2.1. Clinical Presentation
2.2. Utility of Diagnostic Tests
3. Viral and Virus-Induced Immune-Mediated Myocarditis
3.1. Overview of Viruses Associated with Myocarditis
3.2. Virus-Mediated Myocardial Injury
3.3. Immune Response to the Viral Infection
3.4. Clinical Implications of Viral Etiology
4. Myocarditis in the Course of Parasitic Infections
4.1. Parasitic Involvement in Cardiac Disease
4.2. Pathogenesis and Clinical Picture of Chagas’ Disease
4.3. Management of Patients with Chagas’ Cardiomyopathy
4.4. Cardiac Involvement in Other Parasitic Diseases
5. Bacterial Myocarditis
Overview
6. Autoimmune Myocarditis and Drug-Induced Myocarditis
6.1. Overview
6.2. Pathogenesis and Genetic Predisposition to Autoimmune Myocarditis
6.3. Pathogenesis of Drug-Associated Myocarditis
6.4. Clinical Implications of Autoimmune and Drug-Associated Myocarditis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, F.; Kühl, U.; Pieske, B.; Garcia-Pavia, P.; Tschöpe, C. Update on Myocarditis and Inflammatory Cardiomyopathy: Reemergence of Endomyocardial Biopsy. Rev. Esp. Cardiol. Engl. Ed. 2016, 69, 178–187. [Google Scholar] [CrossRef]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Cipriani, M.; Moro, C.; Raineri, C.; Pini, D.; Sormani, P.; Mantovani, R.; Varrenti, M.; Pedrotti, P.; Conca, C.; et al. Clinical Presentation and Outcome in a Contemporary Cohort of Patients With Acute Myocarditis: Multicenter Lombardy Registry. Circulation 2018, 138, 1088–1099. [Google Scholar] [CrossRef]
- Ammirati, E.; Veronese, G.; Bottiroli, M.; Wang, D.W.; Cipriani, M.; Garascia, A.; Pedrotti, P.; Adler, E.D.; Frigerio, M. Update on acute myocarditis. Trends Cardiovasc. Med. 2021, 31, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Kociol, R.D.; Cooper, L.T.; Fang, J.C.; Moslehi, J.J.; Pang, P.S.; Sabe, M.A.; Shah, R.V.; Sims, D.B.; Thiene, G.; Vardeny, O. Recognition and Initial Management of Fulminant Myocarditis: A Scientific Statement From the American Heart Association. Circulation 2020, 141, e69–e92. [Google Scholar] [CrossRef]
- Tymińska, A.; Ozierański, K.; Caforio, A.L.P.; Marcolongo, R.; Marchel, M.; Kapłon-Cieślicka, A.; Baritussio, A.; Filipiak, K.J.; Opolski, G.; Grabowski, M. Myocarditis and inflammatory cardiomyopathy in 2021: An update. Pol. Arch. Intern. Med. 2021, 131, 594–606. [Google Scholar] [CrossRef]
- Caforio, A.L.; Calabrese, F.; Angelini, A.; Tona, F.; Vinci, A.; Bottaro, S.; Ramondo, A.; Carturan, E.; Iliceto, S.; Thiene, G.; et al. A prospective study of biopsy-proven myocarditis: Prognostic relevance of clinical and aetiopathogenetic features at diagnosis. Eur. Heart J. 2007, 28, 1326–1333. [Google Scholar] [CrossRef] [Green Version]
- Ammirati, E.; Veronese, G.; Brambatti, M.; Merlo, M.; Cipriani, M.; Potena, L.; Sormani, P.; Aoki, T.; Sugimura, K.; Sawamura, A.; et al. Fulminant Versus Acute Nonfulminant Myocarditis in Patients With Left Ventricular Systolic Dysfunction. J. Am. Coll. Cardiol. 2019, 74, 299–311. [Google Scholar] [CrossRef]
- Ammirati, E.; Cipriani, M.; Lilliu, M.; Sormani, P.; Varrenti, M.; Raineri, C.; Petrella, D.; Garascia, A.; Pedrotti, P.; Roghi, A.; et al. Survival and Left Ventricular Function Changes in Fulminant Versus Nonfulminant Acute Myocarditis. Circulation 2017, 136, 529–545. [Google Scholar] [CrossRef]
- Błyszczuk, P. Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 2019, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Dennert, R.; Crijns, H.J.; Heymans, S. Acute viral myocarditis. Eur. Heart J. 2008, 29, 2073–2082. [Google Scholar] [CrossRef]
- Cooper, L.T.J. Myocarditis. N. Engl. J. Med. 2009, 360, 1526–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindermann, I.; Barth, C.; Mahfoud, F.; Ukena, C.; Lenski, M.; Yilmaz, A.; Klingel, K.; Kandolf, R.; Sechtem, U.; Cooper, L.T.; et al. Update on myocarditis. J. Am. Coll. Cardiol. 2012, 59, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Eriksson, U.; Lehtonen, J.; Cooper, L.T.J. The Quest for New Approaches in Myocarditis and Inflammatory Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2348–2364. [Google Scholar] [CrossRef]
- Lasrado, N.; Reddy, J. An overview of the immune mechanisms of viral myocarditis. Rev. Med. Virol. 2020, 30, 1–14. [Google Scholar] [CrossRef]
- Schultheiss, H.-P.; Kühl, U.; Cooper, L.T. The management of myocarditis. Eur. Heart J. 2011, 32, 2616–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquaro, G.D.; Perfetti, M.; Camastra, G.; Monti, L.; Dellegrottaglie, S.; Moro, C.; Pepe, A.; Todiere, G.; Lanzillo, C.; Scatteia, A.; et al. Cardiac MR With Late Gadolinium Enhancement in Acute Myocarditis With Preserved Systolic Function: ITAMY Study. J. Am. Coll. Cardiol. 2017, 70, 1977–1987. [Google Scholar] [CrossRef]
- White, J.A.; Hansen, R.; Abdelhaleem, A.; Mikami, Y.; Peng, M.; Rivest, S.; Satriano, A.; Dykstra, S.; Flewitt, J.; Heydari, B.; et al. Natural History of Myocardial Injury and Chamber Remodeling in Acute Myocarditis. Circ. Cardiovasc. Imaging 2019, 12, e008614. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Klingel, K.; Hohenadl, C.; Canu, A.; Albrecht, M.; Seemann, M.; Mall, G.; Kandolf, R. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: Quantitative analysis of virus replication, tissue damage, and inflammation. Proc. Natl. Acad. Sci. USA 1992, 89, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.G.; Sechtem, U.; Schulz-Menger, J.; Holmvang, G.; Alakija, P.; Cooper, L.T.; White, J.A.; Abdel-Aty, H.; Gutberlet, M.; Prasad, S.; et al. Cardiovascular magnetic resonance in myocarditis: A JACC White Paper. J. Am. Coll. Cardiol. 2009, 53, 1475–1487. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, V.M.; Schulz-Menger, J.; Holmvang, G.; Kramer, C.M.; Carbone, I.; Sechtem, U.; Kindermann, I.; Gutberlet, M.; Cooper, L.T.; Liu, P.; et al. Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation: Expert Recommendations. J. Am. Coll. Cardiol. 2018, 72, 3158–3176. [Google Scholar] [CrossRef]
