
sTREM2 Differentially Affects Cytokine Expression in Myeloid-Derived Cell Models via MAPK–JNK Signaling Pathway


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Treatment of THP-1 Cells with sTREM2
2.3. Differentiation and Polarization of THP-1 Monocytes into Macrophages
2.4. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Statistical Analysis
3. Results
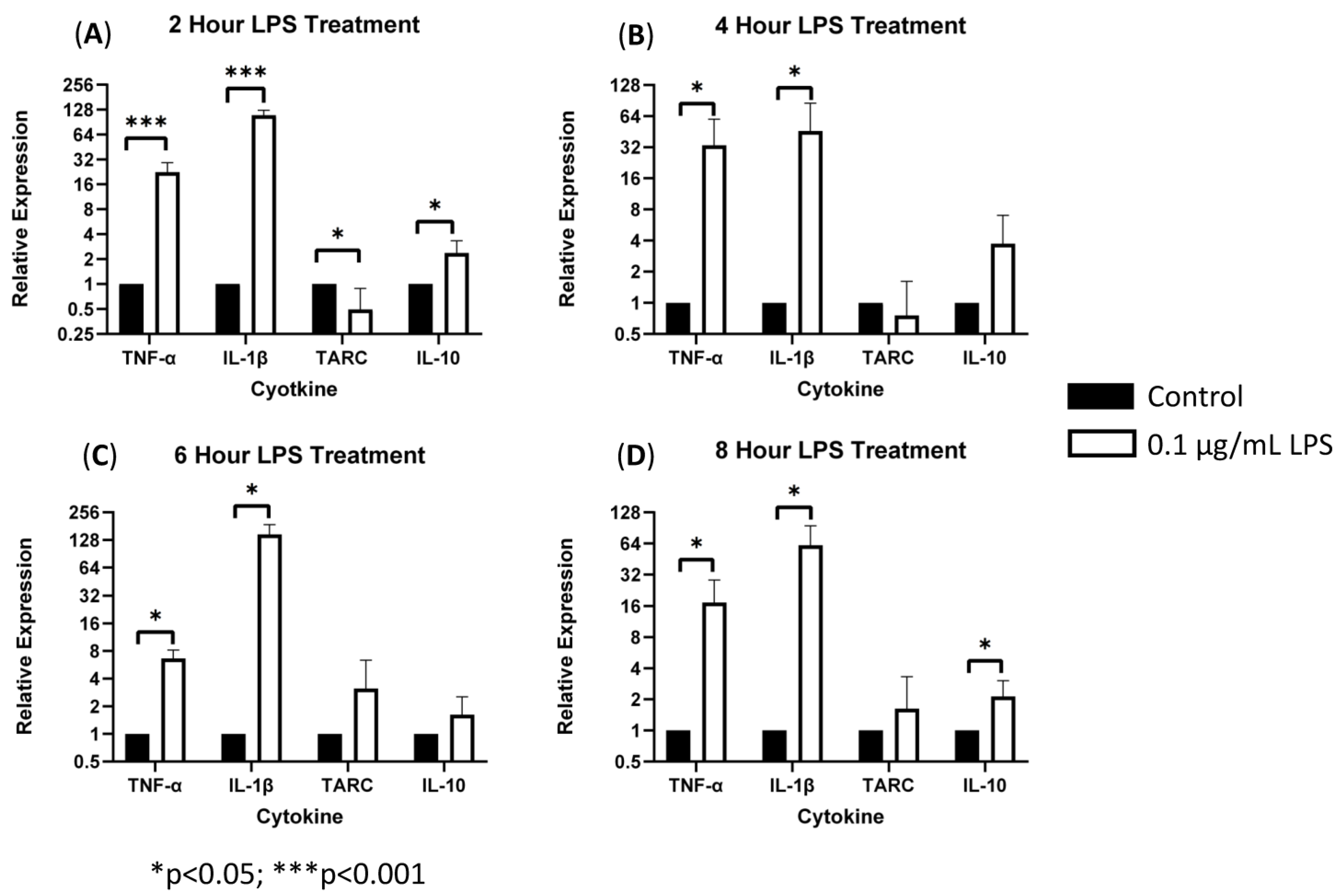
3.1. sTREM2-Induced Expression of Cytokines in THP-1 Cells
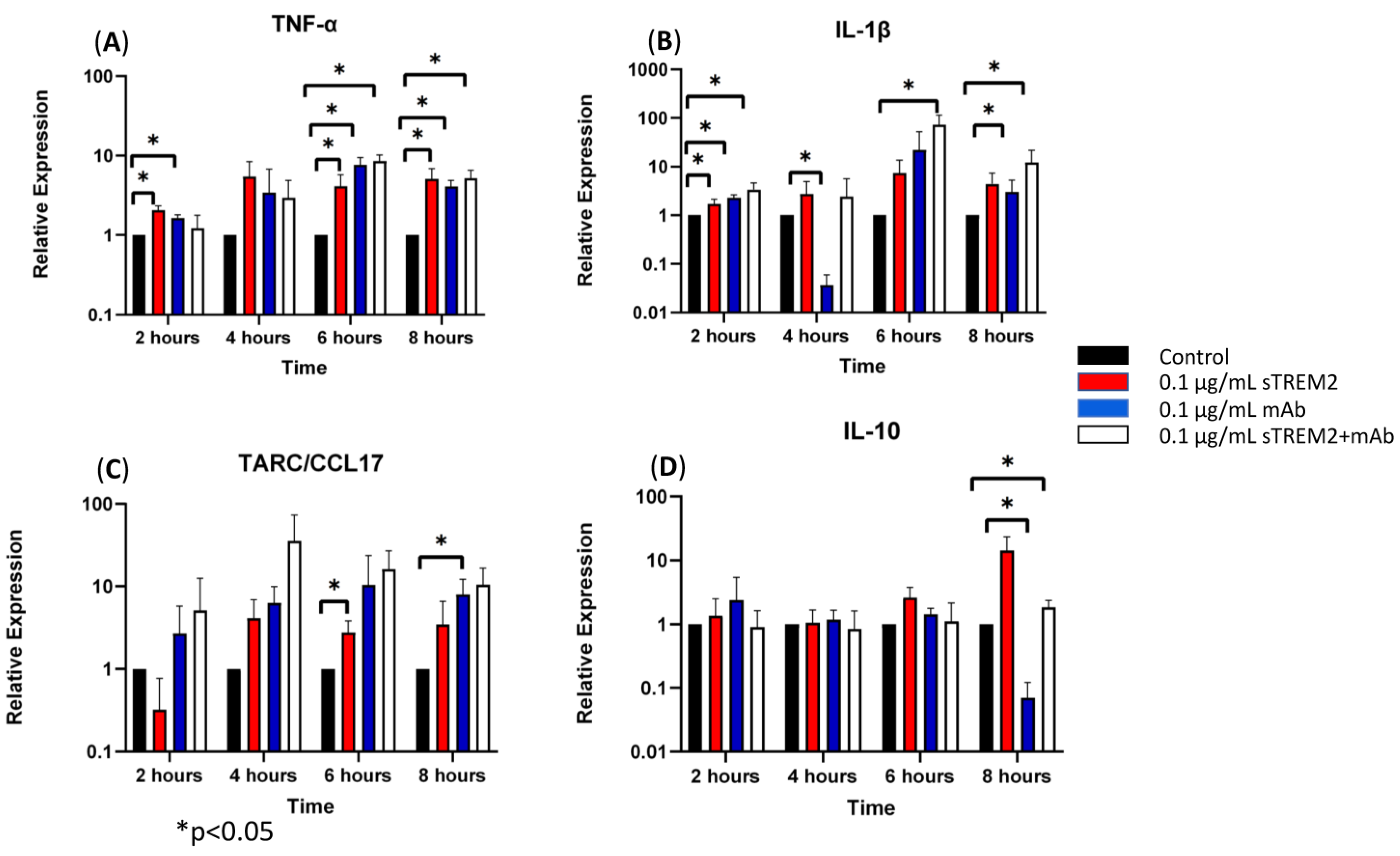
3.2. TREM2 Monoclonal Antibody (mAb) Partially Alleviated sTREM2-Induced Expression of Cytokines
3.3. sTREM2 Induced Expression of Cytokines in Macrophages Differentiated and Polarized from THP-1 Cells
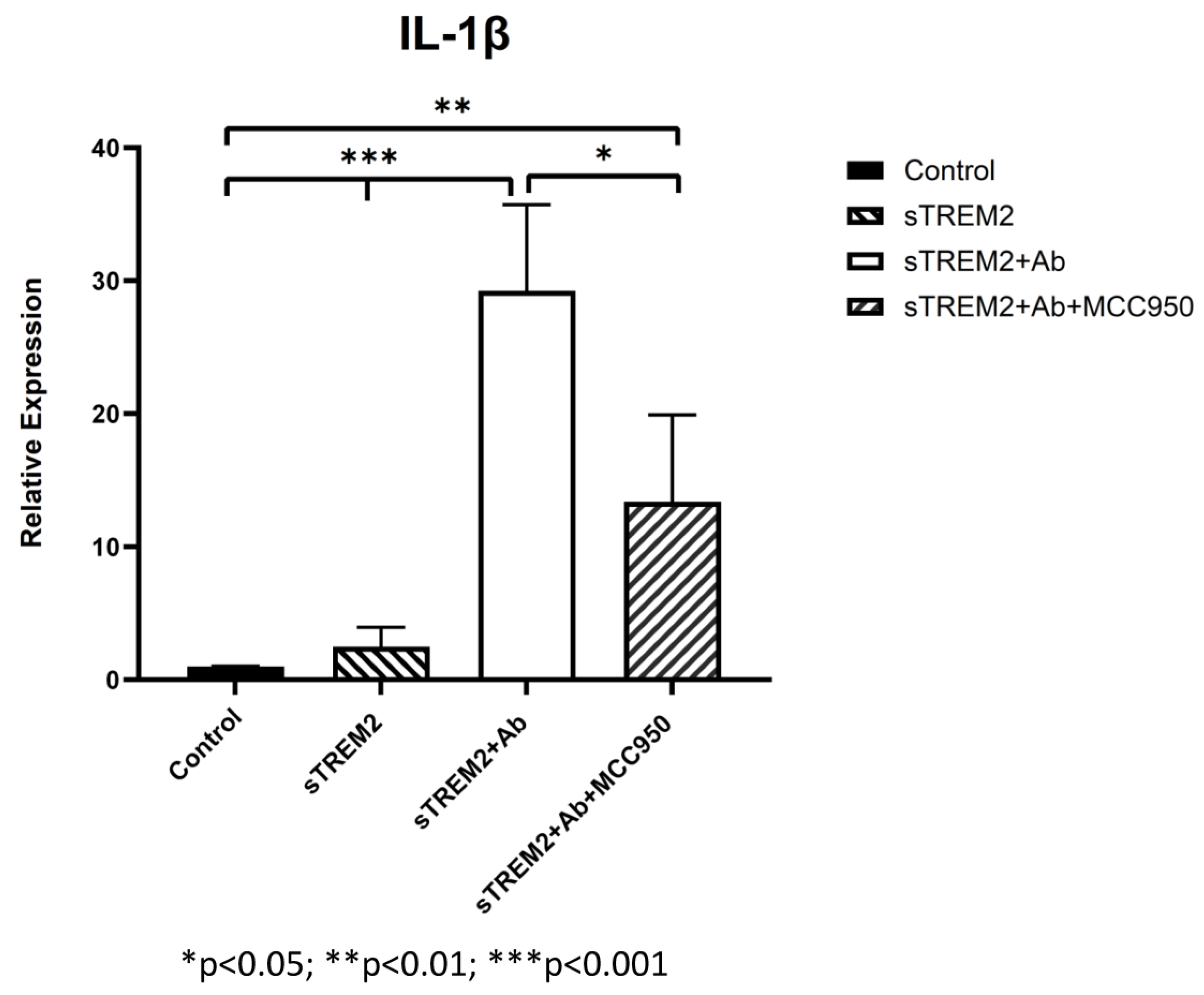
3.4. Inhibition of JNK Blocks sTREM2-Induced Expression of Cytokines in THP-1 Cells
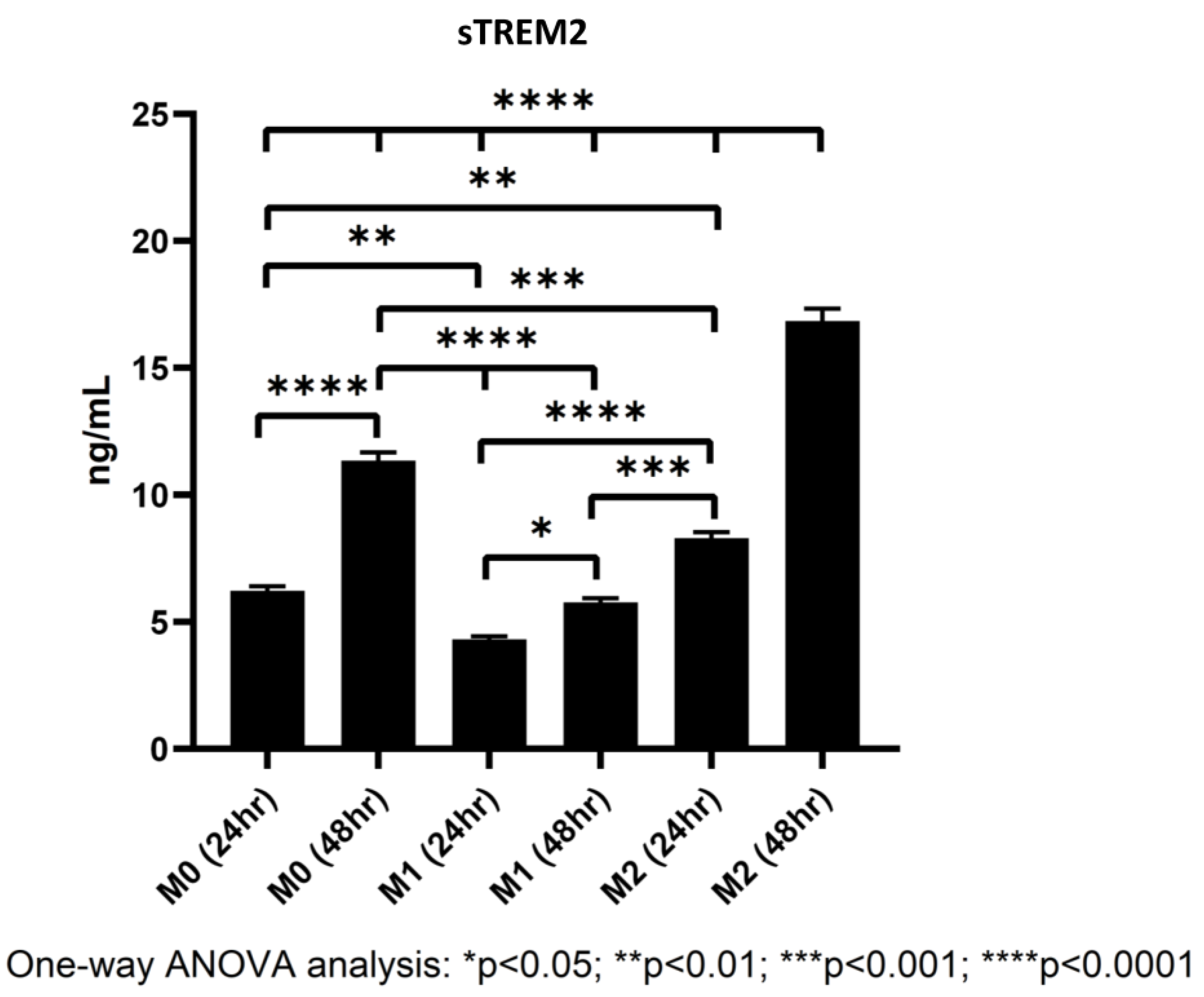
3.5. Polarized Macrophages Differentially Shed sTREM2
4. Discussion
4.1. sTREM2-Induced Cytokine Expression in THP-1 Monocytes
4.2. The Effects of TREM2 Antibody on sTREM2-Induced Cytokine Expression
4.3. The Effects of sTREM2 on Cytokine Expression in Polarized Macrophages
4.4. The Signaling Pathways Involved in sTREM2-Induced Cytokine Expression
