Simple Summary
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) technology has become an important research tool for targeted mutagenesis in plants. However, the construction of mutants in the model legume Medicago truncatula remains challenging as it requires the combination of an efficient CRISPR/Cas9-mediated gene editing system for explants and specific tissue culture techniques for regeneration to whole plants. Here, we describe the construction of M. truncatula mutants using the Agrobacterium tumefaciens-mediated transformation of leaf explants. Regeneration to whole plants was performed at high efficiency (80%), and a gene editing efficiency of up to 70% was reached in the obtained plants. CRISPR/Cas9-mediated gene editing was achieved for three M. truncatula genes. Three mutant lines with knockout mutations in the LysM receptor kinase gene MtLYK10 were further characterized. Inoculation experiments with nitrogen-fixing Sinorhizobium meliloti bacteria indicated that root nodule formation was impaired in the constructed MtLYK10 mutants. In conclusion, we show that the described CRISPR/Cas9 system is suitable for efficient gene editing in M. truncatula and that MtLYK10 plays a role in nodule symbiosis.
Abstract
CRISPR/Cas9 systems are commonly used for plant genome editing; however, the generation of homozygous mutant lines in Medicago truncatula remains challenging. Here, we present a CRISPR/Cas9-based protocol that allows the efficient generation of M. truncatula mutants. Gene editing was performed for the LysM receptor kinase gene MtLYK10 and two major facilitator superfamily transporter genes. The functionality of CRISPR/Cas9 vectors was tested in Nicotiana benthamiana leaves by editing a co-transformed GUSPlus gene. Transformed M. truncatula leaf explants were regenerated to whole plants at high efficiency (80%). An editing efficiency (frequency of mutations at a given target site) of up to 70% was reached in the regenerated plants. Plants with MtLYK10 knockout mutations were propagated, and three independent homozygous mutant lines were further characterized. No off-target mutations were identified in these lyk10 mutants. Finally, the lyk10 mutants and wild-type plants were compared with respect to the formation of root nodules induced by nitrogen-fixing Sinorhizobium meliloti bacteria. Nodule formation was considerably delayed in the three lyk10 mutant lines. Surprisingly, the size of the rare nodules in mutant plants was higher than in wild-type plants. In conclusion, the symbiotic characterization of lyk10 mutants generated with the developed CRISPR/Cas9 protocol indicated a role of MtLYK10 in nodule formation.
1. Introduction
Medicago truncatula (barrel medic) is a model legume and particularly suitable for research on plant–microbe interactions such as the nodule symbiosis with nitrogen-fixing bacteria (rhizobia) and root colonization by symbiotic arbuscular mycorrhizal (AM) fungi. In the nodule symbiosis, Sinorhizobium (Ensifer) bacteria enter roots via root hairs, and nitrogen-fixing bacteroids in formed root nodules supply M. truncatula with fixed nitrogen [1]. In the AM symbiosis, M. truncatula and many other plants benefit from enhanced access to mineral nutrients supplied by the AM fungi, especially phosphorus [2]. M. truncatula is an annual and self-pollinating legume related to the pasture legume M. sativa (alfalfa) [3,4]. The genome of M. truncatula is diploid and has been sequenced for ecotypes such as Jemalong A17 [5] and R108 [6,7,8,9]. Various research tools such as mutant collections containing Tnt1 retrotransposon insertions [10,11] and gene expression data for different tissues and mutants (e.g., MtExpress gene expression atlas [12]) are available. M. truncatula is amenable to genetic transformation using Agrobacterium tumefaciens carrying a given binary vector. The construction of stable transformants usually includes the regeneration of transformed leaf explants to whole plants [13,14,15,16], whereas floral dip transformation provides suboptimal results in M. truncatula [17]. Leaf explants of ecotype R108 exhibit high regeneration capacity and are thus most suitable for obtaining whole plants [13,14]. Furthermore, transgenic M. truncatula roots (hairy roots) can be obtained through transformation with the help of Agrobacterium rhizogenes strains [18,19].
Many bacteria and archaea possess an adaptive defense system that can act against viruses and other foreign DNA. Clustered regularly interspaced short palindromic repeats (CRISPR) derived from foreign DNA are incorporated into the prokaryotic genome and then transcribed. Then, the produced RNAs undergo processing steps resulting in short CRISPR RNAs (crRNAs) that guide CRISPR-associated (Cas) protein(s) to homologous nucleic acid target sequences. As a result, the prokaryotes can efficiently degrade undesired viral and other foreign DNA [20]. Taken advantage of these CRISPR-Cas systems, eukaryotic genomes can now be genetically modified, e.g., by the Cas9 nuclease gene of Streptococcus pyogenes in combination with a designed single guide RNA (sgRNA) construct. The sgRNA molecule consists of the target-specific crRNA fused to the trans-activating CRISPR RNA (tracrRNA). The Cas9-sgRNA complex is then guided to the desired target DNA region in the genome which contains a protospacer adjacent motif (PAM) required for cleavage by Cas9 [21]. The generated double-stranded DNA breaks are then repaired by cellular DNA repair mechanisms, often resulting in short deletions and/or insertions at the cleavage site [22]. CRISPR/Cas9-mediated gene editing has become a tool for targeted mutagenesis in plants [23], including M. truncatula transformed with the help of A. tumefaciens [24,25,26,27,28,29,30,31,32] or A. rhizogenes [33,34,35,36,37,38]. However, the generation of homozygous M. truncatula mutant lines remains challenging as it requires the combination of an efficient CRISPR/Cas9-mediated gene editing system for explants and specific tissue culture techniques for regeneration to whole plants.
Here, we describe the construction of M. truncatula mutants using the transformation of leaf explants with A. tumefaciens carrying constructed CRISPR/Cas9 binary vectors. Regeneration to whole plants was performed at high efficiency. Obtained M. truncatula lines with knockout mutations in the LysM receptor kinase gene MtLYK10 [39] were further characterized. Inoculation experiments with nitrogen-fixing S. meliloti bacteria indicated that the described CRISPR/Cas9 system is suitable for efficient gene editing in M. truncatula and that MtLYK10 plays a role in nodule symbiosis.
2. Materials and Methods
2.1. Bacterial Strains and Growth Media
Escherichia coli DH5α, Top10, and DH10B were grown in LB medium (5 g·L−1 yeast extract, 10 g·L−1 tryptone, 10 g·L−1 NaCl) at 37 °C. Strain DH5α was used for the construction of plasmids pUC-tracrRNA-PMtU6-1 and pUC-tracrRNA-PMtU6-26. Strain Top10 allowed the maintenance of plasmids containing the ccdB gene (pBS-Cas9-ccdB, pISV-Cas9P35S, and pISV-Cas9PGmUbi). Constructed CRISPR/Cas9 binary vectors were transformed into strain DH10B suitable for harboring large plasmids. Agrobacterium tumefaciens EHA105 [40] was grown in LB medium at 27 °C. Sinorhizobium (Ensifer) meliloti strain 1021 was cultured in TY medium (5 g·L−1 tryptone, 3 g·L−1 yeast extract, 0.4 g·L−1 CaCl2) at 27 °C. Details on plasmids used in this study are shown in Supplementary Materials (Table S1). Bacterial culture media were supplemented with appropriate antibiotics (100 mg·L−1 ampicillin, 50 mg·L−1 kanamycin, 25 mg·L−1 rifampicin, and 50 mg·L−1 streptomycin).
2.2. Plant Material, Seed Germination, and Plant Growth Conditions
Medicago truncatula Gaertn. ecotype R108 (also known as R108-1 and spp. tricycla R108), Nicotiana benthamiana Domin, and Glycine max (L.) Merr. cv. Willams 82 (soybean) were used in this study. For germination, M. truncatula seed coats were scarified with a needle. The seeds were surface-sterilized through immersion in diluted sodium hypochlorite (commercial bleach; ~1.0% (w/v) active chlorine) on a shaker for 10 min. After incubation, the seeds were washed several times with sterilized H2O. The seeds were then placed on 0.8% (w/v) water agar plates. The inverted plates were incubated at 4 °C in the dark for 48 h and then at 22 ± 1 °C for 18–20 h. The germinated seedlings were then planted into 2.5 L pots filled with soil containing vermiculite, expanded clay, and peat-based potting soil (Jiffy substrate TPS fine pH 5.8; Jiffy, Moerdijk, The Netherlands) at a ratio of 3:1:1 (v/v/v). The plants were grown in a growth room at 22 ± 1 °C under 16/8 h light/dark conditions (2000 lx light intensity; Philips Lifemax TL-D 36 W/54-765 and TL-D 36 W/29–530 daylight fluorescent tubes at a ratio of 3:1) [41]. N. benthamiana seeds were surface-sterilized with 70% (v/v) ethanol. The seedlings were planted into the Jiffy substrate and plants were kept in a temperature-controlled greenhouse at 25 ± 1 °C under 12/12 h light/dark conditions. G. max seeds were immersed in a 75% (v/v) ethanol solution for 2 min, washed several times with sterilized H2O, and then incubated in a 20% (v/v) H2O2 solution for 5 min. After washing with sterilized H2O, the seeds were incubated overnight at 4 °C and then placed onto 0.8% (w/v) water agar plates. The plates were incubated in the dark at 25 ± 1 °C for 3–4 days. The germinated seedlings were transferred into 8 L pots filled with vermiculite, expanded clay, and Jiffy substrate at a ratio of 3:1:2 (v/v/v). The plants were grown in a temperature-controlled greenhouse at 25 ± 1 °C under 16/8 h light/dark conditions.
2.3. Isolation of Genomic DNA from Plants
Plant DNA was extracted from leaves using the cetyltrimethylammonium bromide (CTAB) method [42]. Harvested leaf tissue (30–50 mg) was frozen with liquid nitrogen and ground in 1.5-mL microtubes using a plastic pestle. The powder was then mixed with 600 μL prewarmed (65 °C) CTAB extraction buffer (50 mM Tris-HCl pH 8.0, 0.6 M NaCl, 10 mM EDTA, 1% (w/v) CTAB, 0.1% (v/v) β-mercaptoethanol). The samples were then incubated at 65 °C for 30 min with gentle shaking. An equal volume of chloroform was then added and the microtubes were shaken well. Subsequently, a 2/3 volume of pre-chilled isopropanol was applied, and the samples were mixed gently. After incubation at −20 °C for 10 min, the microtubes were centrifuged (12,000 rpm; 10 min). The supernatant was discarded, and the precipitated DNA was washed twice with 70% (v/v) ethanol. Finally, the DNA was air-dried and then dissolved in H2O.
2.4. Construction of CRISPR/Cas9 Vectors
The constructed plasmids are shown in Supplementary Materials (Table S1) and the primers used are listed in Supplementary Materials (Table S2). Firstly, a DNA fragment containing Cas9p (plant codon optimized Cas9 gene of S. pyogenes with an increased GC content) was PCR-amplified from pYLCRISPR/Cas9P35S-B [43]. The PCR mixtures (50 μL) contained 50 ng template DNA, 0.25 μM of primers 1 and 2, 0.2 mM dNTP, 1.5 mM MgSO4, 5 μL 10 × PCR Buffer, and 1 unit of KOD-Plus-Neo polymerase (Toyobo, Osaka, Japan). The PCR conditions were as follows: (i) 94 °C for 2 min; (ii) 35 cycles: 98 °C for 10 s, 68 °C for 2.5 min; and (iii) 68 °C for 10 min. The amplicon consisted of an enhancer sequencing, the coding region of Cas9p with N-terminal and C-terminal nuclear localization signal (NLS) sequences [44], and the cauliflower mosaic virus (CaMV) terminator. In parallel, primers 3 and 4 were used to amplify the ccdB expression cassette (with the lac promoter of E. coli) from pYLCRISPR/Cas9P35S-B. The CcdB toxin targets DNA gyrase and thus can be used for the positive selection of recombinant DNA [45]. The amplified Cas9 and ccdB cassettes were then inserted into pBluescript II SK (+) (Stratagene, La Jolla, CA, USA), which was digested with SmaI and NotI using the One Step Cloning Kit from Novoprotein (Suzhou, China). The plasmid was named pBS-Cas9-ccdB and maintained in E. coli Top10.
To drive Cas9p expression under the control of a strong plant promoter, a soybean ubiquitin gene promoter sequence, known to be active in M. truncatula [24,27], was cloned from genomic G. max DNA (GenBank accession number EU310508.1; abbreviated as PGmUbi in plasmids of this study). The PCR mixture (50 μL) contained 25 ng template DNA, 0.2 μM of primers 5 and 6, 200 mM dNTP, and 2.5 units of LA Taq polymerase (Takara Shuze, Osaka, Japan). The PCR conditions were as follows: (i) 95 °C for 5 min; (ii) 35 cycles: 95 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min; and (iii) 72 °C for 10 min. The amplicon was inserted into the T-vector pMD19-T (Takara Shuze, Osaka, Japan) using the DNA ligase provided by the supplier. The resulting plasmid was named pMD19-PGmUbi.
DsRed1 encoding a red fluorescent protein of the mushroom coral Discosoma sp. [46] was used to visualize the transformed M. truncatula plants. DNA was PCR-amplified from pRT104-DsRed1 [47] with primers 7 and 8, and the reaction conditions were identical to those for the construction of pMD19-PGmUbi. The amplicon, consisting of a CaMV 35S promoter, DsRed1, and the polyA signal sequence of pRT104 [48], was cloned into pMD19-T. The BsaI restriction site in DsRed1 was then removed through site-directed mutagenesis [49] using KOD-Plus-Neo polymerase and the primers 9 and 10. DpnI was then used to eliminate the template DNA. The obtained plasmid was named pMD19-DsRed1(ΔB). Subsequently, the DsRed1(ΔB) expression cassette was ligated into the HindIII site of the binary vector pISV2678, constructed by Michael Schultze at the CNRS (Gif-sur-Yvette, France). This vector is a derivative of pGPTV-BAR [50] and contains a Bar (bilaphos resistance) gene under the control of a nopaline synthase promoter for glufosinate selection in transformed plants. The pISV2678 vector carrying the DsRed1(ΔB) expression cassette was named pISV-DsRed1(ΔB). Except for the missing BsaI site, pISV-DsRed1(ΔB) is identical to pISV-DsRed1 constructed previously [47] (GenBank accession number MW701373; Addgene ID 171024).
The pISV-DsRed1(ΔB) vector was further modified to construct a vector containing Cas9p and ccdB. DNA from pBS-Cas9-ccdB was amplified with primers 11 (with a PacI sequence) and 12, and the amplicon was cloned into the EcoRI site of pISV-DsRed1(ΔB). The resulting binary vector, named pISV-CRISPR/Cas9P35S, contained the double CaMV promoter from pISV-DsRed1(ΔB), a translational enhancer sequence (5′ leader of tobacco etch virus), the Cas9p gene sequence with a CaMV 35S terminator, and the ccdB expression cassette flanked by BsaI restriction sites.