- Francone, M.; Chimenti, C.; Galea, N.; Scopelliti, F.; Verardo, R.; Galea, R.; Carbone, I.; Catalano, C.; Fedele, F.; Frustaci, A. CMR sensitivity varies with clinical presentation and extent of cell necrosis in biopsy-proven acute myocarditis. JACC Cardiovasc. Imaging 2014, 7, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Bowles, N.E.; Ni, J.; Kearney, D.L.; Pauschinger, M.; Schultheiss, H.P.; McCarthy, R.; Hare, J.; Bricker, J.T.; Bowles, K.R.; Towbin, J.A. Detection of viruses in myocardial tissues by polymerase chain reaction. evidence of adenovirus as a common cause of myocarditis in children and adults. J. Am. Coll. Cardiol. 2003, 42, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühl, U.; Pauschinger, M.; Noutsias, M.; Seeberg, B.; Bock, T.; Lassner, D.; Poller, W.; Kandolf, R.; Schultheiss, H.P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005, 111, 887–893. [Google Scholar] [CrossRef] [Green Version]
- Klingel, K.; Sauter, M.; Bock, C.T.; Szalay, G.; Schnorr, J.J.; Kandolf, R. Molecular pathology of inflammatory cardiomyopathy. Med. Microbiol. Immunol. 2004, 193, 101–107. [Google Scholar] [CrossRef]
- Bock, C.T.; Klingel, K.; Kandolf, R. Human parvovirus B19-associated myocarditis. N. Engl. J. Med. 2010, 362, 1248–1249. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.; Liu, P.P.; Cooper, L.T.J. Myocarditis. Lancet 2012, 379, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Schultheiss, H.P.; Baumeier, C.; Aleshcheva, G.; Bock, C.T.; Escher, F. Viral Myocarditis-From Pathophysiology to Treatment. J. Clin. Med. 2021, 10, 5240. [Google Scholar] [CrossRef]
- Kindermann, I.; Kindermann, M.; Kandolf, R.; Klingel, K.; Bültmann, B.; Müller, T.; Lindinger, A.; Böhm, M. Predictors of outcome in patients with suspected myocarditis. Circulation 2008, 118, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, R.; Sakamoto, A.; Kawai, K.; Gianatti, A.; Pellegrini, D.; Nasr, A.; Kutys, B.; Guo, L.; Cornelissen, A.; Mori, M.; et al. Pathological Evidence for SARS-CoV-2 as a Cause of Myocarditis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 314–325. [Google Scholar] [CrossRef]
- Ozieranski, K.; Tyminska, A.; Jonik, S.; Marcolongo, R.; Baritussio, A.; Grabowski, M.; Filipiak, K.J.; Opolski, G.; Caforio, A.L.P. Clinically Suspected Myocarditis in the Course of Severe Acute Respiratory Syndrome Novel Coronavirus-2 Infection: Fact or Fiction? J. Card. Fail. 2021, 27, 92–96. [Google Scholar] [CrossRef]
- Pauschinger, M.; Phan, M.D.; Doerner, A.; Kuehl, U.; Schwimmbeck, P.L.; Poller, W.; Kandolf, R.; Schultheiss, H.P. Enteroviral RNA replication in the myocardium of patients with left ventricular dysfunction and clinically suspected myocarditis. Circulation 1999, 99, 889–895. [Google Scholar] [CrossRef] [Green Version]
- McManus, B.M.; Chow, L.H.; Wilson, J.E.; Anderson, D.R.; Gulizia, J.M.; Gauntt, C.J.; Klingel, K.E.; Beisel, K.W.; Kandolf, R. Direct myocardial injury by enterovirus: A central role in the evolution of murine myocarditis. Clin. Immunol. Immunopathol. 1993, 68, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Kallewaard, N.L.; Zhang, L.; Chen, J.W.; Guttenberg, M.; Sanchez, M.D.; Bergelson, J.M. Tissue-specific deletion of the coxsackievirus and adenovirus receptor protects mice from virus-induced pancreatitis and myocarditis. Cell Host Microbe 2009, 6, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Garmaroudi, F.S.; Marchant, D.; Hendry, R.; Luo, H.; Yang, D.; Ye, X.; Shi, J.; McManus, B.M. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015, 10, 629–653. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Zhao, W.; Wang, H.T.; Wu, K.Y.; Li, T.; Guo, X.K.; Tong, S.Q. Coxsackievirus B3-induced apoptosis and caspase-3. Cell Res. 2003, 13, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colston, J.T.; Chandrasekar, B.; Freeman, G.L. Expression of apoptosis-related proteins in experimental coxsackievirus myocarditis. Cardiovasc. Res. 1998, 38, 158–168. [Google Scholar] [CrossRef] [Green Version]
- Xiong, D.; Yajima, T.; Lim, B.-K.; Stenbit, A.; Dublin, A.; Dalton, N.D.; Summers-Torres, D.; Molkentin, J.D.; Duplain, H.; Wessely, R.; et al. Inducible Cardiac-Restricted Expression of Enteroviral Protease 2A Is Sufficient to Induce Dilated Cardiomyopathy. Circulation 2007, 115, 94–102. [Google Scholar] [CrossRef]
- Badorff, C.; Lee, G.-H.; Lamphear, B.J.; Martone, M.E.; Campbell, K.P.; Rhoads, R.E.; Knowlton, K.U. Enteroviral protease 2A cleaves dystrophin: Evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat. Med. 1999, 5, 320–326. [Google Scholar] [CrossRef]
- Liu, P.; Aitken, K.; Kong, Y.-Y.; Opavsky, M.A.; Martino, T.; Dawood, F.; Wen, W.-H.; Kozieradzki, I.; Bachmaier, K.; Straus, D.; et al. The tyrosine kinase p56lck is essential in coxsackievirus B3-mediated heart disease. Nat. Med. 2000, 6, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Mombaerts, P.; Iacomini, J.; Johnson, R.S.; Herrup, K.; Tonegawa, S.; Papaioannou, V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992, 68, 869–877. [Google Scholar] [CrossRef]
- Young, N.S.; Brown, K.E. Parvovirus B19. N. Engl. J. Med. 2004, 350, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Landry, M.L. Parvovirus B19. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Bültmann, B.D.; Klingel, K.; Sotlar, K.; Bock, C.T.; Baba, H.A.; Sauter, M.; Kandolf, R. Fatal parvovirus B19-associated myocarditis clinically mimicking ischemic heart disease: An endothelial cell-mediated disease. Hum. Pathol. 2003, 34, 92–95. [Google Scholar] [CrossRef]
- Bültmann, B.D.; Sotlar, K.; Klingel, K. Parvovirus B19. N. Engl. J. Med. 2004, 350, 2006–2007. [Google Scholar] [PubMed]
- Zakrzewska, K.; Cortivo, R.; Tonello, C.; Panfilo, S.; Abatangelo, G.; Giuggioli, D.; Ferri, C.; Corcioli, F.; Azzi, A. Human parvovirus B19 experimental infection in human fibroblasts and endothelial cells cultures. Virus Res. 2005, 114, 1–5. [Google Scholar] [CrossRef]
- Brown, K.E.; Anderson, S.M.; Young, N.S. Erythrocyte P antigen: Cellular receptor for B19 parvovirus. Science 1993, 262, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Munakata, Y.; Saito-Ito, T.; Kumura-Ishii, K.; Huang, J.; Kodera, T.; Ishii, T.; Hirabayashi, Y.; Koyanagi, Y.; Sasaki, T. Ku80 autoantigen as a cellular coreceptor for human parvovirus B19 infection. Blood 2005, 106, 3449–3456. [Google Scholar] [CrossRef]