4.5. Shedding of sTREM2 by M0, M1, and M2 Macrophages
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting edge: Inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 2000, 164, 4991–4995. [Google Scholar] [CrossRef] [PubMed]
- Kober, D.L.; Brett, T.J. TREM2-ligand interactions in health and disease. J. Mol. Biol. 2017, 11, 1607–1629. [Google Scholar] [CrossRef]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 2013, 288, 33027–33036. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [PubMed]
- Painter, M.M.; Atagi, Y.; Liu, C.; Rademakers, R.; Xu, H.; Fryer, J.D.; Bu, G. TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener. 2015, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef]
- Atagi, Y.; Liu, C.C.; Painter, M.M.; Chen, X.F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The microglial innate immune receptor TREM2 is required for synapse elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991. [Google Scholar] [CrossRef]
- Jay, T.R.; von Saucken, V.E.; Munoz, B.; Codocedo, J.F.; Atwood, B.K.; Lamb, B.T.; Landreth, G.E. TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 2019, 67, 1873–1892. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Coppola, K.; Schweig, J.E.; Crawford, F.; Mullan, M.; Paris, D. Distinct signaling pathways regulate trem2 phagocytic and NFΚB antagonistic activities. Front. Cell. Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef] [PubMed]
- Vilalta, A.; Zhou, Y.; Sevalle, J.; Griffin, J.K.; Satoh, K.; Allendorf, D.H.; De, S.; Puigdellívol, M.; Bruzas, A.; Burguillos, M.A.; et al. Wildtype sTREM2 blocks Abeta aggregation and neurotoxicity, but the Alzheimer’s R47h mutant increases Abeta aggregation. J. Biol. Chem. 2021, 296, 100631. [Google Scholar] [CrossRef]
- Zhong, L.; Xu, Y.; Zhuo, R.; Wang, T.; Wang, K.; Huang, R.; Wang, D.; Gao, Y.; Zhu, Y.; Sheng, X.; et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 1365. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble trem2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Calvet, M.; Morenas-Rodríguez, E.; Kleinberger, G.; Schlepckow, K.; Araque Caballero, M.Á.; Franzmeier, N.; Capell, A.; Fellerer, K.; Nuscher, B.; Eren, E.; et al. Early increase of CSF STREM2 in alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 2019, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chen, X.