To obtain a similar vector with Cas9p driven by a ubiquitin gene promoter, the GmUbi promoter sequence amplified from pMD19-PGmUbi (primers 5 and 6) was cloned into the generated PacI site of pISV-CRISPR/Cas9P35S. The resulting plasmid was named pISV-CRISPR/Cas9PGmUbi.
Next, pUC18 [51] derivatives containing tracrRNA and small nuclear RNA (snRNA) promoter sequences of M. truncatula (PMtU6-1 and PMtU6-26) were constructed. DNA containing the pUC18 plasmid backbone and the tracrRNA was PCR-amplified from pYLsgRNA-AtU6-1 [43] using primers 13 and 14. The PMtU6-1 sequence was obtained from a previous publication [26]. The PMtU6-26 sequence of M. truncatula R108 was the result of a BLAST search at the Medicago truncatula Genome Database [52,53] using the PAtU6-26 sequence of A. thaliana [54] as a query sequence. The promoter sequences were amplified from the genomic DNA of M. truncatula R108 (primers 15 and 16 for PMtU6-1; primers 17 and 18 for PMtU6-26). The PCR mixtures (50 μL) contained 50 ng template DNA, 0.25 μM of each primer, 0.2 mM dNTP, 1.5 mM MgSO4, 5 μL 10 × PCR Buffer, and 1 unit of KOD-Plus-Neo polymerase. The PCR conditions were as follows: (i) 94 °C for 2 min; (ii) 98 °C for 10 s, 68 °C for 1.5 min; 35 cycles; and (iii) 68 °C for 10 min. The amplicons were then fused using a Gibson Assembly Cloning Kit (Novoprotein, Suzhou, China). The resulting plasmids containing two BsaI restriction sites were named pUC-tracrRNA-PMtU6-1 and pUC-tracrRNA-PMtU6-26, respectively.
To design sgRNA sequences for a specific gene to be mutated, target sequences upstream of the PAM sequence (5′-NGG-3′) in a given gene sequence were identified using CRISPR-P 2.0 [55,56] and targetDesign [57,58]. In this study, target sequences were used for the β-glucuronidase (GUSPlus) gene of pCAMBIA1305 and three M. truncatula R108 genes [7]. In this article, these genes were named MtLYK10 (M. truncatula lysin-motif receptor-like kinase 10, Medtr5g033490), MtMFS1 (M. truncatula major facilitator superfamily transporter 1, Medtr3g093270), and MtMFS2 (M. truncatula major facilitator superfamily transporter 2, Medtr3g093290). The synthesized target-specific 20-bp oligonucleotides (crRNA), are listed in Supplementary Materials (Table S3). The oligonucleotide pairs (1 μM in 20 μL water) were then annealed to form a double strand with ATTG/CAAA overhangs (samples heated to 90 °C for 1 min and then cooled to room temperature).
The sgRNA expression cassettes were constructed as follows. (i) In a restriction-ligation reaction, the double-stranded oligonucleotide (crRNA) with the ATTG/CAAA overhangs was fused with tracrRNA-PMtU6-1 or tracrRNA-PMtU6-26 DNA digested with BsaI. The mixture (10 μL) contained 5 U BsaI-HFV2 (NEB, Ipswich, MA, USA), 1 μL 10 × BsaI reaction buffer, 20 U T4 ligase (Genestar, Beijing, China), 1 μL 10 × T4 ligation buffer, 20 ng plasmid DNA (pUC-tracrRNA-PMtU6-1 or pUC-tracrRNA-PMtU6-26), and 0.1 μM of a double-stranded oligonucleotide. The mixture was subject to 5 thermal cycles (37 °C for 5 min; 20 °C for 5 min). (ii) Then, the ligation product (3 μL) served as a template for a PCR to amplify a given sgRNA expression cassette containing the PMtU6-1 or PMtU6-26 promoter, the tracrRNA sequence, and crRNA sequence. The reaction mixture (25 μL) contained 0.2 μM of primers 19 and 20, 0.5 U of KOD-Plus-Neo polymerase, 2.5 μL 10 × PCR Buffer, 0.2 mM dNTPs, and 1.5 mM MgSO4. The PCR was performed for 20 cycles (98 °C for 15 s, 58 °C for 15 s, 68 °C for 10 s). (iii) A total of 1 μL of the PCR product was then directly used as template for a second PCR (50 μL) to produce sgRNA expression cassettes flanked by BsaI restriction sites (primers 21 and 22 for sgRNA expression cassettes containing the PMtU6-1 promoter; primers 23 and 24 for sgRNA expression cassettes containing the PMtU6-26 promoter). The reaction mixture contained 0.2 μM of each primer, 1 U of KOD-Plus-Neo polymerase; 5 μL 10 × PCR Buffer, 0.2 mM dNTPs, and 1.5 mM MgSO4 (98 °C for 15 s, 58 °C for 15 s, 68 °C for 10 s; 25 cycles). (iv) Using Golden Gate cloning [59], the constructed sgRNA expression cassettes with terminal BsaI restriction sites were assembled and inserted into a constructed binary vector containing a Cas9p expression cassette (pISV-CRISPR/Cas9P35S and pISV-CRISPR/Cas9PGmUbi). For comparison, pYLCRISPR/Cas9P35S-B containing the pCAMBIA1300 backbone, Cas9p, under the control of a double CaMV promoter [43] was also used in initial studies. Each reaction mixture (10 μL), containing 60 ng DNA of the binary vector, 15 ng DNA of a given sgRNA expression cassette, 10 U BsaI-HFV2, and 1 μL 10 × BsaI reaction buffer, was incubated at 37 °C for 30 min. Subsequently, 35 U of T4 DNA ligase and 1 μL 10 × T4 ligation buffer were added and the samples were incubated at 37 °C for 5 min, 10 °C for 5 min and 20 °C for 5 min for a total of 15 cycles. Using the described procedure, two sgRNA expression cassettes, each with different promoters (PMtU6-1 and PMtU6-26, respectively), were assembled and ligated into the binary vector. Finally, the ligation products were transformed into competent E. coli DH10B cells which do not grow when transformed, with plasmids containing the ccdB expression cassette. The insertion of the sgRNA expression cassettes into the binary vectors was confirmed through sequencing (pYLCRISPR/Cas9P35S-sgRNA(GUS), pISV-CRISPR/Cas9P35S-sgRNA(GUS), pISV-CRISPR/Cas9PGmUbi-sgRNA(GUS), pISV-CRISPR/Cas9PGmUbi-sgRNA(LYK10), pISV-CRISPR/Cas9P35S-sgRNA(MFS1/MFS2), and pISV-CRISPR/Cas9PGmUbi-sgRNA(MFS1/MFS2)).
2.5. Electroporation of CRISPR/Cas9 Binary Vectors into A. tumefaciens
The constructed CRISPR/Cas9 binary vectors were transformed into competent A. tumefaciens EHA105 cells (Weidi, Shanghai, China) with the help of an electroporator (Ding Guo, Guangzhou, China). Pre-cooled cells (100 μL) were carefully supplemented with vector DNA (≈0.5 μg) in a sterilized 0.2 cm electroporation cuvette. After electroporation at 1500 V for 10 ms, the cells were manually mixed with 1 mL of LB medium (27 °C) and transferred to 1.5 mL plastic tubes. The bacteria were placed on a shaker (200 rpm, 27 °C) for 3 h, and 100 μL of each bacterial suspension was then spread on LB agar plates containing appropriate antibiotics (50 mg·L−1 kanamycin and 25 mg·L−1 rifampicin). After incubation (27 °C) for two days, single colonies were picked from the plates. The presence of the vectors in the bacteria was confirmed via PCR using primers listed in Supplementary Materials (Table S2).
2.6. Agrobacterium-Mediated Transformation of N. benthamiana Leaves
To test the functionality of the different CRISPR/Cas9 vectors binary vectors, they were examined for their capacity to induce mutations in the GUSPlus gene expressed in N. benthamiana leaf cells transformed with agrobacteria. The binary vector pCAMBIA1305 containing GUSPlus with a CaMV 35S promoter was used in these experiments. GUS activity in transformed tissue was visualized using histochemical staining with 5-bromo-4-chloro-3-indolyl β-D-glucuronide (X-Gluc) [60]. For Agrobacterium-mediated transformation, individual colonies of A. tumefaciens EHA105 containing pCAMBIA1305, pYLCRISPR/Cas9P35S-sgRNA(GUS), pISV-CRISPR/Cas9P35S-sgRNA(GUS), and pISV-CRISPR/Cas9PGmUbi-sgRNA(GUS) were grown in 5 mL of LB medium (supplemented with 50 mg L−1 kanamycin and 25 mg L−1 rifampicin) overnight at 27 °C. The cultures were then harvested using centrifugation (6000 rpm for 5 min), washed with 10 mM MgCl2, and then resuspended in the infiltration solution (10 mM MgCl2 supplemented with 200 μM acetosyringone) to obtain an OD600 ≈ 0.1. Next, the resuspended culture was shaken at 50 rpm (27 °C, in the dark) for 3 h. Each strain containing a CRISPR/Cas9 binary vector was then mixed with an equal volume of the strain carrying pCAMBIA1305. The bacterial suspensions were then infiltrated into the underside of 4–5-week-old N. benthamiana leaves using a 1-mL syringe (without needle). The plants were kept in a temperature-regulated greenhouse and leaves were harvested 7 days post infiltration [61].
2.7. Transformation of M. truncatula Leaf Explants and Regeneration to Whole Plants
M. truncatula R108 leaf explants were transformed with A. tumefaciens EHA105 carrying pISV-CRISPR/Cas9PGmUbi-sgRNA(LYK10), pISV-CRISPR/Cas9P35S-sgRNA(MFS1/MFS2), or pISV-CRISPR/Cas9PGmUbi-sgRNA(MFS1/MFS2), according to previously published procedures with various modifications [13,14,15,16]. A single bacterial colony carrying a given CRISPR/Cas9 binary vector was inoculated into 4 mL of LB medium supplemented with 50 mg·L−1 kanamycin and 25 mg·L−1 rifampicin. The bacteria were cultured on a shaker (200 rpm, 27 °C) for 24 h, and 1 mL of the preculture was transferred into 30 mL LB medium containing the same antibiotics. The cells were then cultured on the same shaker for 12–16 h until the cells reached an OD600 ≈ 1. The cells were centrifuged at 4000 rpm for 15 min and then resuspended (OD600 = 0.3) in liquid SH-1 medium, which is defined as SH medium (0.185 g·L−1 MgSO4·7H2O, 2.83 g·L−1 KNO3, 0.463 g·L−1 (NH4)2SO4, 0.4 g·L−1 KH2PO4, 0.166 g·L−1 CaCl2·2H2O, 1 mg·L−1 MnSO4·H2O, 0.5 mg·L−1 H3BO3, 0.1 mg·L−1 ZnSO4·7H2O, 0.1 mg·L−1 KI, 0.1 mg·L−1 Na2MO4·2H2O, 0.2 mg·L−1 CuSO4·5H2O, 0.1 mg·L−1 CoCl2·6H2O, 0.2 mg·L−1 CuSO4·5H2O, 5 mg·L−1 nicotinic acid, 5 mg·L−1 thiamine HCl, 5 mg·L−1 pyridoxine HCl, 140 mg·L−1 FeNa-EDTA, and 100 mg·L−1 myo-inositol; [62]) supplemented with 30 g·L−1 sucrose, 4 mg·L−1 2,4-dichlorophenoxyacetic acid (2,4-D), and 0.5 mg·L−1 6-benzylaminopurine (BAP). The SH-1 medium was adjusted to pH 5.8 with KOH. Healthy leaves of 4–6-week-old M. truncatula R108 plants were surface-sterilized with diluted sodium hypochlorite solution (≈0.6% (w/v) active chlorine) for 15 min and then washed several times with sterilized H2O. The leaves were cut into square pieces (≈1 cm2) with a sharp scalpel blade and immersed in the prepared A. tumefaciens suspension. For agroinfiltration, the bacterial suspension with the immersed leaf explants was exposed to a vacuum (≈−0.085 MPa) for 10 min. The explants were then gently shaken (60 rpm, room temperature) for 1 h, dried with autoclaved filter papers, and transferred to solid SH-1 medium plate containing 3 g·L−1 phytagel. After 3 days of co-cultivation, the explants were washed in liquid SH-1 medium for 20 min, dried with a sterilized filter paper, and finally transferred to solid SH-2 medium, which is defined as SH-1 medium supplemented with 300 mg·L−1 timetin (to suppress the growth of agrobacteria) and 3 mg·L−1 of the herbicide glufosinate (phosphinothricin) ammonium salt. Glufosinate allows selection for transgenic M. truncatula cells expressing the Bar gene, which confers resistance to glufosinate. The plates were kept at 24 °C in the dark. The explants were transferred every two weeks to identical plates that were freshly prepared, and the formation of calli was observed at 6–7 weeks post agroinfiltration. About 4–5 weeks later, the calli were transferred to plates containing solid MSBK plates (pH 5.8), which is defined as 4.43 g·L−1 Murashige and Skoog (MS) basal medium (Coolaber Beijing, China; [63]) supplemented with 30 g·L−1 sucrose, 1 mg·L−1 kinetin, 0.5 mg·L−1 BAP, 2 mg·L−1 glufosinate, 300 mg·L−1 timetin, and 3 g·L−1 phytagel [16]. The plates were incubated at 22 ± 1 °C under 16/8 h light/dark conditions (2000 lx light intensity; Philips Lifemax TL-D 36 W/54-765 and TL-D 36 W/29–530 daylight fluorescent tubes at a ratio of 3:1). About two weeks later, green somatic pro-embryos were observed and transferred to plates filled with phytohormone-free SH-3 medium (SH medium supplemented with 20 g·L−1 sucrose, 150 mg·L−1 timetin, 2 mg·L−1 glufosinate, and 3 g·L−1 phytagel). The plates were refreshed every two weeks. Plantlets with fully expanded trifoliate leaves were observed after 7–8 weeks growth on these plates. The plantlets were then transferred to 350-mL glass vessels which were partially filled with ≈150 mL SH-3 medium. To ensure gas exchange, the lids of the glass vessels had a hole of 0.79 cm2, which was covered with filter paper. The plantlets were placed into fresh glass vessels every two weeks. Fully regenerated plants with well-developed roots were obtained after about 10 weeks growth in these vessels. Finally, the plants were transferred to sterilized 300-mL plastic jar units linked with a cotton wick (0.5 cm in diameter). The upper jar contained vermiculite and expanded clay (3:1 v/v), and the lower jar was filled with half-strength B&D [64] nutrient solution (125 μM CaCl2·2H2O, 62.5 μM KH2PO4, 1.25 μM Fe-citrate, 31.25 μM MgSO4·7H2O, 187.5 μM K2SO4, 0.125 μM MnSO4·7H2O, 0.25 μM H3BO3, 0.025 μM CuSO4·5H2O, 0.0125 μM CoSO4·7H2O, 0.0125 μM Na2MoO4·2H2O, and 0.0625 μM ZnSO4·H2O) supplemented with 1 mM KNO3. The upper jar was covered with a light transparent lid for several days to acclimatize the plants to the new environment. The jar units were kept in a temperature-controlled growth room as described above.