- Verdonschot, J.; Hazebroek, M.; Merken, J.; Debing, Y.; Dennert, R.; Brunner-La Rocca, H.P.; Heymans, S. Relevance of cardiac parvovirus B19 in myocarditis and dilated cardiomyopathy: Review of the literature. Eur. J. Heart Fail. 2016, 18, 1430–1441. [Google Scholar] [CrossRef]
- Weigel-Kelley, K.A.; Yoder, M.C.; Srivastava, A. Alpha5beta1 integrin as a cellular coreceptor for human parvovirus B19: Requirement of functional activation of beta1 integrin for viral entry. Blood 2003, 102, 3927–3933. [Google Scholar] [CrossRef] [PubMed]
- Duechting, A.; Tschöpe, C.; Kaiser, H.; Lamkemeyer, T.; Tanaka, N.; Aberle, S.; Lang, F.; Torresi, J.; Kandolf, R.; Bock, C.T. Human parvovirus B19 NS1 protein modulates inflammatory signaling by activation of STAT3/PIAS3 in human endothelial cells. J. Virol. 2008, 82, 7942–7952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Ishii, K.K.; Munakata, Y.; Saitoh, T.; Kaku, M.; Sasaki, T. Regulation of tumor necrosis factor alpha promoter by human parvovirus B19 NS1 through activation of AP-1 and AP-2. J. Virol. 2002, 76, 5395–5403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, T.C.; Tzang, B.S.; Huang, C.N.; Lee, Y.J.; Liu, G.Y.; Chen, M.C.; Tsay, G.J. Increased expression and secretion of interleukin-6 in human parvovirus B19 non-structural protein (NS1) transfected COS-7 epithelial cells. Clin. Exp. Immunol. 2006, 144, 152–157. [Google Scholar] [CrossRef]
- Lupescu, A.; Geiger, C.; Zahir, N.; Aberle, S.; Lang, P.A.; Kramer, S.; Wesselborg, S.; Kandolf, R.; Foller, M.; Lang, F.; et al. Inhibition of Na+/H+ exchanger activity by parvovirus B19 protein NS1. Cell. Physiol. Biochem. 2009, 23, 211–220. [Google Scholar] [CrossRef]
- Dorsch, S.; Liebisch, G.; Kaufmann, B.; von Landenberg, P.; Hoffmann, J.H.; Drobnik, W.; Modrow, S. The VP1 unique region of parvovirus B19 and its constituent phospholipase A2-like activity. J. Virol. 2002, 76, 2014–2018. [Google Scholar] [CrossRef] [Green Version]
- Lupescu, A.; Bock, C.T.; Lang, P.A.; Aberle, S.; Kaiser, H.; Kandolf, R.; Lang, F. Phospholipase A2 activity-dependent stimulation of Ca2+ entry by human parvovirus B19 capsid protein VP1. J. Virol. 2006, 80, 11370–11380. [Google Scholar] [CrossRef] [Green Version]
- Pozzuto, T.; von Kietzell, K.; Bock, T.; Schmidt-Lucke, C.; Poller, W.; Zobel, T.; Lassner, D.; Zeichhardt, H.; Weger, S.; Fechner, H. Transactivation of human parvovirus B19 gene expression in endothelial cells by adenoviral helper functions. Virology 2011, 411, 50–64. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.; Wong, S.; Zhi, N.; Qiu, J. The Genome of Human Parvovirus B19 Can Replicate in Nonpermissive Cells with the Help of Adenovirus Genes and Produces Infectious Virus. J. Virol. 2009, 83, 9541–9553. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, U.; Lassner, D.; Dorner, A.; Rohde, M.; Escher, F.; Seeberg, B.; Hertel, E.; Tschope, C.; Skurk, C.; Gross, U.M.; et al. A distinct subgroup of cardiomyopathy patients characterized by transcriptionally active cardiotropic erythrovirus and altered cardiac gene expression. Basic Res. Cardiol. 2013, 108, 372. [Google Scholar] [CrossRef]
- Pietsch, H.; Escher, F.; Aleshcheva, G.; Lassner, D.; Bock, C.T.; Schultheiss, H.P. Detection of parvovirus mRNAs as markers for viral activity in endomyocardial biopsy-based diagnosis of patients with unexplained heart failure. Sci. Rep. 2020, 10, 22354. [Google Scholar] [CrossRef]
- Escher, F.; Aleshcheva, G.; Pietsch, H.; Baumeier, C.; Gross, U.M.; Schrage, B.N.; Westermann, D.; Bock, C.T.; Schultheiss, H.P. Transcriptional Active Parvovirus B19 Infection Predicts Adverse Long-Term Outcome in Patients with Non-Ischemic Cardiomyopathy. Biomedicines 2021, 9, 1898. [Google Scholar] [CrossRef]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Luo, M.H. Herpesviruses: Epidemiology, pathogenesis, and interventions. Virol. Sin. 2017, 32, 347–348. [Google Scholar] [CrossRef] [Green Version]
- Häusler, M.; Sellhaus, B.; Scheithauer, S.; Gaida, B.; Kuropka, S.; Siepmann, K.; Panek, A.; Berg, W.; Teubner, A.; Ritter, K.; et al. Myocarditis in newborn wild-type BALB/c mice infected with the murine gamma herpesvirus MHV-68. Cardiovasc. Res. 2007, 76, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankuweit, S.; Klingel, K. Viral myocarditis: From experimental models to molecular diagnosis in patients. Heart Fail. Rev. 2013, 18, 683–702. [Google Scholar] [CrossRef]
- Lenzo, J.C.; Fairweather, D.; Cull, V.; Shellam, G.R.; Lawson, C.M.J. Characterisation of murine cytomegalovirus myocarditis: Cellular infiltration of the heart and virus persistence. J. Mol. Cell. Cardiol. 2002, 34, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Eliassen, E.; Krueger, G.R.; Das, B.B. Human herpesvirus 6-induced inflammatory cardiomyopathy in immunocompetent children. Ann. Pediatr. Cardiol. 2017, 10, 259–268. [Google Scholar] [PubMed]
- Kühl, U.; Lassner, D.; Wallaschek, N.; Gross, U.M.; Krueger, G.R.; Seeberg, B.; Kaufer, B.B.; Escher, F.; Poller, W.; Schultheiss, H.P. Chromosomally integrated human herpesvirus 6 in heart failure: Prevalence and treatment. Eur. J. Heart Fail. 2015, 17, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, T.J.; Bhave, N.M.; Allen, L.A.; Chung, E.H.; Spatz, E.S.; Ammirati, E.; Baggish, A.L.; Bozkurt, B.; Cornwell, W.K., III; Harmon, K.G.; et al. 2022 ACC Expert Consensus Decision Pathway on Cardiovascular Sequelae of COVID-19 in Adults: Myocarditis and Other Myocardial Involvement, Post-Acute Sequelae of SARS-CoV-2 Infection, and Return to Play: A Report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2022, 79, 1717–1756. [Google Scholar] [PubMed]
- Lala, A.; Johnson, K.W.; Januzzi, J.L.; Russak, A.J.; Paranjpe, I.; Richter, F.; Zhao, S.; Somani, S.; Van Vleck, T.; Vaid, A.; et al. Prevalence and Impact of Myocardial Injury in Patients Hospitalized With COVID-19 Infection. J. Am. Coll. Cardiol. 2020, 76, 533–546. [Google Scholar] [CrossRef]