-F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Vukic, V.; Callaghan, D.; Walker, D.; Lue, L.-H.; Liu, Q.-Y.; Stanimirovic, D.B.; Zhang, W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 2009, 34, 95–106. [Google Scholar] [CrossRef]
- Bamji-Mirza, M.; Callaghan, D.; Najem, D.; Shen, S.; Hasim, M.S.; Yang, Z.; Zhang, W. Stimulation of insulin signaling and inhibition of JNK-AP1 activation protect cells from Aβ-induced signaling dysregulation and inflammatory response. J. Alzheimer’s Dis. 2014, 40, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef]
- London, A.; Cohen, M.; Schwartz, M. Microglia and monocyte-derived macrophages: Functionally distinct populations that act in concert in CNS plasticity and repair. Front. Cell. Neurosci. 2013, 7, 34. [Google Scholar] [CrossRef]
- Ferri, E.; Rossi, P.D.; Geraci, A.; Ciccone, S.; Cesari, M.; Arosio, B. The sTREM2 Concentrations in the Blood: A Marker of Neurodegeneration? Front. Mol. Biosci. 2021, 7, 627931. [Google Scholar] [CrossRef] [PubMed]
- Knapskog, A.B.; Henjum, K.; Idland, A.V.; Eldholm, R.S.; Persson, K.; Saltvedt, I.; Watne, L.O.; Engedal, K.; Nilsson, L.N.G. Cerebrospinal fluid sTREM2 in Alzheimer’s disease: Comparisons between clinical presentation and AT classification. Sci. Rep. 2020, 10, 15886. [Google Scholar] [CrossRef] [PubMed]