Plants of the T0 generation were examined for the emission of red fluorescent signals. For seed production, the transgenic plants were transferred to 2.5 L pots filled with vermiculite and expanded clay (3:1 v/v). The plants were watered with half-strength B&D nutrient solution containing 0.5 mM KNO3 (≈300 mL per pot every 3 days).
2.8. Detection of Red Fluorescent Signals in Transformed M. truncatula Plants
A Zeiss ImagerZ1 fluoresce microscope (Carl Zeiss, Oberkochen, Germany) was used to detect red fluorescent signals in transgenic M. truncatula plants expressing DsRed1 [46]. The excitation wavelengths were 540–552 nm and the emission window 575–640 nm. Typically, small root pieces were harvested from T0 generation plants before they were transferred from jar units to pots.
2.9. Identification of Mutations in Transformed Plants
Genomic DNA was extracted from the infiltration zone of N. benthamina leaves (5 leaves from 5 different plants per A. tumefaciens strain) to identify sgRNA-mediated mutations in GUSPlus sequences. Likewise, leaves of 10 transformed and regenerated M. truncatula plants were used for the extraction of genomic DNA to detect mutations in the MtLYK10, MtMFS1, and MtMFS2 genes. The DNA was then used as a template for a PCR (primers 31 and 32 for GUSPlus; primers 33 and 34 for MtMFS1; primers 35 and 36 for MtMFS2; and primers 37 and 38 for MtLYK10). The reaction mixture (50 μL) contained 200 ng template DNA, 0.25 μM of gene-specific primers, 0.8 mM dNTP, 5 mM MgSO4, 5 μL 10 × PCR Buffer, and 2.5 U of LA Taq (2 × LA taq mix; Takara, Osaka, Japan). The annealing temperature was 60 °C and the extension time was 1 min. PCR products amplified from M. truncatula DNA were then directly subjected to Sanger sequencing. When the sequencing results showed double peak signals, the corresponding amplicon was inserted into pMD19-T and sequencing data with single peak signals were obtained from transformed E. coli colonies. PCR products amplified from N. benthamiana leaves were directly cloned into pMD19-T. For each transformed leaf, three randomly sampled E. coli colonies were sequenced to estimate the mutation frequency at the GUSPlus target sites.
2.10. Analysis of Generated lyk10 Mutant Plants
T0 generation plants with identified mutations in MtLYK10 were propagated by selfing. The resulting T1 generation plants were then subjected to PCRs (primers 37 and 38; Supplementary Materials, Table S2) and sequencing to identify homozygous (biallelic) mutant plants in three independent lyk10 mutant lines (lyk10-1, lyk10-1, and lyk10-1). The prediction of possible off-target mutations in the lyk10 mutants was performed with offTarget software [57,65] using the corresponding crRNA-PAM sequences as query sequences. The nucleotide sequences of possible off-target regions in various genes (MTR_2g063570, MTR_7g029350, MTR_4g127480, MTR_2g069950, MTR_5g024910, MTR_3g112020, MTR_1g054795, MTR_4g116370, and MTR_7g056073) were analyzed by sequencing the amplicons of PCRs using primers 41–58.
Furthermore, the nodule formation of homozygous lyk10 mutant plants was analyzed. T1 generation plants (25–30 per identified mutation) and wild-type plants were grown in sterilized 300 mL plastic jar units (two jars were connected with a cotton wick). The upper jar was filled with vermiculite and expanded clay at a ratio of 3:1 (v/v). The lower jar contained B&D nutrient solution [64] (250 μM CaCl2·2H2O, 125 μM KH2PO4, 2.5 μM Fe-citrate, 62.5 μM MgSO4·7H2O, 375 μM K2SO4, 0.25 μM MnSO4·7H2O, 0.5 μM H3BO3, 0.05 μM CuSO4·5H2O, 0.025 μM CoSO4·7H2O, 0.025 μM Na2MoO4·2H2O, and 0.125 μM ZnSO4·H2O) supplemented with 1 mM KNO3. The growth conditions were the same as described above. After one week, plants (1 plant per jar unit) were inoculated with S. meliloti 1021. The bacteria were centrifuged and resuspended in 10 mM MgSO4 (OD600 adjusted to 0.2). Each jar system was inoculated with 2 mL bacteria. Genomic DNA was isolated from a harvested leaf at 7 days post inoculation (dpi) to identify homozygous lyk10 mutant plants using sequencing, as described above. The roots of three homozygous mutant lines were harvested at two different time points (10 and 21 dpi) to determine the number of formed nodules for each plant. Longitudinal sections of nodules (harvested at 21 dpi) were analyzed using a Zeiss Lumar.V12 microscope (Carl Zeiss, Oberkochen, Germany). Image J2 software [66] was used to calculate the area of the sectioned nodules. Statistical analysis (Student’s t-test) was performed with GraphPad Prism (version 8.0.2 (263); Prism software, Vestal, NY, USA).
3. Results
3.1. Construction of CRISPR/Cas9 Binary Vectors Suitable for M. truncatula Transformation
In previous studies, pISV2678, a binary vector with a Bar gene, was found to be very effective for the A. tumefaciens-mediated transformation of M. truncatula leaf explants [41,67,68,69]. We therefore aimed to construct a CRISPR/Cas9 vector with a pISV2678 backbone and a DsRed1 expression cassette for the visual confirmation of transformants. The Cas9p gene with N-terminal and C-terminal NLS sequences was PCR-amplified from pYLCRISPR/Cas9P35S-B [43] along with the transcriptional enhancer 5’UTR of Tobacco Etch Virus and the CaMV terminator. Furthermore, a ccdB expression cassette was amplified from pYLCRISPR/Cas9P35S-B. Both amplicons were then cloned into pBluescript II SK (+), resulting in pBS-Cas9-ccdB (Figure 1a). Next, the binary vector pISV2678 was modified by inserting a DsRed1 expression cassette (lacking the undesired BsaI restriction site) into the HindIII site of pISV2678, resulting in pISV-DsRed1(ΔB) (Figure 1b). The insert of pBS-Cas9-ccdB was then cloned into the EcoRI site of pISV-DsRed1(ΔB), yielding pISV-CRISPR/Cas9P35S (Figure 1c). In this vector, the Cas9p gene expression is under the control of a double CaMV promoter, while the ccdB expression cassette is flanked by unique BsaI restriction sites. In addition, a similar binary vector, pISV-CRISPR/Cas9PGmUbi, was constructed in which Cas9p gene expression is driven by a strong ubiquitin gene promoter of soybean (Figure 1d). Furthermore, plasmids containing tracrRNA and snRNA promoter sequences of M. truncatula (PMtU6-1 or PMtU6-26) were constructed. The pUC18 plasmid backbone and the tracrRNA sequence were PCR-amplified from pYLsgRNA-AtU6-1 [43] and the PMtU6-1 or PMtU6-26 promoter sequences from genomic DNA of M. truncatula. The plasmids containing the amplified DNA fragments were named pUC-tracrRNA-PMtU6-1 and pUC-tracrRNA-PMtU6-26, respectively (Figure 1e).
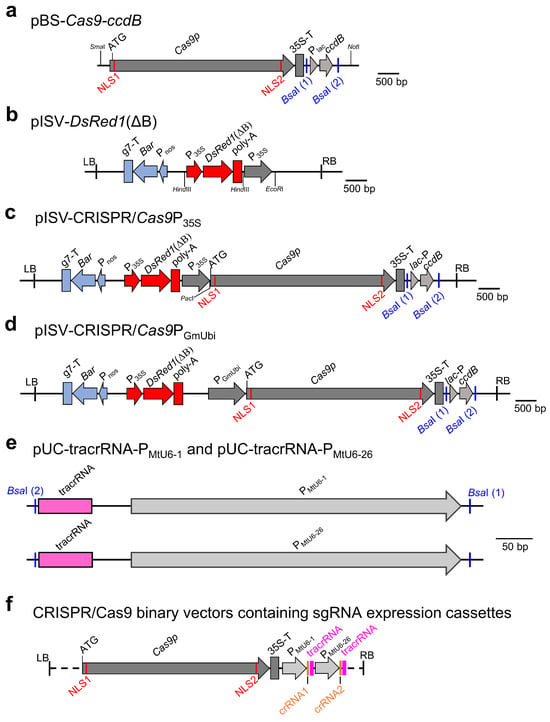
Figure 1.
CRISPR/Cas9 plasmids constructed in this study. (a) Insert of pBS-Cas9-ccdB containing the Cas9p gene and the ccdB expression cassette flanked by BsaI restriction sites. (b) T-DNA region of pISV-DsRed1(ΔB) containing the Bar gene expression cassette, the DsRed1(ΔB) expression cassette, and a double CaMV promoter. (c) T-DNA region of pISV-CRISPR/Cas9P35S which is a pISV-DsRed1(ΔB) derivative containing Cas9p driven by a double CaMV promoter and the ccdB expression cassette flanked by BsaI restriction sites. (d) T-DNA region of pISV-CRISPR/Cas9PGmUbi which is a pISV-DsRed1(ΔB) derivative containing Cas9p driven by a GmUbi promoter and the ccdB expression cassette flanked by BsaI restriction sites. (e) Inserts of pUC plasmids containing the tracrRNA sequence and a snRNA gene promoter of M. truncatula (PMtU6-1 or PMtU6-26) flanked by BsaI restriction sites. (f) Schematic view of CRISPR/Cas9 binary vectors containing two sgRNA expression cassettes, each consisting of a tracrRNA and a crRNA sequence. Abbreviations: NLS, nuclear localization sequence; P: promoter; T: terminator; ccdB: suicide gene; Bar, phosphotransferase (PPT) gene; tracrRNA: trans-activating crRNA; crRNA: CRISPR RNA.
Using CRISPR-P 2.0 and targetDesign software, target sequences upstream of the PAM sequence (5′-NGG-3′) were identified for the GUSPlus gene in the binary vector pCAMBIA1305 and the M. truncatula genes MtLYK10, MtMFS1, and MtMFS2. Synthesized oligonucleotides (listed in Supplementary Material, Table S3) were then annealed to form double strands with ATTG/CAAA overhangs (crRNA), which were then cloned into pUC-tracrRNA-PMtU6-1 or pUC-tracrRNA-PMtU6-26 digested with BsaI. The obtained DNA was used as a template for a PCR to amplify a given sgRNA expression cassette consisting of the PMtU6-1 or PMtU6-26 promoter, the tracrRNA, and a synthesized crRNA. In a second PCR, terminal sequences with a BsaI restriction site were introduced into the sgRNA expression cassette. Finally, using a restriction-ligation reaction with BsaI, the sgRNA expression cassettes were inserted into pISV-CRISPR/Cas9P35S (sgRNA expression cassettes to edit GUSPlus, MtMFS1, and MtMFS2) and pISV-Cas9PGmUbi (sgRNA cassettes to edit GUSPlus, MtLYK10, MtMFS1, and MtMFS2), respectively. For comparison, pYLCRISPR/Cas9P35S-B [43], a pCAMBIA1300 derivative containing Cas9p under the control of a double CaMV 35S promoter, was also used in initial GUSPlus experiments. The procedure for cloning sgRNA expression cassettes and inserting them into the binary vectors is illustrated in Supplementary Materials (Figure S1). A schematic view of the obtained CRISPR/Cas9 binary vectors is shown in Figure 1f. All constructed binary vectors contained two sgRNA expression cassettes with different promoters (PMtU6-1 and PMtU6-26, respectively).
3.2. Testing for the Functionality of Constructed CRISPR/Cas9 Binary Vectors in N. benthamiana
Transient gene expression experiments with N. benthamiana plants were performed to examine the functionality of the constructed CRISPR/Cas9 binary vectors. The vectors (pISV-CRISPR/Cas9P35S-sgRNA(GUS) and pISV-CRISPR/Cas9PGmUbi-sgRNA(GUS)) contained two sgRNA expression cassettes to induce mutations at two different target sites of a co-transformed GUSPlus gene. For comparison, the two sgRNA expression cassettes targeting GUSPlus were also introduced into pYLCRISPR/Cas9P35S-B [43] (vector pYLCRISPR/Cas9P35S-sgRNA(GUS)). Prior to agroinfiltration, suspensions of A. tumefaciens EHA105 harboring a given CRISPR/Cas9 binary vector were mixed with an equal volume of EHA105 carrying pCAMBIA1305, which contains a GUSPlus expression cassette. Each bacterial suspension was injected into five leaves of five different N. benthamiana plants and leaves were harvested 7 days post infiltration. GUS activity tests with X-Gluc showed blue coloration in the infiltration zones, indicating the successful transformation of the GUSPlus gene. Subsequently, DNA was isolated from the infiltration zones and used as a template to amplify a GUSPlus gene fragment containing the two target sites. As the sequencing of amplicons resulted in double peaks, the amplicons were cloned into pMD19-T, which was then transformed into E. coli cells to obtain single peaks from isolated plasmid DNA. Sequence comparisons showed that all the CRISPR/Cas9 binary vectors tested can cause mutations at the GUSPlus target sites. In a number of sequences, gene knockout was observed due to a frameshift mutation at a single target site. Mutations at two target sites led to DNA deletion between the target sites. Examples of obtained GUSPlus sequence variants are shown in Figure 2a and the corresponding Sanger sequencing chromatograms in Figure 2b. The editing efficiency was then quantified for each binary vector using data from a total of 194 sequences. The use of the pISV-CRISPR/Cas9PGmUbi-sgRNA(GUS) vector resulted in the highest frequency of simultaneous mutations at both target sites (Figure 2c). These data indicated that the constructed CRISPR/Cas9 binary vectors were functional and thus could be used for experiments to edit specific genes in the M. truncatula genome.