- Basso, C.; Leone, O.; Rizzo, S.; De Gaspari, M.; van der Wal, A.C.; Aubry, M.C.; Bois, M.C.; Lin, P.T.; Maleszewski, J.J.; Stone, J.R. Pathological features of COVID-19-associated myocardial injury: A multicentre cardiovascular pathology study. Eur. Heart J. 2020, 41, 3827–3835. [Google Scholar] [CrossRef]
- Collet, J.P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2021, 42, 1289–1367. [Google Scholar] [CrossRef]
- Artico, J.; Shiwani, H.; Moon, J.C.; Gorecka, M.; McCann, G.P.; Roditi, G.; Morrow, A.; Mangion, K.; Lukaschuk, E.; Shanmuganathan, M.; et al. Myocardial Involvement After Hospitalization for COVID-19 Complicated by Troponin Elevation: A Prospective, Multicenter, Observational Study. Circulation 2023, 147, 364–374. [Google Scholar] [CrossRef]
- Castiello, T.; Georgiopoulos, G.; Finocchiaro, G.; Claudia, M.; Gianatti, A.; Delialis, D.; Aimo, A.; Prasad, S. COVID-19 and myocarditis: A systematic review and overview of current challenges. Heart Fail. Rev. 2022, 27, 251–261. [Google Scholar] [CrossRef]
- Gauchotte, G.; Venard, V.; Segondy, M.; Cadoz, C.; Esposito-Fava, A.; Barraud, D.; Louis, G. SARS-CoV-2 fulminant myocarditis: An autopsy and histopathological case study. Int. J. Legal. Med. 2021, 135, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Albert, C.L.; Carmona-Rubio, A.E.; Weiss, A.J.; Procop, G.G.; Starling, R.C.; Rodriguez, E.R. The Enemy Within. Circulation 2020, 142, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.L.; Dmytrenko, O.; Greenberg, L.; Bredemeyer, A.L.; Ma, P.; Liu, J.; Penna, V.; Winkler, E.S.; Sviben, S.; Brooks, E.; et al. SARS-CoV-2 Infects Human Engineered Heart Tissues and Models COVID-19 Myocarditis. JACC Basic Transl. Sci. 2021, 6, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Sala, S.; Peretto, G.; Gramegna, M.; Palmisano, A.; Villatore, A.; Vignale, D.; De Cobelli, F.; Tresoldi, M.; Cappelletti, A.M.; Basso, C.; et al. Acute myocarditis presenting as a reverse Tako-Tsubo syndrome in a patient with SARS-CoV-2 respiratory infection. Eur. Heart J. 2020, 41, 1861–1862. [Google Scholar] [CrossRef] [Green Version]
- Bojkova, D.; Wagner, J.U.G.; Shumliakivska, M.; Aslan, G.S.; Saleem, U.; Hansen, A.; Luxán, G.; Günther, S.; Pham, M.D.; Krishnan, J.; et al. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. Cardiovasc. Res. 2020, 116, 2207–2215. [Google Scholar] [CrossRef]
- Pollack, A.; Kontorovich, A.R.; Fuster, V.; Dec, G.W. Viral myocarditis—Diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 2015, 12, 670–680. [Google Scholar] [CrossRef]
- Opavsky, M.A.; Penninger, J.; Aitken, K.; Wen, W.H.; Dawood, F.; Mak, T.; Liu, P. Susceptibility to myocarditis is dependent on the response of alphabeta T lymphocytes to coxsackieviral infection. Circ. Res. 1999, 85, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Fukuoka, M.; Li, G.; Liu, Y.; Chen, M.; Konviser, M.; Chen, X.; Opavsky, M.A.; Liu, P.P. Regulatory T Cells Protect Mice Against Coxsackievirus-Induced Myocarditis Through the Transforming Growth Factor β–Coxsackie-Adenovirus Receptor Pathway. Circulation 2010, 121, 2624–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caforio, A.L.; Marcolongo, R.; Jahns, R.; Fu, M.; Felix, S.B.; Iliceto, S. Immune-mediated and autoimmune myocarditis: Clinical presentation, diagnosis and management. Heart Fail. Rev. 2013, 18, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.R. Learning from myocarditis: Mimicry, chaos and black holes. F1000Prime Rep. 2014, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Maisch, B. Cardio-Immunology of Myocarditis: Focus on Immune Mechanisms and Treatment Options. Front. Cardiovasc. Med. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Fu, Z. Roles of Host Immunity in Viral Myocarditis and Dilated Cardiomyopathy. J. Immunol. Res. 2018, 2018, 5301548. [Google Scholar] [CrossRef]
- Xuan, Y.; Chen, C.; Wen, Z.; Wang, D.W. The Roles of Cardiac Fibroblasts and Endothelial Cells in Myocarditis. Front. Cardiovasc. Med. 2022, 9, 882027. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Li, J.; Savvatis, K.; Klingel, K.; Blankenberg, S.; Tschöpe, C.; Westermann, D. Cardiac fibroblasts aggravate viral myocarditis: Cell specific coxsackievirus B3 replication. Mediat. Inflamm. 2014, 2014, 519528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühl, U.; Pauschinger, M.; Seeberg, B.; Lassner, D.; Noutsias, M.; Poller, W.; Schultheiss, H.P. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 2005, 112, 1965–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figulla, H.R.; Stille-Siegener, M.; Mall, G.; Heim, A.; Kreuzer, H. Myocardial enterovirus infection with left ventricular dysfunction: A benign disease compared with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 1995, 25, 1170–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Why, H.J.; Meany, B.T.; Richardson, P.J.; Olsen, E.G.; Bowles, N.E.; Cunningham, L.; Freeke, C.A.; Archard, L.C. Clinical and prognostic significance of detection of enteroviral RNA in the myocardium of patients with myocarditis or dilated cardiomyopathy. Circulation 1994, 89, 2582–2589. [Google Scholar] [CrossRef] [Green Version]
- Schultheiss, H.-P.; Piper, C.; Sowade, O.; Waagstein, F.; Kapp, J.-F.; Wegscheider, K.; Groetzbach, G.; Pauschinger, M.; Escher, F.; Arbustini, E.; et al. Betaferon in chronic viral cardiomyopathy (BICC) trial: Effects of interferon-β treatment in patients with chronic viral cardiomyopathy. Clin. Res. Cardiol. 2016, 105, 763–773. [Google Scholar] [CrossRef]
- Kühl, U.; Lassner, D.; von Schlippenbach, J.; Poller, W.; Schultheiss, H.-P. Interferon-Beta Improves Survival in Enterovirus-Associated Cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 1295–1296. [Google Scholar] [CrossRef] [Green Version]
- Kühl, U.; Pauschinger, M.; Schwimmbeck, P.L.; Seeberg, B.; Lober, C.; Noutsias, M.; Poller, W.; Schultheiss, H.P. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 2003, 107, 2793–2798. [Google Scholar] [CrossRef] [Green Version]