- Henjum, K.; Almdahl, I.S.; Årskog, V.; Minthon, L.; Hansson, O.; Fladby, T.; Nilsson, L.N. Cerebrospinal fluid soluble Trem2 in aging and Alzheimer’s disease. Alzheimers Res. Ther. 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef]
- Chen, S.; Peng, J.; Sherchan, P.; Ma, Y.; Xiang, S.; Yan, F.; Zhao, H.; Jiang, Y.; Wang, N.; Zhang, J.H.; et al. TREM2 activation attenuates neuroinflammation and neuronal apoptosis via PI3K/Akt pathway after intracerebral hemorrhage in mice. J. Neuroinflammation. 2020, 17, 168. [Google Scholar] [CrossRef]
- Peng, Q.; Malhotra, S.; Tochia, J.A.; Kerr, W.G.; Coggeshall, K.M.; Humphrey, M.B. TREM2- and DAP12 dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci. Signal. 2010, 3, ra38. [Google Scholar] [CrossRef]
- Hamerman, J.A.; Jarioura, J.R.; Humphrey, M.B.; Nakamura, M.C.; Seaman, W.E.; Lanier, L.L. Cutting edge: Inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells-2 (TREM2) and DAP12. J. Immunol. 2006, 177, 2051–2055. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Hwang, J.C.; Choi, D.Y.; Park, M.H.; Hong, J.T. NF-κB as a Key Mediator of Brain Inflammation in Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2019, 18, 3–10. [Google Scholar] [CrossRef]