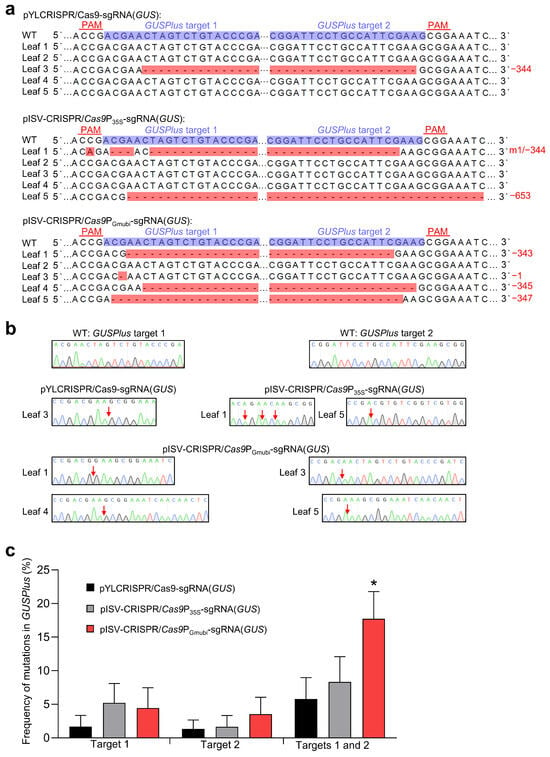
Figure 2.
Testing A. tumefaciens strains carrying different CRISPR/Cas9 vectors in N. benthamiana leaves. The vectors pYLCRISPR/Cas9P35S-sgRNA(GUS), pISV-CRISPR/Cas9P35S-sgRNA(GUS), and pISV-CRISPR/Cas9PGmUbi-sgRNA(GUS) were examined for their capacity to induce mutations at two target sites in the co-transformed GUSPlus gene. (a) Sequencing results of DNA from five transformed leaves. The target sites besides the PAM sequence are shown in the wild-type (WT) sequence. Mutations at both target sites resulted in large deletions. Abbreviations: −, deletion; m, mutation. (b) Sanger sequencing chromatograms obtained from clones transformed into E. coli. The arrows mark the mutation sites. (c) Frequency of mutations at target sites 1 and 2 in GUSPlus. In total, 194 GUSPlus sequences were analyzed. Data indicate means ± SE (n = 5). The asterisk indicates a significantly increased mutation frequency induced by pISV-CRISPR/Cas9PGmUbi-sgRNA(GUS) as compared to the other binary vectors (Student’s t-test, p < 0.05).
3.3. Efficient CRISPR/Cas9-Mediated Generation of Mutant Lines in M. truncatula
To determine whether specific M. truncatula genes can be mutated using the established CRISPR/Cas9 system, we constructed binary vectors to edit three M. truncatula genes, namely, the LysM-type receptor kinase gene MtLYK10 [39] and two MFS transporter genes related to N-acetylglucosamine transporters in rice [70]. The exon/intron structure and selected target sites in these three M. truncatula genes are illustrated in Figure 3a. The constructed CRISPR/Cas9 binary vectors (pISV-CRISPR/Cas9P35S-sgRNA(MFS1/MFS2), pISV-CRISPR/Cas9PGmUbi-sgRNA(MFS1/MFS2), and pISV-CRISPR/Cas9PGmUbi-sgRNA(LYK10)) were then used for the A. tumefaciens-mediated transformation of leaf explants. Pictures illustrating the different cultivation steps from leaf disks to whole transgenic plants are shown in Figure 3b–g. The whole procedure lasted 6–9 months. The expression of transformed DsRed1 in regenerated glufosinate-resistant plants (T0 generation) was confirmed through the fluorescence microscopy analysis of the roots (Figure 3h). A PCR analysis confirmed that the transformed plants contained the Bar gene (Figure 3i). A quantitative analysis indicated that most leaf disks could form a callus (callus formation efficiency of about 95%). The regeneration efficiency from calli to whole plants was also high. On average, transgenic plants could be obtained from about 85% of the calli (Figure 3j). The sequencing results of PCR-amplified DNA from the leaves of T0 generation plants indicated double peaks in the target regions, indicating that one allele of the target gene was successfully edited (Supplementary Materials, Figure S2). For confirmation, the amplicons were cloned into pMD19-T and sequenced. Only single peaks were observed in chromatograms obtained from the plasmid DNA of individual E. coli colonies.
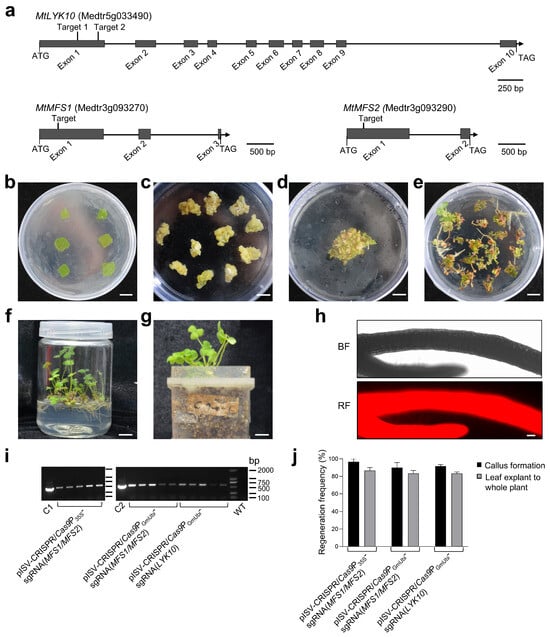
Figure 3.
Transformation of leaf explants and regeneration to whole M. truncatula plants. (a) Schematic diagram illustrating the exon/intron structure of the M. truncatula genes MtLYK10, MtMFS1, and MtMFS2. The target sites designed for gene editing are in exon 1. (b) Explants from M. truncatula leaves after transformation with A. tumefaciens bacteria on SH-1 medium. (c) Six-week-old calli kept on SH-2 medium. (d) Calli placed on MSBK medium for 2 weeks. (e) Plantlets grown on SH-3 medium for 7 weeks. (f) Plants grown in glass vessels containing SH-3 medium for 10 weeks. (g) Plants grown in plastic jar units filled with vermiculite and expanded clay. DNA from individual plants was used for PCRs and sequencing. (h) Analysis for red fluorescent signals in roots using a Zeiss ImagerZ1 fluorescence microscope. Root pieces were taken from regenerated plants before transfer into plastic jar units. (i) PCR analysis of the Bar gene in plants transformed with the indicated binary vectors. The leaf DNA of M. truncatula wild-type plants (WT) served as a negative control and the plasmid DNA of pISV-CRISPR/Cas9P35S and pISV-CRISPR/Cas9PGmUbi as a positive control (lanes C1 and C2, respectively). (j) Regeneration efficiency of leaf explants transformed with the indicated binary vectors to form a callus or a whole plant. Scale bars: 1 cm in (b–g) and 100 μm in (h).
The variations in MtLYK10 sequences (single peaks obtained from the plasmid DNA of a single E. coli colony) are shown in Figure 4. These sequences were obtained from 10 regenerated plants derived from the transformation of 10 different leaf explants with pISV-CRISPR/Cas9PGmUbi-sgRNA(LYK10). This vector contains sgRNA expression cassettes to edit MtLYK10 at two different target sites. Mutations were observed at target site 1 for two plants and at target site 2 for four plants. In three plants (P1, P8, and P9), the open reading frame of MtLYK10 (exon 1) was disrupted. In one plant (P3), mutations at both target sites caused a deletion of 654 bp. Accordingly, the analysis of amplified MtLYK10 DNA from P3 by agarose gel electrophoresis revealed two bands (Supplementary Materials, Figure S3). Overall, six mutations in the MtLYK10 gene were obtained at the two target sites, resulting in an editing efficiency of 2/10 (=20%) for target site 1 and 4/10 (=40%) for target site 2.
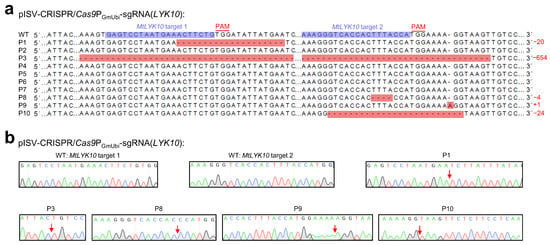
Figure 4.
CRISPR/Cas9-mediated editing of MtLYK10. M. truncatula leaf explants were transformed with A. tumefaciens carrying pISV-CRISPR/Cas9PGmUbi-sgRNA(LYK10). Leaf DNA was extracted from wild-type plants and regenerated plants (T0 generation) at 32 weeks post transformation. (a) The MtLYK10 sequences (region of target sites) obtained from the Sanger sequencing of wild-type (WT) plants and from 10 regenerated plants (P1 to P10) were aligned. The two target sites next to the PAM sequences are shown in the wild-type sequence. Mutations at both target sites in P3 resulted in a large deletion. Abbreviations: +, insertion; −, deletion; m, mutation. (b) Mutations were identified by cloning the MtLYK10 amplicons into pMD19-T and Sanger-sequencing plasmid DNA from individual E. coli colonies. The arrows in the chromatograms mark the mutation sites.
The variations in single-peak sequences of the two examined MFS transporter genes in 10 regenerated plants transformed with pISV-CRISPR/Cas9P35S-sgRNA(MFS1/MFS2) and pISV-CRISPR/Cas9PGmUbi-sgRNA(MFS1/MFS2) are shown in Figure 5. The two vectors contain identical sgRNA sequences (one target site in the MtMFS1 gene and another target site in the MtMFS2 gene), but differ with respect to the promoter controlling Cas9p expression (CaMV 35S promoter and GmUbi promoter, respectively). For the plants transformed with pISV-CRISPR/Cas9P35S-sgRNA(MFS1/MFS2), no mutation was observed at the target site in MtMFS1 while mutations at the target site of MtMFS2 were identified in five plants (50% editing efficiency). Four of these mutations (plants P3, P4, P6, and P8) resulted in a disrupted frameshift of MtMFS2 (exon 1) (Figure 5a,b). For the plants transformed with pISV-CRISPR/Cas9PGmUbi-sgRNA(MFS1/MFS2), two plants showed mutations at the target site of MtMFS1 (20% editing efficiency) and seven plants at the target site of MtMFS2 (70% editing efficiency). In one plant (P1), the open reading frame of MtMFS1 (exon 1) was altered. Four plants (P4, P5, P8, and P9) exhibited sequences with a disrupted open reading frame of MtMFS2 (exon 1). Furthermore, one plant (P2) showed simultaneous mutations in MtMFS1 and MtMFS2. In this plant, the insertion of a nucleotide disrupted the open reading frame of MtMFS1 while the mutated MtMFS2 sequence exhibited a deletion of 18 bp that caused the elimination of six amino acids while the reading frame remained unaffected (Figure 5c,d).

Figure 5.
CRISPR/Cas9-mediated editing of MtMFS1 and MtMFS2. M. truncatula leaf explants were transformed with A. tumefaciens carrying either pISV-CRISPR/Cas9P35S-sgRNA(MFS1/MFS2) or pISV-CRISPR/Cas9PGmUbi-sgRNA(MFS1/MFS2). Leaf DNA was extracted from wild-type plants and regenerated plants (T0 generation) at 32 weeks post transformation. (a) MtMFS1 and MtMFS2 sequences (region of target sites) obtained from wild-type (WT) plants and from 10 regenerated plants (P1 to P10) transformed with pISV-CRISPR/Cas9P35S-sgRNA(MFS1/MFS2). The sequences obtained from Sanger sequencing results (double peaks in mutant plants) were aligned. The target sites next to the PAM sequence are shown in the wild-type sequences. (b) Confirmation of identified mutations by cloning the MtMFS1 and MtMFS2 amplicons into pMD19-T and Sanger-sequencing plasmid DNA from individual E. coli colonies. The arrows in the chromatograms mark the mutation sites. (c,d) Similar sequence analysis was performed for plants transformed with pISV-CRISPR/Cas9PGmUbi-sgRNA(MFS1/MFS2). Abbreviations: +, insertion; −, deletion; m, mutation.
3.4. Analysis of Homozygous lyk10 Mutants
For T0 generation plants, the sequencing analysis of MtLYK10 indicated the presence of frameshift mutations in three independent plants (P1, P3, and P8 in Figure 4). After selfing of these plants, seeds were collected and used for further analysis of the T1 generation. MtLYK10 sequencing data were obtained from leaf DNA of 2-week-old plants. In about one quarter of T1 generation plants, no wild-type allele sequences could be observed, indicating the generation of homozygous MtLYK10 mutant plants (Figure 6a). The mutations in these plants were identical to those of the T0 generation plants P1, P3, and P8 (Figure 4). The mutant lines were named lyk10-1 (derived from P1), lyk10-2 (derived from P3), and lyk10-3 (derived from P8). No off-target mutations were identified in these three lyk10 mutant lines when the DNA of nine genes with predicted off-target sites was sequenced (Supplementary Materials, Table S4). Finally, the identified homozygous lyk10 mutant and wild-type plants were compared with respect to the formation of root nodules induced by S. meliloti. In contrast to wild-type plants, no nodules were observed on the roots of the three mutant lines at 10 dpi (Figure 6b). At 21 dpi, nodules on the roots of mutant plants were observed but their numbers were statistically lower than in wild-type plants (Figure 6c). Surprisingly, the size of the rare nodules in mutant plants was higher than in wild-type plants (Figure 6d). In conclusion, nodule formation was considerably delayed in the three constructed lyk10 mutant lines.
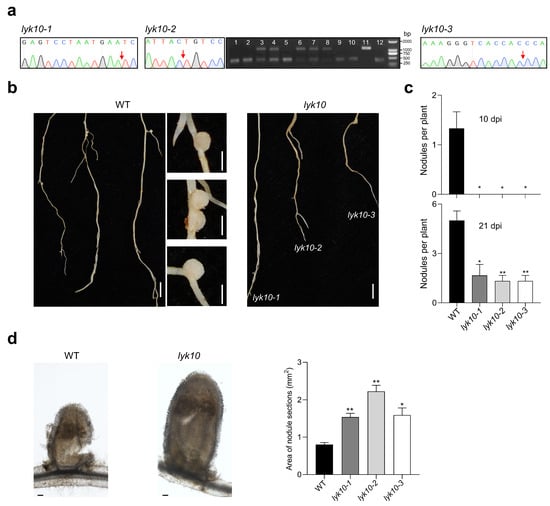
Figure 6.
Analysis of nodule formation on the roots of three independent lyk10 mutant lines. Mutant plants (T1 generation of mutant lines lyk10-1, lyk10-2, and lyk10-3) and M. truncatula R108 wild-type (WT) plants were inoculated with S. meliloti 1021. (a) Homozygous mutant plants were identified through the sequencing of MtLYK10 amplicons (no double peaks at target sites; arrows mark the mutation sites). The amplified DNA of the lyk10-2 line (derived from P3; see Figure 4) contains a large deletion that could also be visualized using agarose gel electrophoresis. Heterozygous mutant plants showed two bands, whereas for homozygous plants, only a single lower DNA band was detected (lanes 1, 2, 5, 9, 10, and 12). (b) Photographs of wild-type and lyk10 mutant plants harvested at 10 dpi (bar = 1 cm). At this time point, nodules (right images) were formed only on wild-type roots (bar = 1 mm). (c) Quantitative analysis of nodule formation at 10 dpi and 21 dpi. (d) Left: Example of a longitudinal section of a wild-type nodule and a nodule of the lyk10-1 mutant (bar = 100 μm). Right: Nodule size as determined by photographing cross-sections of nodules and quantifying the areas with Image J2 software. Data in (c,d) are presented as mean ± SE (n = 3). The asterisks indicate that lyk10 mutant lines and wild-type plants are significantly different (Student’s t-test, * p < 0.05, ** p < 0.01).