- McNamara, D.M.; Holubkov, R.; Starling, R.C.; Dec, G.W.; Loh, E.; Torre-Amione, G.; Gass, A.; Janosko, K.; Tokarczyk, T.; Kessler, P.; et al. Controlled Trial of Intravenous Immune Globulin in Recent-Onset Dilated Cardiomyopathy. Circulation 2001, 103, 2254–2259. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Sun, Y.; Su, G.; Li, Y.; Shuai, X. Intravenous Immunoglobulin Therapy for Acute Myocarditis in Children and Adults A Meta-Analysis. Int. Heart J. 2019, 60, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Yen, C.-Y.; Hung, M.-C.; Wong, Y.-C.; Chang, C.-Y.; Lai, C.-C.; Wu, K.-G. Role of intravenous immunoglobulin therapy in the survival rate of pediatric patients with acute myocarditis: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 10459. [Google Scholar] [CrossRef] [Green Version]
- Krueger, G.R.; Ablashi, D.V. Human herpesvirus-6: A short review of its biological behavior. Intervirology 2003, 46, 257–269. [Google Scholar] [CrossRef]
- Tschöpe, C.; Elsanhoury, A.; Schlieker, S.; Van Linthout, S.; Kühl, U. Immunosuppression in inflammatory cardiomyopathy and parvovirus B19 persistence. Eur. J. Heart Fail. 2019, 21, 1468–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Developed by the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) Endorsed by the Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [PubMed]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Circulation 2018, 138, e272–e391. [Google Scholar] [PubMed] [Green Version]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef]
- Hidron, A.; Vogenthaler, N.; Santos-Preciado, J.I.; Rodriguez-Morales, A.J.; Franco-Paredes, C.; Rassi, A.J. Cardiac involvement with parasitic infections. Clin. Microbiol. Rev. 2010, 23, 324–349. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.L.; Nunes, M.P.; Teixeira, M.M.; Rocha, M.O.C. Diagnosis and management of Chagas disease and cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 576–589. [Google Scholar] [CrossRef]
- World Health Organization. Chagas Disease (Also Known as American Trypanosomiasis). Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 12 October 2022).
- Blum, J.A.; Zellweger, M.J.; Burri, C.; Hatz, C. Cardiac involvement in African and American trypanosomiasis. Lancet Infect. Dis. 2008, 8, 631–641. [Google Scholar] [CrossRef]
- Moolani, Y.; Bukhman, G.; Hotez, P.J. Neglected Tropical Diseases as Hidden Causes of Cardiovascular Disease. PLOS Negl. Trop. Dis. 2012, 6, e1499. [Google Scholar] [CrossRef]
- Nunes, M.C.P.; Beaton, A.; Acquatella, H.; Bern, C.; Bolger, A.F.; Echeverría, L.E.; Dutra, W.O.; Gascon, J.; Morillo, C.A.; Oliveira-Filho, J.; et al. Chagas Cardiomyopathy: An Update of Current Clinical Knowledge and Management: A Scientific Statement From the American Heart Association. Circulation 2018, 138, e169–e209. [Google Scholar] [CrossRef] [PubMed]
- Bonney, K.M.; Luthringer, D.J.; Kim, S.A.; Garg, N.J.; Engman, D.M. Pathology and Pathogenesis of Chagas Heart Disease. Annu. Rev. Pathol. 2019, 14, 421–447. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.C.; Andrews, N.W. Host cell invasion by Trypanosoma cruzi: A unique strategy that promotes persistence. FEMS Microbiol. Rev. 2012, 36, 734–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Añez, N.; Carrasco, H.; Parada, H.; Crisante, G.; Rojas, A.; Fuenmayor, C.; Gonzalez, N.; Percoco, G.; Borges, R.; Guevara, P.; et al. Myocardial parasite persistence in chronic chagasic patients. Am. J. Trop. Med. Hyg. 1999, 60, 726–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochitte, C.E.; Oliveira, P.F.; Andrade, J.M.; Ianni, B.M.; Parga, J.R.; Avila, L.F.; Kalil-Filho, R.; Mady, C.; Meneghetti, J.C.; Lima, J.A.; et al. Myocardial delayed enhancement by magnetic resonance imaging in patients with Chagas’ disease: A marker of disease severity. J. Am. Coll. Cardiol. 2005, 46, 1553–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acquatella, H.; Asch, F.M.; Barbosa, M.M.; Barros, M.; Bern, C.; Cavalcante, J.L.; Echeverria Correa, L.E.; Lima, J.; Marcus, R.; Marin-Neto, J.A.; et al. Recommendations for Multimodality Cardiac Imaging in Patients with Chagas Disease: A Report from the American Society of Echocardiography in Collaboration With the InterAmerican Association of Echocardiography (ECOSIAC) and the Cardiovascular Imaging Department of the Brazilian Society of Cardiology (DIC-SBC). J. Am. Soc. Echocardiogr. 2018, 31, 3–25. [Google Scholar]
- Cianciulli, T.F.; Lax, J.A.; Saccheri, M.C.; Papantoniou, A.; Morita, L.A.; Prado, N.G.; Dorelle, A.N.; Riarte, A.R.; Prezioso, H.A. Early detection of left ventricular diastolic dysfunction in Chagas’ disease. Cardiovasc. Ultrasound 2006, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Rassi, A.J.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Rossi, M.A.; Ramos, S.G.; Bestetti, R.B. Chagas’ heart disease: Clinical-pathological correlation. Front. Biosci.-Landmark 2003, 8, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Marin-Neto, J.A.; Cunha-Neto, E.; Maciel, B.C.; Simões, M.V. Pathogenesis of Chronic Chagas Heart Disease. Circulation 2007, 115, 1109–1123. [Google Scholar] [CrossRef]
- Cunha-Neto, E.; Bilate, A.M.; Hyland, K.V.; Fonseca, S.G.; Kalil, J.; Engman, D.M.; Cunha-Neto, E.; Bilate, A.M.; Hyland, K.V.; Fonseca, S.G.; et al. Induction of cardiac autoimmunity in Chagas heart disease: A case for molecular mimicry. Autoimmunity 2006, 39, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Villagómez, D.; McCurley, T.L.; Vnencak-Jones, C.L.; Correa-Oliveira, R.; Colley, D.G.; Carter, C.E. Polymerase chain reaction amplification of three different Trypanosoma cruzi DNA sequences from human chagasic cardiac tissue. Am. J. Trop. Med. Hyg. 1998, 59, 563–570. [Google Scholar] [CrossRef] [Green Version]
- Palomino, S.A.; Aiello, V.D.; Higuchi, M.L. Systematic mapping of hearts from chronic chagasic patients: The association between the occurrence of histopathological lesions and Trypanosoma cruzi antigens. Ann. Trop. Med. Parasitol. 2000, 94, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.O.C.; Ribeiro, A.L.P.; Teixeira, M.M. Clinical management of chronic Chagas cardiomyopathy. Front. Biosci.-Landmark 2003, 8, 44–54. [Google Scholar]
- Bestetti, R.B.; Otaviano, A.P.; Fantini, J.P.; Cardinalli-Neto, A.; Nakazone, M.A.; Nogueira, P.R. Prognosis of patients with chronic systolic heart failure: Chagas disease versus systemic arterial hypertension. Int. J. Cardiol. 2013, 168, 2990–2991. [Google Scholar] [CrossRef]
- Vilas Boas, L.G.; Bestetti, R.B.; Otaviano, A.P.; Cardinalli-Neto, A.; Nogueira, P.R. Outcome of Chagas cardiomyopathy in comparison to ischemic cardiomyopathy. Int. J. Cardiol. 2013, 167, 486–490. [Google Scholar] [CrossRef]
- Pereira Nunes Mdo, C.; Barbosa, M.M.; Ribeiro, A.L.; Amorim Fenelon, L.M.; Rocha, M.O. Predictors of mortality in patients with dilated cardiomyopathy: Relevance of chagas disease as an etiological factor. Rev. Esp. Cardiol. 2010, 63, 788–797. [Google Scholar]
- Morillo, C.A.; Marin-Neto, J.A.; Avezum, A.; Sosa-Estani, S.; Rassi, A.; Rosas, F.; Villena, E.; Quiroz, R.; Bonilla, R.; Britto, C.; et al. Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. N. Engl. J. Med. 2015, 373, 1295–1306. [Google Scholar] [CrossRef] [Green Version]
- Koten, J.W.; De Raadt, P. Myocarditis in Trypanosoma rhodesiense infections. Trans. R. Soc. Trop. Med. Hyg. 1969, 63, 485–489. [Google Scholar] [CrossRef]
- Adams, J.H.; Haller, L.; Boa, F.Y.; Doua, F.; Dago, A.; Konian, K. Human African trypanosomiasis (T.b. gambiense): A study of 16 fatal cases of sleeping sickness with some observations on acute reactive arsenical encephalopathy. Neuropathol. Appl. Neurobiol. 1986, 12, 81–94. [Google Scholar] [CrossRef]
- Blum, J.A.; Burri, C.; Hatz, C.; Kazumba, L.; Mangoni, P.; Zellweger, M.J. Sleeping hearts: The role of the heart in sleeping sickness (human African trypanosomiasis). Trop. Med. Int. Health 2007, 12, 1422–1432. [Google Scholar] [CrossRef]
- Blum, A.; Mudji, J.; Grize, L.; Burri, C.; Zellweger, M.J.; Blum, J. Sleeping hearts: 12 years after a follow up study on cardiac findings due to sleeping sickness. One Health 2020, 11, 100182. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.C.; Constantine, M.; Ahmadi, A.; Shiau, C.; Chen, L.Y.C. Eosinophilic Myocarditis. Am. J. Med. Sci. 2017, 354, 486–492. [Google Scholar] [CrossRef]
- Brambatti, M.; Matassini, M.V.; Adler, E.D.; Klingel, K.; Camici, P.G.; Ammirati, E. Eosinophilic Myocarditis: Characteristics, Treatment, and Outcomes. J. Am. Coll. Cardiol. 2017, 70, 2363–2375. [Google Scholar] [CrossRef]
- Ferrero, P.; Piazza, I.; Lorini, L.F.; Senni, M. Epidemiologic and clinical profiles of bacterial myocarditis. Report of two cases and data from a pooled analysis. Indian Heart J. 2020, 72, 82–92. [Google Scholar] [CrossRef]
- Canter, C.E.; Simpson, K.E. Diagnosis and Treatment of Myocarditis in Children in the Current Era. Circulation 2014, 129, 115–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, C.; Baranchuk, A. Diagnosis and Treatment of Lyme Carditis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 717–726. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Adler, Y.; Agostini, C.; Allanore, Y.; Anastasakis, A.; Arad, M.; Böhm, M.; Charron, P.; Elliott, P.M.; Eriksson, U.; et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur. Heart J. 2017, 38, 2649–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruestle, K.; Hackner, K.; Kreye, G.; Heidecker, B. Autoimmunity in Acute Myocarditis: How Immunopathogenesis Steers New Directions for Diagnosis and Treatment. Curr. Cardiol. Rep. 2020, 22, 28. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Baldeviano, G.C.; Ligons, D.L.; Talor, M.V.; Barin, J.G.; Rose, N.R.; Cihakova, D. Susceptibility to autoimmune myocarditis is associated with intrinsic differences in CD4(+) T cells. Clin. Exp. Immunol. 2012, 169, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Bracamonte-Baran, W.; Čiháková, D. Cardiac Autoimmunity: Myocarditis. Adv. Exp. Med. Biol. 2017, 1003, 187–221. [Google Scholar]
- Baldeviano, G.C.; Barin, J.G.; Talor, M.V.; Srinivasan, S.; Bedja, D.; Zheng, D.; Gabrielson, K.; Iwakura, Y.; Rose, N.R.; Cihakova, D. Interleukin-17A Is Dispensable for Myocarditis but Essential for the Progression to Dilated Cardiomyopathy. Circ. Res. 2010, 106, 1646–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammirati, E.; Raimondi, F.; Piriou, N.; Sardo Infirri, L.; Mohiddin, S.A.; Mazzanti, A.; Shenoy, C.; Cavallari, U.A.; Imazio, M.; Aquaro, G.D.; et al. Acute Myocarditis Associated With Desmosomal Gene Variants. JACC Heart Fail. 2022, 10, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Lota, A.S.; Hazebroek, M.R.; Theotokis, P.; Wassall, R.; Salmi, S.; Halliday, B.P.; Tayal, U.; Verdonschot, J.; Meena, D.; Owen, R.; et al. Genetic Architecture of Acute Myocarditis and the Overlap With Inherited Cardiomyopathy. Circulation 2022, 146, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Narula, N.; Giuliani, L.; Di Toro, A. Genetic basis of myocarditis: Myth or reality? In Myocarditis: Pathogenesis, Diagnosis and Treatment; Caforio, A.L.P., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 45–89. [Google Scholar]
- Baggio, C.; Gagno, G.; Porcari, A.; Paldino, A.; Artico, J.; Castrichini, M.; Dal Ferro, M.; Bussani, R.; Merlo, M. Myocarditis: Which Role for Genetics? Curr. Cardiol. Rep. 2021, 23, 58. [Google Scholar] [CrossRef] [PubMed]
- Gil-Cruz, C.; Perez-Shibayama, C.; De Martin, A.; Ronchi, F.; van der Borght, K.; Niederer, R.; Onder, L.; Lütge, M.; Novkovic, M.; Nindl, V.; et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science 2019, 366, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, E.D.; Zucker, M.J.; Kramer, N. Giant cell myocarditis: Most fatal of autoimmune diseases. Semin. Arthritis Rheum. 2000, 30, 1–16. [Google Scholar] [CrossRef]
- Bang, V.; Ganatra, S.; Shah Sachin, P.; Dani Sourbha, S.; Neilan Tomas, G.; Thavendiranathan, P.; Resnic Frederic, S.; Piemonte Thomas, C.; Barac, A.; Patel, R.; et al. Management of Patients With Giant Cell Myocarditis. J. Am. Coll. Cardiol. 2021, 77, 1122–1134. [Google Scholar] [CrossRef]