4. Discussion
In this study, we used CRISPR/Cas9 technology to optimize gene editing in the model legume M. truncatula. The procedure is based on the transformation of leaf explants with the help of A. tumefaciens EHA105 carrying a constructed derivative of pISV-CRISPR/Cas9PGmUbi. The binary vectors contained a plant codon-optimized Cas9 gene (Cas9p), Bar, DsRed1, and sgRNA expression cassettes with snRNA promoter sequences of M. truncatula (PMtU6-1 and PMtU6-26). We found that the high transformation and regeneration efficiency of leaf explants under glufosinate selection is probably critical for the construction of mutant plants. Small technical details such as the preparation of surface-sterilized leaf explants (≈1 cm2) from healthy leaves and the use of timetin to suppress the growth of agrobacteria were found to be crucial for the regeneration to whole plants. Furthermore, we observed that kinetin (at a concentration of 1 mg·L−1 [16]) and the used light conditions [41] are important factors for efficient somatic pro-embryogenesis.
The efficiency of CRISPR/Cas9 systems also depends on strong promoters. Ubiquitin promoters are constitutive promoters that are frequently active in a number of different plants. For example, a ubiquitin promoter from Lotus japonicus was routinely used to overexpress genes in M. truncatula in previous studies [71,72,73]. When comparing different binary vectors in transformation experiments with N. benthamiana, we found the highest mutation frequency at target sites of the co-transformed GUSPlus gene for pISV-CRISPR/Cas9PGmUbi-sgRNA(GUS), which contains Cas9p under the control of a soybean ubiquitin gene promoter [24,27]. Likewise, the editing efficiency in M. truncatula was considerably higher for this vector as compared to the same vector with a CaMV 35S promoter controlling Cas9p expression (Figure 5). We therefore recommend using pISV-CRISPR/Cas9PGmUbi for future gene editing studies with M. truncatula.
To ensure high gene editing efficiency in M. truncatula, our sgRNA expression cassettes contained PMtU6-1 and PMtU6-26, which are snRNA promoters of M. truncatula. The PMtU6-1 promoter has been used in previous works [26,30,31], while PMtU6-26 was found to be as active as PMtU6-1 in the current study. The combination of these promoters allowed the simultaneous editing of two target sites in M. truncatula. With these promoters, an editing efficiency of up to 70% (per target site) was achieved (Figure 5), which is comparable to or higher than the mutation frequencies reported in previous publications on CRISPR/Cas9-mediated gene editing in M. truncatula (e.g., references [24,26,29,31]). Future gene editing in M. truncatula could be performed with pISV-CRISPR/Cas9PGmUbi containing more than two sgRNA expression cassettes (e.g., by assembling them with the help of Esp3I in addition to BsaI). For such constructs, additional M. truncatula promoters such as PMtU6-6 [31,38] and PMtU6-8 [36] could be used. Alternatively, multiple sgRNAs, each separated by RNA cleavage sequences, could be expressed from a single promoter and then processed to sgRNAs, e.g., with the endoribonuclease Csy4 [25].
In general, the probability to obtain a gene knockout mutant increases when the binary vector contains multiple sgRNA sequences for several targets in the same gene. This, however, may also increase the risk of mutations in other genes. In our study, off-target mutations were not identified in the obtained lyk10 mutant plants. To minimize the risk of off-target mutations, we recommend mutating a given M. truncatula gene using a maximum of two sgRNA sequences. Off-target effects could be also reduced by other means, including the use of CRISPR/Cas9 systems with a nuclease-deactivated variant of Cas9 (dCas9) [74].
In our transient transformation experiments with N. benthamiana, co-transformation with the two A. tumefaciens strains carrying pISV-CRISPR/Cas9PGmUbi-sgRNA(GUS) and pCAMBIA1305 (containing GUSPlus) resulted in a surprisingly high number of mutations in the GUSPlus gene. It is therefore possible to use N. benthamiana leaves as a test system to rapidly verify the functionality and gene editing activity of pISV-CRISPR/Cas9PGmUbi derivatives containing sgRNA expression cassettes. M. truncatula DNA fragments with target sites could be cloned into pCAMBIA1305 and co-transformation experiments could be performed as described for GUSPlus in our study. Results from such a pre-test could provide information on the editing activity of a given sgRNA within a few days (post infiltration with agrobacteria), while the regeneration of leaf explants to whole M. truncatula plants takes several months. Indeed, our study shows that the sgRNA sequence chosen for editing MtMFS1 was suboptimal for unknown reasons and that preliminary transformation experiments with N. benthamiana plants are particularly recommended when the mutation of multiple genes is envisaged. The pre-testing of binary vectors with sgRNA expression cassettes could also be performed through the CRISPR/Cas9-mediated reconstitution of a fluorescence protein in N. benthamiana [75].
The constructed CRISPR/Cas9 binary vectors contained the fluorescence marker gene DsRed1. Accordingly, all regenerated plants were found to be fluorescent when analyzed under red fluorescence conditions. To simplify the removal of the Cas9p gene in regenerated M. truncatula plants, the DsRed1 marker will be helpful as non-fluorescent plants can be selected conveniently to obtain transgene-free (T-DNA-free) mutant plants through outcrossing. Furthermore, the DsRed1 marker allows the convenient identification of transgenic hairy roots obtained through A. rhizogenes-mediated transformation. However, the efficiency of CRISPR/Cas9-mediated gene editing in hairy roots and their regeneration to whole M. truncatula plants appears to be relatively low in various studies published so far [33,35,36,37,76]. According to our experience, the large-scale regeneration of hairy roots to whole M. truncatula plants under glufosinate selection conditions is challenging and less efficient than the regeneration procedure with A. tumefaciens-transformed leaf explants described in this study (unpublished observations).
The MtMFS1 and MtMFS2 genes of M. truncatula mutated in this study encode major facilitator superfamily transporters [77], which are predicted to import or secrete N-acetylglucosamine. This sugar has been hypothesized to act as a symbiotic signal in the association between AM fungi and monocots (maize and rice), in which N-acetylglucosamine transporters (homologous to MtMFS1 and MtMFS2) have been identified [70]. It is worth mentioning in this context that M. truncatula mutants carrying a Tnt1 retrotransposon in a β-N-acetylhexosaminidase gene (MtHEXO2) exhibit reduced colonization by AM fungi. MtHEXO2 is an extracellular enzyme that releases N-acetylglucosamine from fungal chitooligosaccharide signals [69]. Future research will reveal whether MtMFS1 and MtMFS2 are N-acetylglucosamine transporters and whether they are required for the establishment of AM symbiosis.
The MtLYK10 gene encodes a LysM-type receptor kinase showing homology to exopolysaccharide/glycan receptors of L. japonicus involved in nodule formation (LjEPR3 receptor; [78,79,80]) and in symbiosis with AM fungi (LjEPR3a receptor [81]). Our results that homozygous lyk10 mutants exhibit delayed nodule formation in M. truncatula are consistent with findings on a MtLYK10 mutant reported previously [39]. However, since only a single Tnt1 retrotransposon insertion mutant was analyzed in that earlier study, the conclusions regarding the symbiotic role of MtLYK10 had to be interpreted with caution. In our study, three independent lyk10 mutants inoculated with S. meliloti showed a similar symbiotic phenotype. Thus, our results clearly show that MtLYK10 is a symbiotic gene that positively affects nodule development during early symbiotic stages. Interestingly, when compared to wild-type plants, the size of the rare nodules formed by the three lyk10 mutant plants was increased, suggesting that MtLYK10 negatively affects nodule size in mature nodules. Alternatively, the increased nodule size was the result of a compensation reaction, as the nodules of poorly nodulated root systems may tend to have a larger volume and more infected cells containing nitrogen-fixing bacteroids. Future experiments are required to investigate whether MtLYK10 can recognize rhizobial glycans such as exopolysaccharide (succinoglycan) [82], cyclic β-glucan [83], and mixed-linkage β-glucan [84].
5. Conclusions
This article shows that MtLYK10 plays a role in nodule symbiosis and that the used CRISPR/Cas9 technology is suitable for efficient gene editing in the model legume M. truncatula. The CRISPR/Cas9-mediated generation of mutant lines makes it possible to characterize, with relatively little effort, the function of M. truncatula genes for which no or only one gene knockout mutant was obtained in earlier mutagenesis programs [85]. The constructed pISV-CRISPR/Cas9PGmUbi, pUC-tracrRNA-PMtU6-1, and pUC-tracrRNA-PMtU6-26 vectors were deposited in the Addgene repository [86] and are available to the scientific community. Our data show that the leaf transformation, glufosinate selection, and regeneration procedures from leaf explants to whole M. truncatula plants are highly efficient but require several months to obtain seeds. Future optimization steps should focus on technical parameters that could further standardize and accelerate these processes. Finally, the pre-testing of binary vectors with different sgRNA sequences in N. benthamiana should be considered in future experiments, especially if the simultaneous editing of multiple genes is planned.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biology13010053/s1, Table S1. Plasmids used in this study; Table S2. Primers used in this study; Table S3. Target-specific oligonucleotides used in this study; Table S4. Sequence analysis of possible off-target sites in lyk10 mutants; Figure S1. Illustration of the cloning procedure to generate CRISPR/Cas9 binary vectors with two sgRNA expression cassettes; Figure S2. Identification of M. truncatula plants with mutations in MtLYK10, MtMFS1, and MtMFS2; Figure S3. PCR analysis of the P3 plant containing a deletion in MtLYK10.
Author Contributions
Conceptualization, D.R., Z.-P.X. and C.S.; methodology, C.-X.Z., R.-J.L., D.R., Z.-P.X. and C.S.; validation, C.-X.Z., R.-J.L., Z.-P.X. and C.S.; investigation, C.-X.Z. and R.-J.L.; resources, Z.-P.X. and C.S.; writing—original draft preparation, C.-X.Z.; writing—review and editing, C.-X.Z., R.-J.L., L.B., D.R., Z.-P.X. and C.S.; visualization, Z.-P.X. and C.S.; supervision, Z.-P.X. and C.S.; project administration, D.R., Z.-P.X. and C.S.; funding acquisition, D.R., Z.-P.X. and C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Sino-Swiss Science and Technology Cooperation Program, grant 32161133026 from the National Natural Science Foundation of China to C.S., grant IZLCZ0-206026 from the Swiss National Science Foundation to D.R.; research in Guangzhou was also supported by the National Natural Science Foundation of China (grant 31670241 to C.S.), the Guangzhou Expert Program for Rural Science and Technology (grant 20212100024 from the Guangzhou Municipal Science and Technology Bureau to Z.-P.X.), the Science Foundation of the State Key Laboratory of Biocontrol, and the Guangdong Key Laboratory of Plant Resources.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The snRNA promoter sequences PMtU6-1 and PMtU6-26 have been submitted to GenBank (accession numbers OR568513 and OR568514, respectively); plasmids can be requested from C.S.; the plasmids pISV-CRISPR/Cas9PGmUbi (Addgene ID 209189), pUC-tracrRNA-PMtU6-1 (Addgene ID 209190), and pUC-tracrRNA-PMtU6-26 (Addgene ID 209191) are also available via Addgene (https://www.addgene.org; accessed on 7 October 2023).
Acknowledgments
We are grateful to Qi Sun, Ping Yang, and Yan Wang (Sun Yat-sen University) for their help with various aspects of this study. We thank Yao-Guang Liu (South China Agricultural University, Guangzhou, China) for pYLsgRNA-AtU6-1 and pYLCRISPR/Cas9P35S-B. Eva Kondorosi (Hungarian Academy of Sciences, Szeged, Hungary) and Michael Schultze (University of York, York, UK) kindly provided pISV2678. Kai Yang (Sun Yat-sen University) is thanked for E. coli DH10B.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Perret, X.; Staehelin, C.; Broughton, W.J. Molecular basis of symbiotic promiscuity. Microbiol. Mol. Biol. Rev. 2000, 64, 180–201. [Google Scholar] [CrossRef]
- Smith, S.E.; Read, D.J. Mycorrhizal Symbiosis, 3rd ed.; Academic Press: New York, NY, USA, 2008; pp. 1–768. [Google Scholar]
- Barker, D.G.; Bianchi, S.; Blondon, F.; Dattée, Y.; Duc, G.; Essad, S.; Flament, P.; Gallusci, P.; Génier, G.; Guy, P. Medicago truncatula, a model plant for studying the molecular genetics of the Rhizobium-legume symbiosis. Plant Mol. Biol. Rep. 1990, 8, 40–49. [Google Scholar] [CrossRef]
- Bandyopadhyay, K.; Verdier, J.; Kang, Y. The model legume Medicago truncatula: Past, present, and future. In Plant Biotechnology: Progress in Genomic Era; Khurana, S., Gaur, R., Eds.; Springer: Singapore, 2019; pp. 109–130. [Google Scholar]
- Young, N.D.; Debellé, F.; Oldroyd, G.E.D.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.X.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef]
- Moll, K.M.; Zhou, P.; Ramaraj, T.; Fajardo, D.; Devitt, N.P.; Sadowsky, M.J.; Stupar, R.M.; Tiffin, P.; Miller, J.R.; Young, N.D. Strategies for optimizing BioNano and Dovetail explored through a second reference quality assembly for the legume model, Medicago truncatula. BMC Genom. 2017, 18, 578. [Google Scholar] [CrossRef]
- Kaur, P.; Lui, C.; Dudchenko, O.; Nandety, R.S.; Hurgobin, B.; Pham, M.; Aiden, E.L.; Wen, J.; Mysore, K. Delineating the Tnt1 insertion landscape of the model legume Medicago truncatula cv. R108 at the Hi-C resolution using a chromosome-length genome assembly. Int. J. Mol. Sci. 2021, 22, 4326. [Google Scholar] [CrossRef]
- Medicago truncatula R108 Hi-C Genome Portal (MtrunR108_HiC). Available online: https://medicago.toulouse.inrae.fr/MtrunR108_HiC/ (accessed on 1 November 2021).