- Thuny, F.; Naidoo, J.; Neilan, T.G. Cardiovascular complications of immune checkpoint inhibitors for cancer. Eur. Heart J. 2022, 43, 4458–4468. [Google Scholar] [CrossRef]
- Tan, S.; Day, D.; Nicholls, S.J.; Segelov, E. Immune Checkpoint Inhibitor Therapy in Oncology: Current Uses and Future Directions: JACC: CardioOncology State-of-the-Art Review. JACC Cardio Oncol. 2022, 4, 579–597. [Google Scholar] [CrossRef]
- Wei, S.C.; Meijers, W.C.; Axelrod, M.L.; Anang, N.A.S.; Screever, E.M.; Wescott, E.C.; Johnson, D.B.; Whitley, E.; Lehmann, L.; Courand, P.Y.; et al. A Genetic Mouse Model Recapitulates Immune Checkpoint Inhibitor-Associated Myocarditis and Supports a Mechanism-Based Therapeutic Intervention. Cancer Discov. 2021, 11, 614–625. [Google Scholar] [CrossRef]
- Moslehi, J.; Salem, J.E. Immune Checkpoint Inhibitor Myocarditis Treatment Strategies and Future Directions. JACC Cardio Oncol. 2022, 4, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, M.L.; Meijers, W.C.; Screever, E.M.; Qin, J.; Carroll, M.G.; Sun, X.; Tannous, E.; Zhang, Y.; Sugiura, A.; Taylor, B.C.; et al. T cells specific for α-myosin drive immunotherapy-related myocarditis. Nature 2022, 611, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Zamami, Y.; Niimura, T.; Okada, N.; Koyama, T.; Fukushima, K.; Izawa-Ishizawa, Y.; Ishizawa, K. Factors Associated With Immune Checkpoint Inhibitor-Related Myocarditis. JAMA Oncol. 2019, 5, 1635–1637. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, C.; Li, Y.; Qin, J.; Amancherla, K.; Jing, Y.; Hu, Q.; Liang, K.; Zhang, Z.; Ye, Y.; et al. Hormonal therapies up-regulate MANF and overcome female susceptibility to immune checkpoint inhibitor myocarditis. Sci. Transl. Med. 2022, 14, eabo1981. [Google Scholar] [CrossRef]
- Ameratunga, R.; Woon, S.T.; Sheppard, M.N.; Garland, J.; Ondruschka, B.; Wong, C.X.; Stewart, R.A.H.; Tatley, M.; Stables, S.R.; Tse, R.D. First Identified Case of Fatal Fulminant Necrotizing Eosinophilic Myocarditis Following the Initial Dose of the Pfizer-BioNTech mRNA COVID-19 Vaccine (BNT162b2, Comirnaty): An Extremely Rare Idiosyncratic Hypersensitivity Reaction. J. Clin. Immunol. 2022, 42, 441–447. [Google Scholar] [CrossRef]
- Ilonze, O.J.; Guglin, M.E. Myocarditis following COVID-19 vaccination in adolescents and adults: A cumulative experience of 2021. Heart Fail. Rev. 2022, 27, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.; McCain, J.; King, K.R.; Hong, K.; Aisagbonhi, O.; Adler, E.D.; Urey, M.A. Biopsy-Proven Giant Cell Myocarditis Following the COVID-19 Vaccine. Circ. Heart Fail. 2022, 15, e009321. [Google Scholar] [CrossRef] [PubMed]
- Patone, M.; Mei, X.W.; Handunnetthi, L.; Dixon, S.; Zaccardi, F.; Shankar-Hari, M.; Watkinson, P.; Khunti, K.; Harnden, A.; Coupland, C.A.C.; et al. Risk of Myocarditis After Sequential Doses of COVID-19 Vaccine and SARS-CoV-2 Infection by Age and Sex. Circulation 2022, 146, 743–754. [Google Scholar] [CrossRef]
- Heymans, S.; Cooper, L.T. Myocarditis after COVID-19 mRNA vaccination: Clinical observations and potential mechanisms. Nat. Rev. Cardiol. 2022, 19, 75–77. [Google Scholar] [CrossRef]
- Yonker, L.M.; Swank, Z.; Bartsch, Y.C.; Burns, M.D.; Kane, A.; Boribong, B.P.; Davis, J.P.; Loiselle, M.; Novak, T.; Senussi, Y.; et al. Circulating Spike Protein Detected in Post-COVID-19 mRNA Vaccine Myocarditis. Circulation 2023, 147, 867–876. [Google Scholar] [CrossRef]
- Marrama, D.; Mahita, J.; Sette, A.; Peters, B. Lack of evidence of significant homology of SARS-CoV-2 spike sequences to myocarditis-associated antigens. EBioMedicine 2022, 75, 103807. [Google Scholar] [CrossRef]
- Barmada, A.; Klein, J.; Ramaswamy, A.; Brodsky, N.N.; Jaycox, J.R.; Sheikha, H.; Jones, K.M.; Habet, V.; Campbell, M.; Sumida, T.S.; et al. Cytokinopathy with aberrant cytotoxic lymphocytes and profibrotic myeloid response in SARS-CoV-2 mRNA vaccine-associated myocarditis. Sci. Immunol. 2023, 8, eadh3455. [Google Scholar] [CrossRef]
- Wojnicz, R.; Nowalany-Kozielska, E.; Wojciechowska, C.; Glanowska, G.; Wilczewski, P.; Niklewski, T.; Zembala, M.; Poloński, L.; Rozek, M.M.; Wodniecki, J. Randomized, Placebo-Controlled Study for Immunosuppressive Treatment of Inflammatory Dilated Cardiomyopathy. Circulation 2001, 104, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Frustaci, A.; Chimenti, C.; Calabrese, F.; Pieroni, M.; Thiene, G.; Maseri, A. Immunosuppressive Therapy for Active Lymphocytic Myocarditis. Circulation 2003, 107, 857–863. [Google Scholar] [CrossRef] [Green Version]
- Frustaci, A.; Russo, M.A.; Chimenti, C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: The TIMIC study. Eur. Heart J. 2009, 30, 1995–2002. [Google Scholar] [CrossRef]
- Chimenti, C.; Russo, M.A.; Frustaci, A. Immunosuppressive therapy in virus-negative inflammatory cardiomyopathy: 20-year follow-up of the TIMIC trial. Eur. Heart J. 2022, 43, 3463–3473. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Cheng, G.Y.; Shan, Z.G.; Baritussio, A.; Lorenzoni, G.; Tyminska, A.; Ozieranski, K.; Iliceto, S.; Marcolongo, R.; Gregori, D.; et al. Efficacy of immunosuppressive therapy in myocarditis: A 30-year systematic review and meta analysis. Autoimmun. Rev. 2021, 20, 102710. [Google Scholar] [CrossRef] [PubMed]
- Ozierański, K.; Tymińska, A.; Marchel, M.; Januszkiewicz, Ł.; Maciejewski, C.; Główczyńska, R.; Marcolongo, R.; Caforio, A.L.; Wojnicz, R.; Mizia-Stec, K.; et al. A multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy of immunosuppression in biopsy-proven virus-negative myocarditis or inflammatory cardiomyopathy (IMPROVE-MC). Cardiol. J. 2022, 29, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Marcolongo, R.; Baritussio, A.; Gianstefani, S.; Cheng, C.-Y.; Iliceto, S.; Caforio, A.L.P. Clinical Management and Follow-Up of Myocarditis Patients on Immunosuppressive Therapy. In Myocarditis: Pathogenesis, Diagnosis and Treatment; Caforio, A.L.P., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 285–295. [Google Scholar]