- Li, A.; Liu, A.; Wu, S.; Qu, K.; Hu, H.; Yang, J.; Shrestha, N.; Liu, J.; Ren, G. Comparison of structural variants in the whole genome sequences of two Medicago truncatula ecotypes: Jemalong A17 and R108. BMC Plant Biol. 2022, 22, 77. [Google Scholar] [CrossRef]
- Tadege, M.; Wen, J.; He, J.; Tu, H.; Kwak, Y.; Eschstruth, A.; Cayrel, A.; Endre, G.; Zhao, P.X.; Chabaud, M. Large-scale insertional mutagenesis using the Tnt1 retrotransposon in the model legume Medicago truncatula. Plant J. 2008, 54, 335–347. [Google Scholar] [CrossRef]
- Sun, L.; Gill, U.S.; Nandety, R.S.; Kwon, S.; Mehta, P.; Dickstein, R.; Udvardi, M.K.; Mysore, K.S.; Wen, J. Genome-wide analysis of flanking sequences reveals that Tnt1 insertion is positively correlated with gene methylation in Medicago truncatula. Plant J. 2019, 98, 1106–1119. [Google Scholar] [CrossRef]
- Carrere, S.; Verdier, J.; Gamas, P. MtExpress, a comprehensive and curated RNAseq-based gene expression atlas for the model legume Medicago truncatula. Plant Cell Physiol. 2021, 62, 1494–1500. [Google Scholar] [CrossRef]
- Hoffmann, B.; Trinh, T.H.; Leung, J.; Kondorosi, A.; Kondorosi, E. A new Medicago truncatula line with superior in vitro regeneration, transformation, and symbiotic properties isolated through cell culture selection. Mol. Plant Microbe Interact. 1997, 10, 307–315. [Google Scholar] [CrossRef]
- Trinh, T.H.; Ratet, P.; Kondorosi, E.; Durand, P.; Kamaté, K.; Bauer, P.; Kondorosi, A. Rapid and efficient transformation of diploid Medicago truncatula and Medicago sativa ssp. falcata lines improved in somatic embryogenesis. Plant Cell Rep. 1998, 17, 345–355. [Google Scholar] [CrossRef]
- Cosson, V.; Eschstruth, A.; Ratet, P. Medicago truncatula transformation using leaf explants. Methods Mol. Biol. 2015, 1223, 43–56. [Google Scholar]
- Jiang, Q.; Fu, C.; Wang, Z.Y. A unified Agrobacterium-mediated transformation protocol for alfalfa (Medicago sativa L.) and Medicago truncatula. Methods Mol. Biol. 2019, 1864, 153–163. [Google Scholar]
- Trieu, A.T.; Burleigh, S.H.; Kardailsky, I.V.; Maldonado-Mendoza, I.E.; Versaw, W.K.; Blaylock, L.A.; Shin, H.; Chiou, T.J.; Katagi, H.; Dewbre, G.R.; et al. Transformation of Medicago truncatula via infiltration of seedlings or flowering plants with Agrobacterium. Plant J. 2000, 22, 531–541. [Google Scholar] [CrossRef]
- Boisson-Dernier, A.; Chabaud, M.; Garcia, F.; Bécard, G.; Rosenberg, C.; Barker, D.G. Agrobacterium rhizogenes-transformed roots of Medicago truncatula for the study of nitrogen-fixing and endomycorrhizal symbiotic associations. Mol. Plant Microbe Interact. 2001, 14, 695–700. [Google Scholar] [CrossRef]
- Chabaud, M.; Boisson-Dernier, A.; Zhang, J.; Taylor, C.G.; Yu, O.; Barker, D.G. Agrobacterium rhizogenes-mediated root transformation. In Medicago truncatula Handbook; Mathesius, U., Journet, E.P., Sumner, L.W., Eds.; The Samuel Roberts Noble Foundation: Ardmore, OK, USA, 2006; pp. 1–8. [Google Scholar]
- Barrangou, R. The roles of CRISPR–Cas systems in adaptive immunity and beyond. Curr. Opin. Immunol. 2015, 32, 36–41. [Google Scholar] [CrossRef]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The biology of CRISPR-Cas: Backward and forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef]
- Xue, C.; Greene, E.C. DNA repair pathway choices in CRISPR-Cas9-mediated genome editing. Trends Genet. 2021, 37, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, C.; Gao, C. Applications of CRISPR-Cas in agriculture and plant biotechnology. Nat. Rev. Mol. Cell Biol. 2020, 21, 661–677. [Google Scholar] [CrossRef]
- Curtin, S.J.; Tiffin, P.; Guhlin, J.; Trujillo, D.I.; Burghart, L.T.; Atkins, P.; Baltes, N.J.; Denny, R.; Voytas, D.F.; Stupar, R.M.; et al. Validating genome-wide association candidates controlling quantitative variation in nodulation. Plant Physiol. 2017, 173, 921–931. [Google Scholar] [CrossRef]
- Čermák, T.; Curtin, S.J.; Gil-Humanes, J.; Čegan, R.; Kono, T.J.Y.; Konečná, E.; Belanto, J.J.; Starker, C.G.; Mathre, J.W.; Greenstein, R.L.; et al. A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 2017, 29, 1196–1217. [Google Scholar] [CrossRef]
- Meng, Y.; Hou, Y.; Wang, H.; Ji, R.; Liu, B.; Wen, J.; Niu, L.; Lin, H. Targeted mutagenesis by CRISPR/Cas9 system in the model legume Medicago truncatula. Plant Cell Rep. 2017, 36, 371–374. [Google Scholar] [CrossRef]
- Curtin, S.J.; Xiong, Y.; Michno, J.M.; Campbell, B.W.; Stec, A.O.; Čermák, T.; Starker, C.; Voytas, D.F.; Eamens, A.L.; Stupar, R.M. CRISPR/Cas9 and TALENs generate heritable mutations for genes involved in small RNA processing of Glycine max and Medicago truncatula. Plant Biotechnol. J. 2018, 16, 1125–1137. [Google Scholar] [CrossRef]
- Wolabu, T.W.; Park, J.J.; Chen, M.; Cong, L.; Ge, Y.; Jiang, Q.; Debnath, S.; Li, G.; Wen, J.; Wang, Z. Improving the genome editing efficiency of CRISPR/Cas9 in Arabidopsis and Medicago truncatula. Planta 2020, 252, 15. [Google Scholar] [CrossRef]
- Confalonieri, M.; Carelli, M.; Gianoglio, S.; Moglia, A.; Biazzi, E.; Tava, A. CRISPR/Cas9-mediated targeted mutagenesis of CYP93E2 modulates the triterpene saponin biosynthesis in Medicago truncatula. Front. Plant Sci. 2021, 12, 690231. [Google Scholar] [CrossRef]
- Zhu, F.; Deng, J.; Chen, H.; Liu, P.; Zheng, L.; Ye, Q.; Li, R.; Brault, M.; Wen, J.; Frugier, F.; et al. A CEP peptide receptor-like kinase regulates auxin biosynthesis and ethylene signaling to coordinate root growth and symbiotic nodulation in Medicago truncatula. Plant Cell 2020, 32, 2855–2877. [Google Scholar] [CrossRef]
- Zhu, F.; Ye, Q.; Chen, H.; Dong, J.; Wang, T. Multigene editing reveals that MtCEP1/2/12 redundantly control lateral root and nodule number in Medicago truncatula. J. Exp. Bot. 2021, 72, 3661–3676. [Google Scholar] [CrossRef]
- Luu, T.B.; Carles, N.; Bouzou, L.; Gibelin-Viala, C.; Remblière, C.; Gasciolli, V.; Bono, J.J.; Lefebvre, B.; Pauly, N.; Cullimore, J. Analysis of the structure and function of the LYK cluster of Medicago truncatula A17 and R108. Plant Sci. 2023, 332, 111696. [Google Scholar] [CrossRef]
- Yang, S.; Wang, Q.; Fedorova, E.; Liu, J.; Qin, Q.; Zheng, Q.; Price, P.A.; Pan, H.; Wang, D.; Griffitts, J.S.; et al. Microsymbiont discrimination mediated by a host-secreted peptide in Medicago truncatula. Proc. Natl. Acad. Sci. USA 2017, 114, 6848–6853. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, S.; Liu, J.; Terecskei, K.; Ábrahám, E.; Gombár, A.; Domonkos, Á.; Szűcs, A.; Körmöczi, P.; Wang, T.; et al. Host-secreted antimicrobial peptide enforces symbiotic selectivity in Medicago truncatula. Proc. Natl. Acad. Sci. USA 2017, 114, 6854–6859. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, J.; Li, H.; Yang, S.; Körmöczi, P.; Kereszt, A.; Zhu, H. Nodule-specific cysteine-rich peptides negatively regulate nitrogen-fixing symbiosis in a strain-specific manner in Medicago truncatula. Mol. Plant Microbe Interact. 2018, 31, 240–248. [Google Scholar] [CrossRef]
- Kim, G.B.; Son, S.U.; Yu, H.J.; Mun, J.H. MtGA2ox10 encoding C20-GA2-oxidase regulates rhizobial infection and nodule development in Medicago truncatula. Sci. Rep. 2019, 9, 5952. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, Y.; Zhang, H.; Xu, Y.; Zhou, C.; Liu, W.; Zhu, R.; Shang, C.; Li, J.; Shen, Z.; et al. Efficient generation of CRISPR/Cas9-mediated homozygous/biallelic Medicago truncatula mutants using a hairy root system. Front. Plant Sci. 2020, 11, 294. [Google Scholar] [CrossRef] [PubMed]
- Lacchini, E.; Erffelinck, M.L.; Mertens, J.; Marcou, S.; Molina-Hidalgo, F.J.; Tzfadia, O.; Venegas-Molina, J.; Cárdenas, P.D.; Pollier, J.; Tava, A.; et al. The saponin bomb: A nucleolar-localized β-glucosidase hydrolyzes triterpene saponins in Medicago truncatula. New Phytol. 2023, 239, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Maillet, F.; Fournier, J.; Mendis, H.C.; Tadege, M.; Wen, J.; Ratet, P.; Mysore, K.S.; Gough, C.; Jones, K. Sinorhizobium meliloti succinylated high-molecular-weight succinoglycan and the Medicago truncatula LysM receptor-like kinase MtLYK10 participate independently in symbiotic infection. Plant J. 2020, 102, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Hood, E.E.; Gelvin, S.B.; Melchers, L.S.; Hoekema, A. New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res. 1993, 2, 208–218. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, L.Y.; Liu, W.; Tian, Y.; Xiong, J.S.; Wang, Y.H.; Li, R.J.; Li, H.M.; Wen, J.; Mysore, K.; et al. Role of the Nod factor hydrolase MtNFH1 in regulating Nod factor levels during rhizobial infection and in mature nodules of Medicago truncatula. Plant Cell 2018, 30, 397–414. [Google Scholar] [CrossRef]
- Agbagwa, I.; Datta, S.; Patil, P.; Singh, P.; Nadarajan, N. A protocol for high-quality genomic DNA extraction from legumes. Genet. Mol. Res. 2012, 11, 4632–4639. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y.G. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Bernard, P. Positive selection of recombinant DNA by CcdB. Biotechniques 1996, 21, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.E.; Tour, O.; Palmer, A.E.; Steinbach, P.A.; Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 7877–7882. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, F.; Zhu, P.F.; Khan, A.; Xie, Z.P.; Staehelin, C. Use of the rhizobial type III effector gene nopP to improve Agrobacterium rhizogenes-mediated transformation of Lotus japonicus. Plant Methods 2021, 17, 66. [Google Scholar] [CrossRef]
- Töpfer, R.; Matzeit, V.; Gronenborn, B.; Schell, J.; Steinbiss, H.H. A set of plant expression vectors for transcriptional and translational fusions. Nucleic Acids Res. 1987, 15, 5890. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, C.; Xiao, W.; Yuan, D.; Wan, G.; Ma, L. Site-directed mutagenesis by combination of homologous recombination and DpnI digestion of the plasmid template in Escherichia coli. Anal. Biochem. 2008, 373, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Kemper, E.; Schell, J.; Masterson, R.J. New plant binary vectors with selectable markers located proximal to the left T-DNA border. Plant Mol. Biol. 1992, 20, 1195–1197. [Google Scholar] [CrossRef]
- Norrander, J.; Kempe, T.; Messing, J. Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene 1983, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Krishnakumar, V.; Bidwell, S.; Rosen, B.; Chan, A.; Zhou, S.; Gentzbittel, L.; Childs, K.L.; Yandell, M.; Gundlach, H.; et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genom. 2014, 15, 312. [Google Scholar] [CrossRef]
- Medicago truncatula Genome Database. Available online: https://medicago.toulouse.inrae.fr/MtrunR108_HiC/ (accessed on 2 July 2020).
- Waibel, F.; Filipowicz, W. RNA-polymerase specificity of transcription of Arabidopsis U snRNA genes determined by promoter element spacing. Nature 1990, 346, 199–202. [Google Scholar] [CrossRef]
- Liu, H.; Ding, Y.; Zhou, Y.; Jin, W.; Xie, K.; Chen, L.L. CRISPR-P 2.0: An improved CRISPR-Cas9 tool for genome editing in plants. Mol. Plant 2017, 10, 530–532. [Google Scholar] [CrossRef]
- CRISPR-P 2.0. Available online: http://crispr.hzau.edu.cn/cgi-bin/CRISPR2/CRISPR (accessed on 4 January 2022).
- Xie, X.; Ma, X.; Zhu, Q.; Zeng, D.; Li, G.; Liu, Y.G. CRISPR-GE: A convenient software toolkit for CRISPR-based genome editing. Mol. Plant 2017, 10, 1246–1249. [Google Scholar] [CrossRef]
- targetDesign. Available online: http://skl.scau.edu.cn/targetdesign/ (accessed on 4 January 2022).
- Bird, J.E.; Marles-Wright, J.; Giachino, A. A user’s guide to golden gate cloning methods and standards. ACS Synth. Biol. 2022, 11, 3551–3563. [Google Scholar] [CrossRef]
- Kosugi, S.; Ohashi, Y.; Nakajima, K.; Arai, Y. An improved assay for β-glucuronidase in transformed cells: Methanol almost completely suppresses a putative endogenous β-glucuronidase activity. Plant Sci. 1990, 70, 133–140. [Google Scholar] [CrossRef]
- Li, J.F.; Norville, J.E.; Aach, J.; McCormack, M.; Zhang, D.; Bush, J.; Church, G.M.; Sheen, J.J. Multiplex and homologous recombination–mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 2013, 31, 688–691. [Google Scholar] [CrossRef]
- Schenk, R.U.; Hildebrandt, A.C. Medium and techniques for induction and growth of monocotyledonous and dicotyledonous plant cell cultures. Can. J. Bot. 1972, 50, 199–204. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F.J. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plant 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Broughton, W.J.; Dilworth, M.J. Control of leghaemoglobin synthesis in snake beans. Biochem. J. 1971, 125, 1075–1080. [Google Scholar] [CrossRef]
- offTarget. Available online: http://skl.scau.edu.cn/offtarget/ (accessed on 13 September 2023).
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.S.; Balland-Vanney, M.; Xie, Z.P.; Schultze, M.; Kondorosi, A.; Kondorosi, E.; Staehelin, C. Molecular cloning of a bifunctional β-xylosidase/α-L-arabinosidase from alfalfa roots: Heterologous expression in Medicago truncatula and substrate specificity of the purified enzyme. J. Exp. Bot. 2007, 58, 2799–2810. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, W.; Cai, J.; Zhang, L.Y.; Wong, K.B.; Feddermann, N.; Boller, T.; Xie, Z.P.; Staehelin, C. The nodulation factor hydrolase of Medicago truncatula: Characterization of an enzyme specifically cleaving rhizobial nodulation signals. Plant Physiol. 2013, 163, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Liu, W.; Cheng, J.; Li, R.J.; Wen, J.; Mysore, K.S.; Xie, Z.P.; Reinhardt, D.; Staehelin, C. An extracellular β-N-acetylhexosaminidase of Medicago truncatula hydrolyzes chitooligosaccharides and is involved in arbuscular mycorrhizal symbiosis but not required for nodulation. New Phytol. 2023, 239, 1954–1973. [Google Scholar] [CrossRef]
- Nadal, M.; Sawers, R.; Naseem, S.; Bassin, B.; Kulicke, C.; Sharman, A.; An, G.; An, K.; Ahern, K.R.; Romag, A.; et al. An N-acetylglucosamine transporter required for arbuscular mycorrhizal symbioses in rice and maize. Nat. Plants 2017, 3, 17073. [Google Scholar] [CrossRef]
- Lei, M.J.; Wang, Q.; Li, X.; Chen, A.; Luo, L.; Xie, Y.; Li, G.; Luo, D.; Mysore, K.S.; Wen, J.; et al. The small GTPase ROP10 of Medicago truncatula is required for both tip growth of root hairs and Nod factor-induced root hair deformation. Plant Cell 2015, 27, 806–822. [Google Scholar] [CrossRef]
- Kovács, S.; Kiss, E.; Jenei, S.; Fehér-Juhász, E.; Kereszt, A.; Endre, G. The Medicago truncatula IEF gene is crucial for the progression of bacterial infection during symbiosis. Mol. Plant Microbe Interact. 2022, 35, 401–415. [Google Scholar] [CrossRef]
- Li, R.J.; Zhang, C.X.; Fan, S.Y.; Wang, Y.H.; Wen, J.; Mysore, K.S.; Xie, Z.P.; Staehelin, C. The Medicago truncatula hydrolase MtCHIT5b degrades Nod factors of Sinorhizobium meliloti and cooperates with MtNFH1 to regulate the nodule symbiosis. Front. Plant Sci. 2022, 13, 1034230. [Google Scholar] [CrossRef]
- Hajiahmadi, Z.; Movahedi, A.; Wei, H.; Li, D.; Orooji, Y.; Ruan, H.; Zhuge, Q. Strategies to increase on-target and reduce off-target effects of the CRISPR/Cas9 system in plants. Int. J. Mol. Sci. 2019, 20, 3719. [Google Scholar] [CrossRef]
- Wang, Z.; Shea, Z.; Li, Q.; Wang, K.; Mills, K.; Zhang, B.; Zhao, B. Evaluate the guide RNA effectiveness via Agrobacterium-mediated transient assays in Nicotiana benthamiana. Front. Plant Sci. 2023, 14, 1111683. [Google Scholar] [CrossRef]