- Cooper, L.T.J.; Berry, G.J.; Shabetai, R. Idiopathic giant-cell myocarditis--natural history and treatment. Multicenter Giant Cell Myocarditis Study Group Investigators. N. Engl. J. Med. 1997, 336, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Ekström, K.; Lehtonen, J.; Kandolin, R.; Räisänen-Sokolowski, A.; Salmenkivi, K.; Kupari, M. Incidence, Risk Factors, and Outcome of Life-Threatening Ventricular Arrhythmias in Giant Cell Myocarditis. Circ. Arrhythm. Electrophysiol. 2016, 9, e004559. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T.J.; Hare, J.M.; Tazelaar, H.D.; Edwards, W.D.; Starling, R.C.; Deng, M.C.; Menon, S.; Mullen, G.M.; Jaski, B.; Bailey, K.R.; et al. Usefulness of immunosuppression for giant cell myocarditis. Am. J. Cardiol. 2008, 102, 1535–1539. [Google Scholar] [CrossRef] [Green Version]
- Baritussio, A.; Giordani, A.S.; Rizzo, S.; Masiero, G.; Iliceto, S.; Marcolongo, R.; Caforio, A.L. Management of myocarditis in clinical practice. Minerva Cardiol. Angiol. 2022, 70, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; Yousaf, N.; Battisti, N.M.L.; Moslehi, J.; Larkin, J. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 2018, 19, e447–e458. [Google Scholar] [CrossRef]
- Awadalla, M.; Mahmood Syed, S.; Groarke John, D.; Hassan Malek, Z.O.; Nohria, A.; Rokicki, A.; Murphy Sean, P.; Mercaldo Nathaniel, D.; Zhang, L.; Zlotoff Daniel, A.; et al. Global Longitudinal Strain and Cardiac Events in Patients With Immune Checkpoint Inhibitor-Related Myocarditis. J. Am. Coll. Cardiol. 2020, 75, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Awadalla, M.; Mahmood, S.S.; Nohria, A.; Hassan, M.Z.O.; Thuny, F.; Zlotoff, D.A.; Murphy, S.P.; Stone, J.R.; Golden, D.L.A.; et al. Cardiovascular magnetic resonance in immune checkpoint inhibitor-associated myocarditis. Eur. Heart J. 2020, 41, 1733–1743. [Google Scholar] [CrossRef]
- Power, J.R.; Alexandre, J.; Choudhary, A.; Ozbay, B.; Hayek, S.; Asnani, A.; Tamura, Y.; Aras, M.; Cautela, J.; Thuny, F.; et al. Electrocardiographic Manifestations of Immune Checkpoint Inhibitor Myocarditis. Circulation 2021, 144, 1521–1523. [Google Scholar] [CrossRef]
- Salem, J.-E.; Manouchehri, A.; Moey, M.; Lebrun-Vignes, B.; Bastarache, L.; Pariente, A.; Gobert, A.; Spano, J.-P.; Balko, J.M.; Bonaca, M.P.; et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: An observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018, 19, 1579–1589. [Google Scholar] [CrossRef]
- Lyon, A.R.; López-Fernández, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on cardio-oncology developed in collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [CrossRef]
- Salem, J.E.; Allenbach, Y.; Vozy, A.; Brechot, N.; Johnson, D.B.; Moslehi, J.J.; Kerneis, M. Abatacept for Severe Immune Checkpoint Inhibitor-Associated Myocarditis. N. Engl. J. Med. 2019, 380, 2377–2379. [Google Scholar] [CrossRef]
- Salem, J.E.; Bretagne, M.; Abbar, B.; Leonard-Louis, S.; Ederhy, S.; Redheuil, A.; Boussouar, S.; Nguyen, L.S.; Procureur, A.; Stein, F.; et al. Abatacept/Ruxolitinib and Screening for Concomitant Respiratory Muscle Failure to Mitigate Fatality of Immune-Checkpoint Inhibitor Myocarditis. Cancer Discov. 2023, 13, 1100–1115. [Google Scholar] [CrossRef]
- Ammirati, E.; Moslehi, J.J. Diagnosis and Treatment of Acute Myocarditis: A Review. JAMA 2023, 329, 1098–1113. [Google Scholar] [CrossRef]
- Kracalik, I.; Oster, M.E.; Broder, K.R.; Cortese, M.M.; Glover, M.; Shields, K.; Creech, C.B.; Romanson, B.; Novosad, S.; Soslow, J.; et al. Outcomes at least 90 days since onset of myocarditis after mRNA COVID-19 vaccination in adolescents and young adults in the USA: A follow-up surveillance study. Lancet Child Adolesc. Health 2022, 6, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Lupi, L.; Palazzini, M.; Ciabatti, M.; Rossi, V.A.; Gentile, P.; Uribarri, A.; Vecchio, C.R.; Nassiacos, D.; Cereda, A.; et al. Outcome and Morphofunctional Changes on Cardiac Magnetic Resonance in Patients With Acute Myocarditis Following mRNA COVID-19 Vaccination. Circ. Heart Fail. 2023, e010315. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh, N.; Steckman, D.A.; Sauer, W.H.; Judson, M.A. Pathophysiology and clinical management of cardiac sarcoidosis. Nat. Rev. Cardiol. 2015, 12, 278–288. [Google Scholar] [CrossRef]
- Trivieri, M.G.; Spagnolo, P.; Birnie, D.; Liu, P.; Drake, W.; Kovacic, J.C.; Baughman, R.; Fayad, Z.A.; Judson, M.A. Challenges in Cardiac and Pulmonary Sarcoidosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1878–1901. [Google Scholar] [CrossRef] [PubMed]
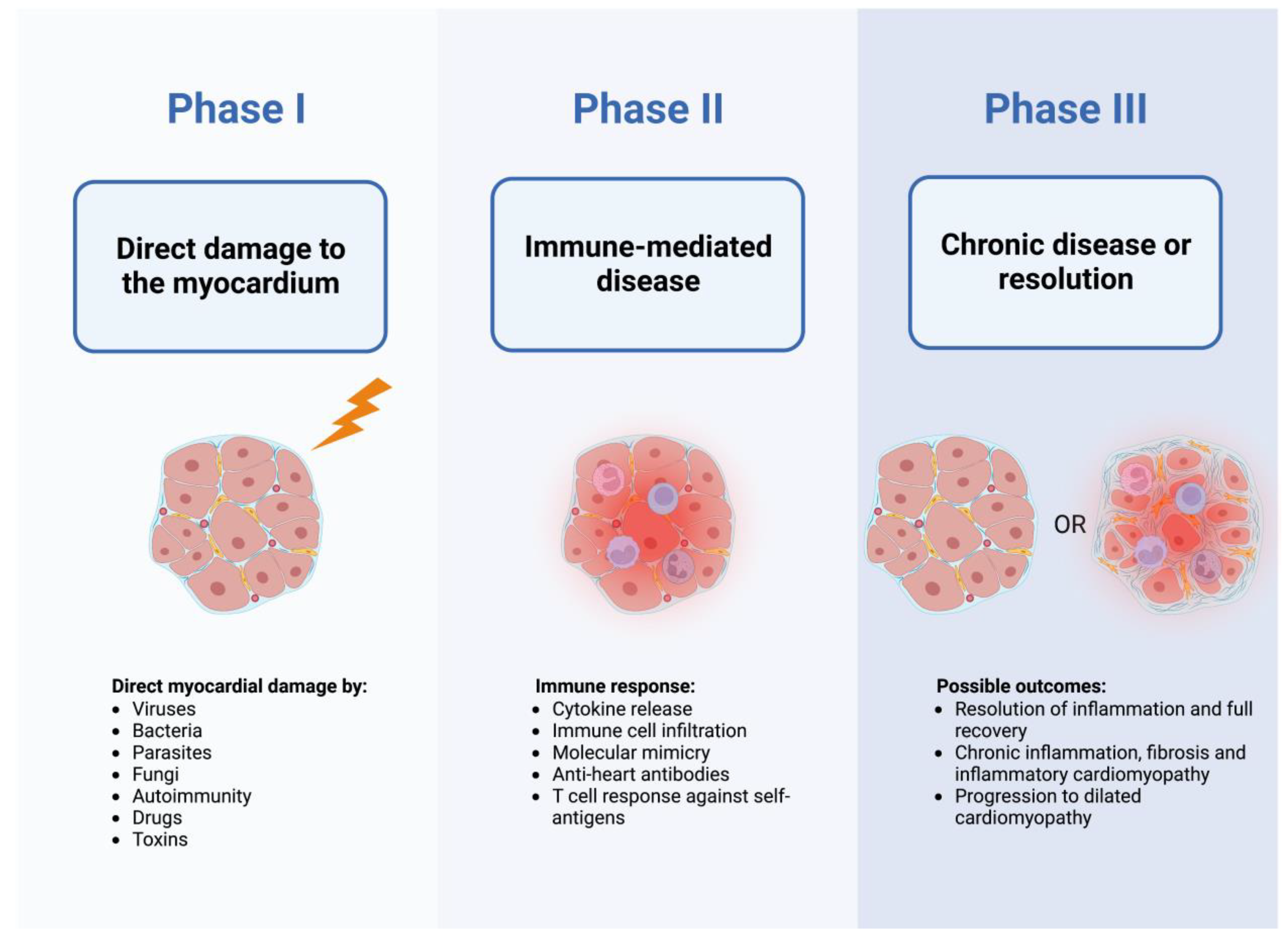


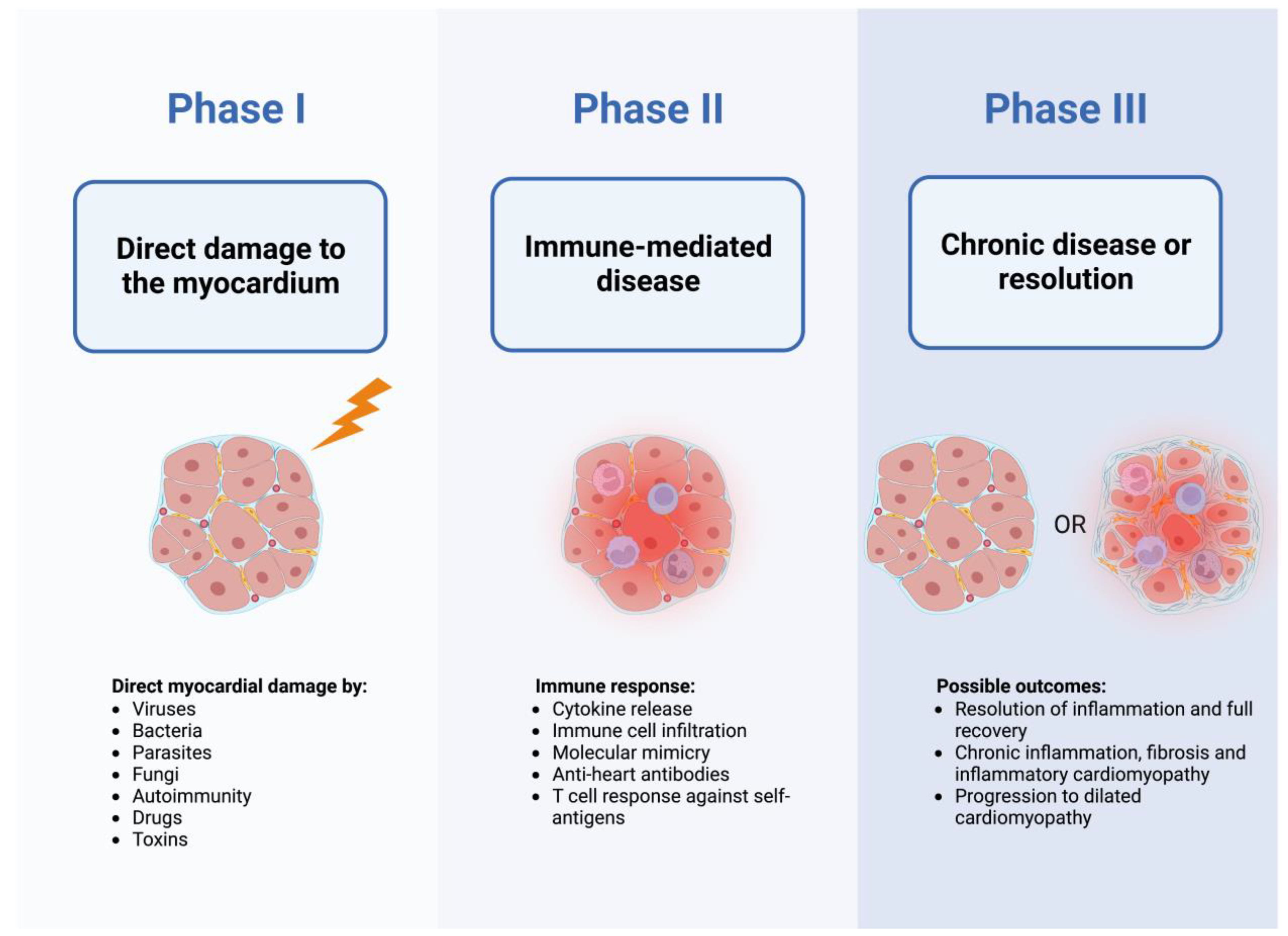{kind=link}
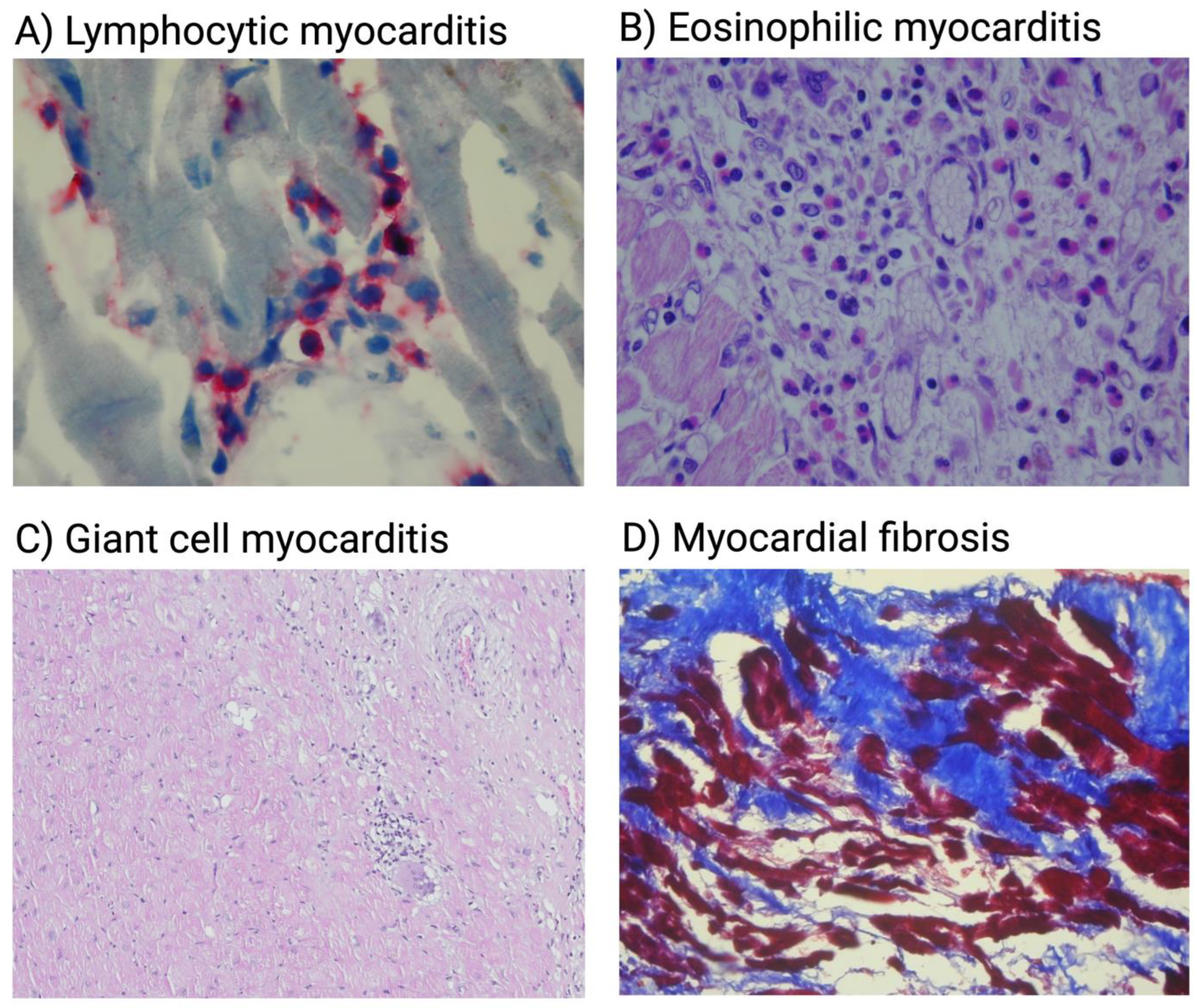{kind=link}
| Virus | Tropism or Tissue Toxicity | Genome |
|---|---|---|
| Parvovirus B19 | Vasculotropic | ssDNA |
| Enteroviruses | Cardiotropic | (+) ssRNA |
| Adenoviruses | dsDNA | |
| Human herpesvirus type 6 | Lymphotropic | dsDNA |
| Epstein–Barr Virus | ||
| Cytomegalovirus | ||
| Hepatitis C virus | Cardiotoxic | (+) ssRNA |
| Human immunodeficiency virus | ||
| Influenza viruses | ||
| SARS-CoV-2 | ACE2-tropic | (+) ssRNA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brociek, E.; Tymińska, A.; Giordani, A.S.; Caforio, A.L.P.; Wojnicz, R.; Grabowski, M.; Ozierański, K. Myocarditis: Etiology, Pathogenesis, and Their Implications in Clinical Practice. Biology 2023, 12, 874. https://doi.org/10.3390/biology12060874
Brociek E, Tymińska A, Giordani AS, Caforio ALP, Wojnicz R, Grabowski M, Ozierański K. Myocarditis: Etiology, Pathogenesis, and Their Implications in Clinical Practice. Biology. 2023; 12(6):874. https://doi.org/10.3390/biology12060874
Chicago/Turabian StyleBrociek, Emil, Agata Tymińska, Andrea Silvio Giordani, Alida Linda Patrizia Caforio, Romuald Wojnicz, Marcin Grabowski, and Krzysztof Ozierański. 2023. "Myocarditis: Etiology, Pathogenesis, and Their Implications in Clinical Practice" Biology 12, no. 6: 874. https://doi.org/10.3390/biology12060874
APA StyleBrociek, E., Tymińska, A., Giordani, A. S., Caforio, A. L. P., Wojnicz, R., Grabowski, M., & Ozierański, K. (2023). Myocarditis: Etiology, Pathogenesis, and Their Implications in Clinical Practice. Biology, 12(6), 874. https://doi.org/10.3390/biology12060874