- Michno, J.M.; Wang, X.; Liu, J.; Curtin, S.J.; Kono, T.J.; Stupar, R. CRISPR/Cas mutagenesis of soybean and Medicago truncatula using a new web-tool and a modified Cas9 enzyme. GM Crops Food 2015, 6, 243–252. [Google Scholar] [CrossRef]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef] [PubMed]
- Kawaharada, Y.; Kelly, S.; Nielsen, M.W.; Hjuler, C.T.; Gysel, K.; Muszyński, A.; Carlson, R.W.; Thygesen, M.B.; Sandal, N.; Asmussen, M.H.; et al. Receptor-mediated exopolysaccharide perception controls bacterial infection. Nature 2015, 523, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Kawaharada, Y.; Nielsen, M.W.; Kelly, S.; James, E.K.; Andersen, K.R.; Rasmussen, S.R.; Füchtbauer, W.; Madsen, L.H.; Heckmann, A.B.; Radutoiu, S.; et al. Differential regulation of the Epr3 receptor coordinates membrane-restricted rhizobial colonization of root nodule primordia. Nat. Commun. 2017, 8, 14534. [Google Scholar] [CrossRef]
- Wong, J.E.; Gysel, K.; Birkefeldt, T.G.; Vinther, M.; Muszyński, A.; Azadi, P.; Laursen, N.S.; Sullivan, J.T.; Ronson, C.W.; Stougaard, J.; et al. Structural signatures in EPR3 define a unique class of plant carbohydrate receptors. Nat. Commun. 2020, 11, 3797. [Google Scholar] [CrossRef]
- Kelly, S.; Hansen, S.B.; Rübsam, H.; Saake, P.; Pedersen, E.B.; Gysel, K.; Madland, E.; Wu, S.; Wawra, S.; Reid, D.; et al. A glycan receptor kinase facilitates intracellular accommodation of arbuscular mycorrhiza and symbiotic rhizobia in the legume Lotus japonicus. PLoS Biol. 2023, 21, e3002127. [Google Scholar] [CrossRef]
- Leigh, J.A.; Signer, E.R.; Walker, G.C. Exopolysaccharide-deficient mutants of Rhizobium meliloti that form ineffective nodules. Proc. Natl. Acad. Sci. USA 1985, 82, 6231–6235. [Google Scholar] [CrossRef]
- Breedveld, M.W.; Miller, K.J. Cyclic β-glucans of members of the family Rhizobiaceae. Microbiol. Rev. 1994, 58, 145–161. [Google Scholar] [CrossRef]
- Pérez-Mendoza, D.; Rodríguez-Carvajal, M.Á.; Romero-Jiménez, L.; de Araujo Farias, G.; Lloret, J.; Gallegos, M.T.; Sanjuán, J. Novel mixed-linkage β-glucan activated by c-di-GMP in Sinorhizobium meliloti. Proc. Natl. Acad. Sci. USA 2015, 112, E757–E765. [Google Scholar] [CrossRef]
- Cheng, X.; Krom, N.; Zhang, S.; Mysore, K.S.; Udvardi, M.; Wen, J. Enabling reverse genetics in Medicago truncatula using high-throughput sequencing for Tnt1 flanking sequence recovery. Methods Mol. Biol. 2017, 1610, 25–37. [Google Scholar]
- Kamens, J.J. The Addgene repository: An international nonprofit plasmid and data resource. Nucleic Acids Res. 2015, 43, D1152–D1157. [Google Scholar] [CrossRef]
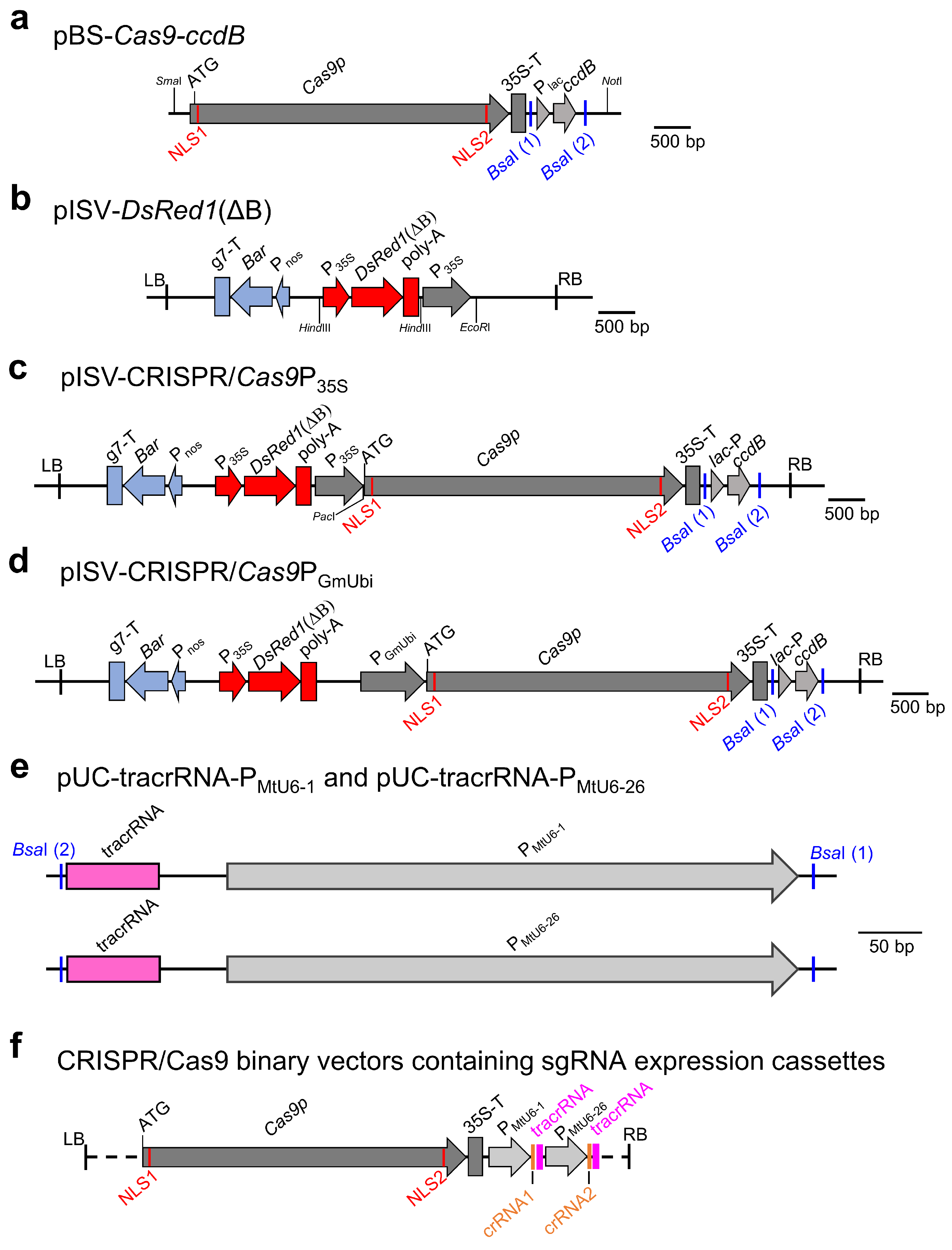
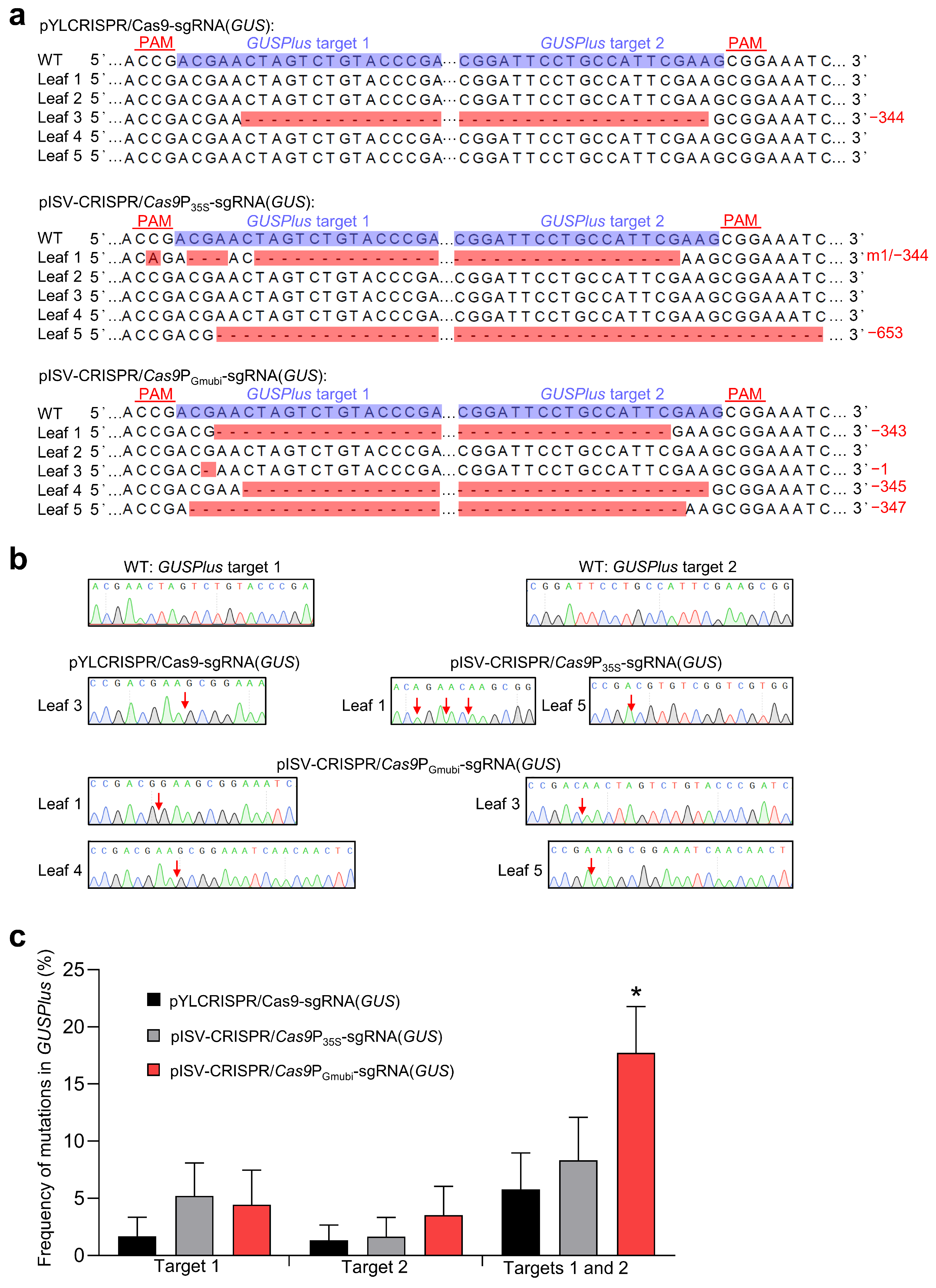
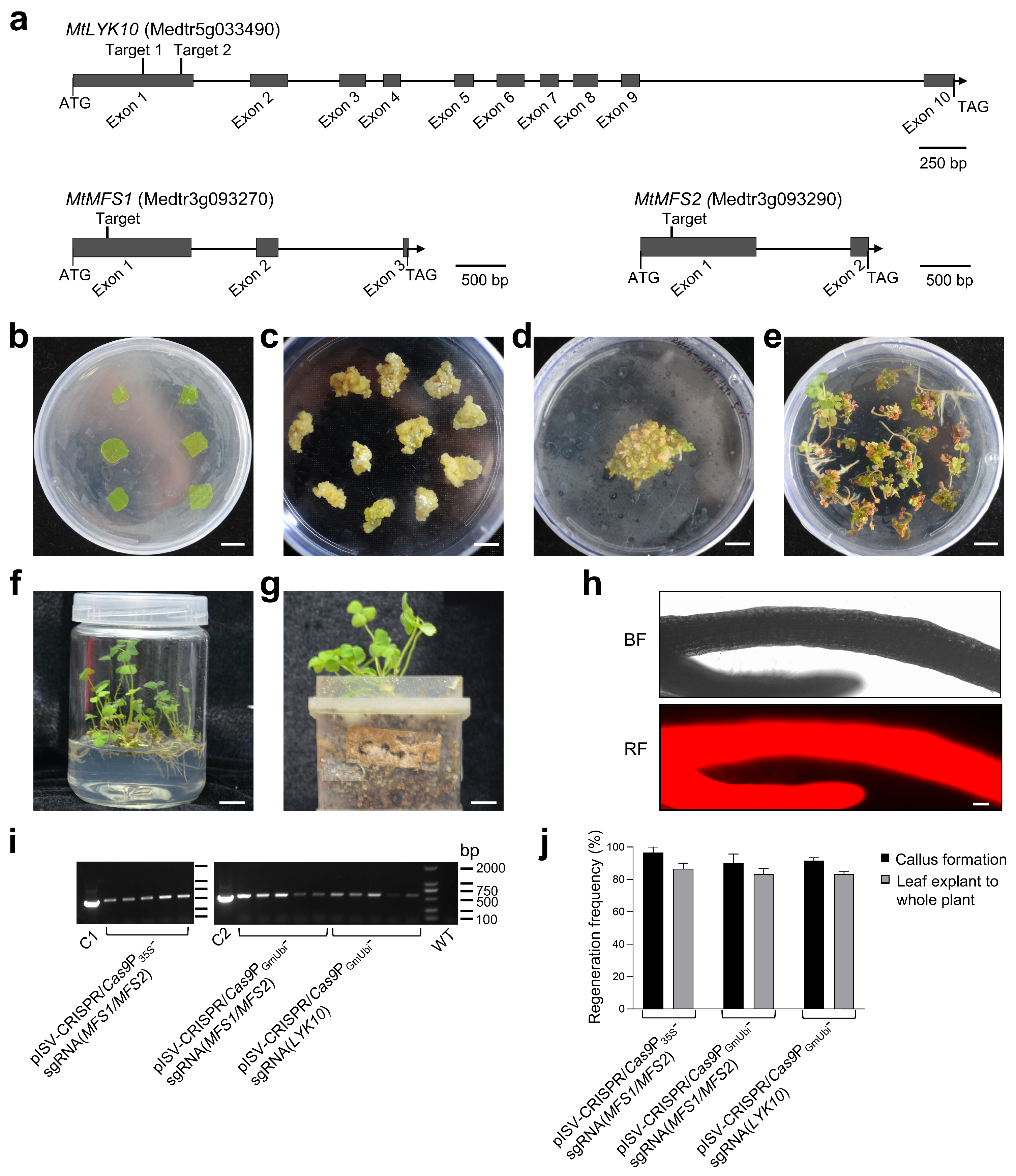
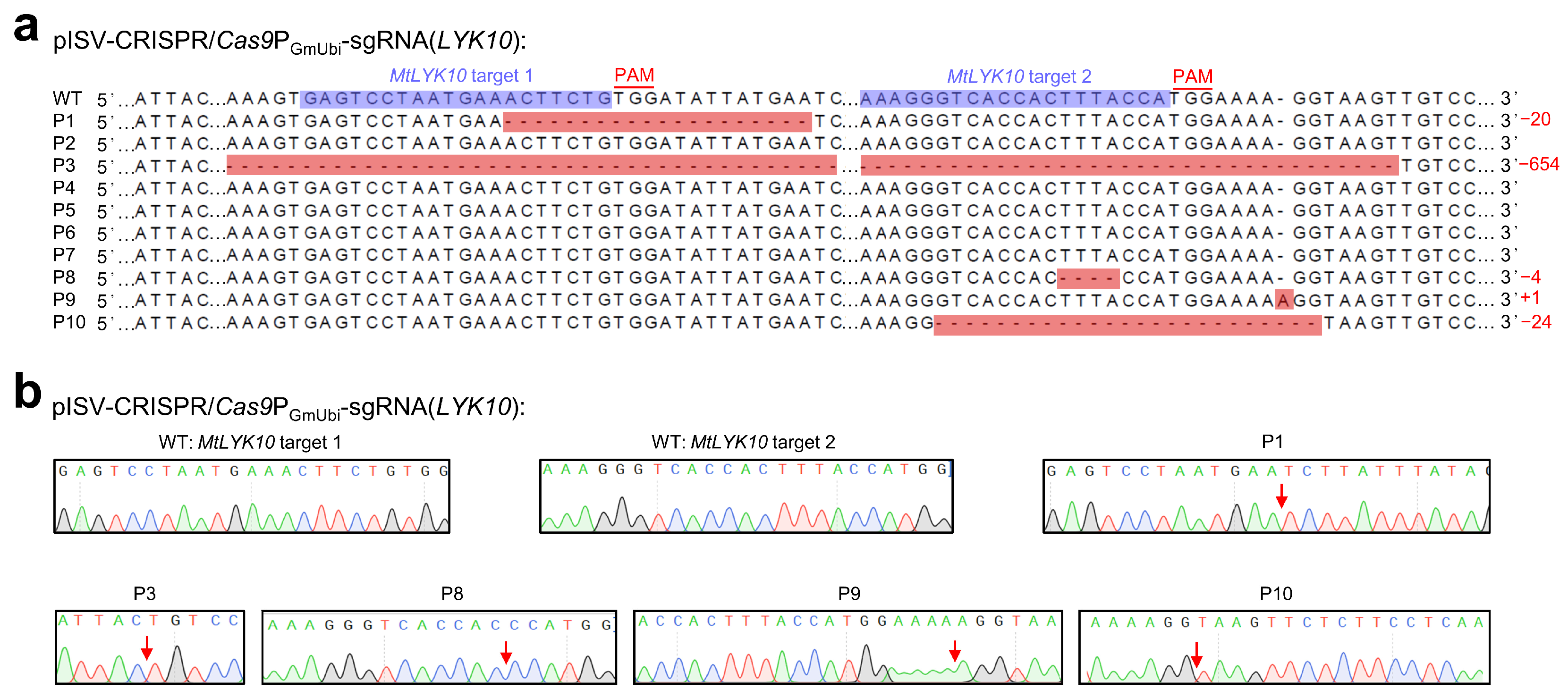
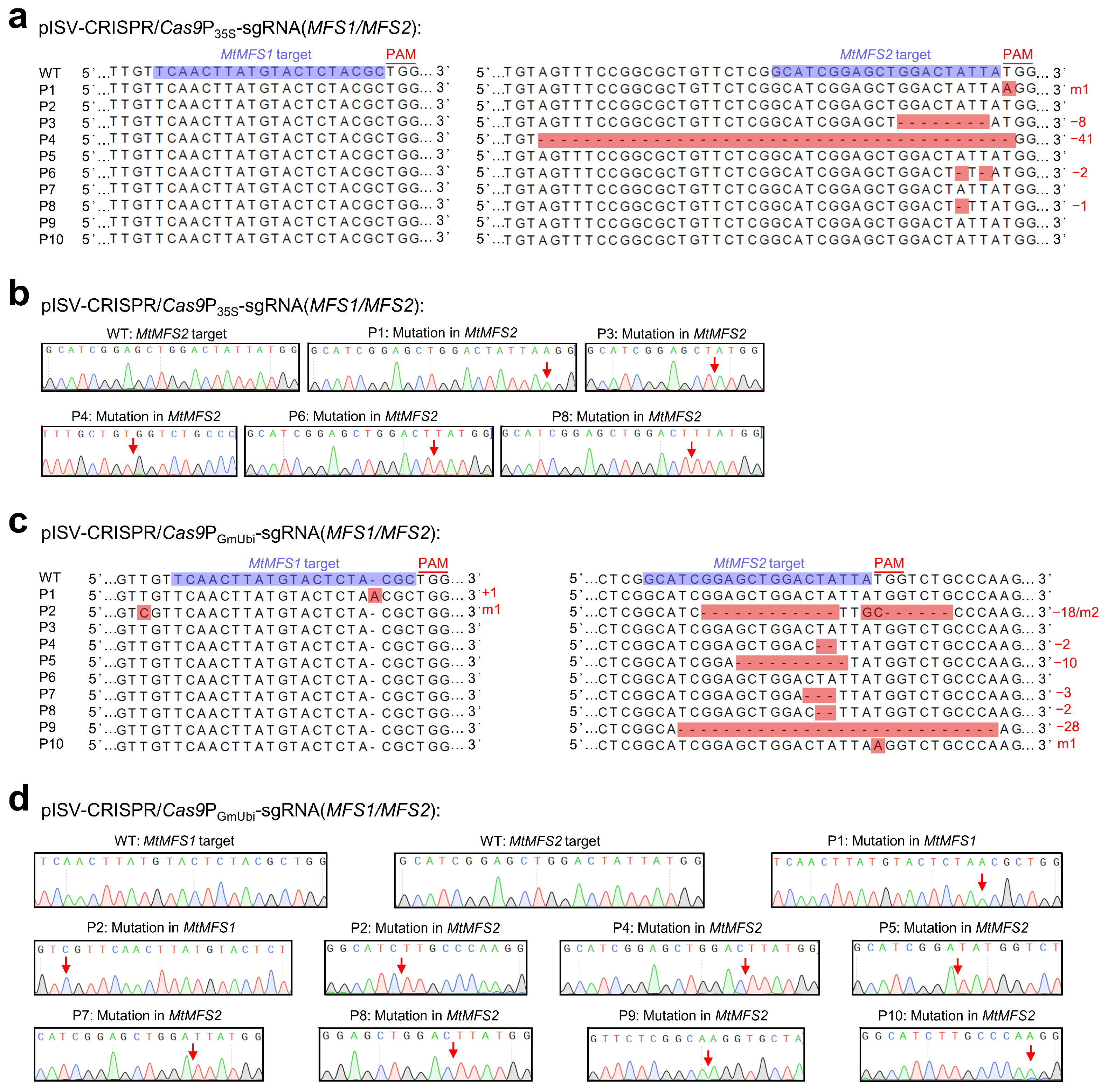
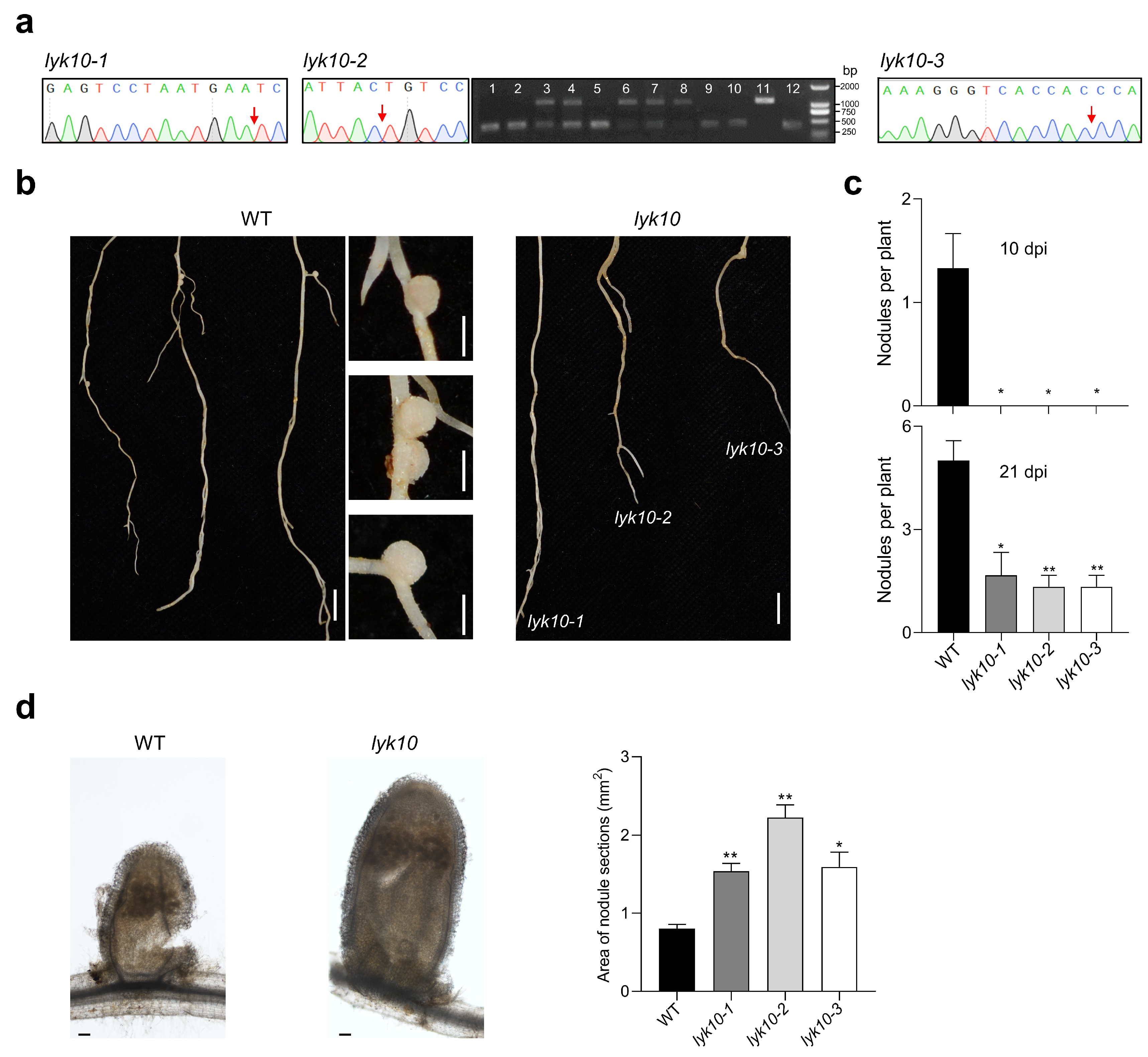
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).