- Koprich, J.B.; Reske-Nielsen, C.; Mithal, P.; Isacson, O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J. Neuroinflamm. 2008, 5, 8. [Google Scholar] [CrossRef]
- Morishima, Y.; Gotoh, Y.; Zieg, J.; Barrett, T.; Takano, H.; Flavell, R.; Davis, R.J.; Shirasaki, Y.; Greenberg, M.E. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 2001, 21, 7551–7560. [Google Scholar] [CrossRef]
- Shoji, M.; Iwakami, N.; Takeuchi, S.; Waragai, M.; Suzuki, M.; Kanazawa, I.; Lippa, C.F.; Ono, S.; Okazawa, H. JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res. Mol. Brain Res. 2000, 85, 221–233. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IL-1α | IL-1β | IL-6 | IL-8 | GM-CSF | IFN-γ | MCAF | TNF-α | |
|---|---|---|---|---|---|---|---|---|
| M0 CTR | 25.58 | 28.01 | 9.47 | 183.83 | 111.45 | 28.14 | 31.08 | 101.89 |
| M0 0.1 µg/mL sTREM2 | 22.97 ± 0.42 | 37.26 ± 2.79 | 12.7 ± 0.42 | 163.96 ± 30.11 | 92.11 ± 18.93 | 41.85 ± 22.71 | 34.63 ± 9.31 | 113.42 ± 8.78 |
| M0 1 µg/mL sTREM2 | 29.64 | 36.09 | 10.93 | 165.38 | 99.53 | 41.45 | 31.58 | 104.44 |
| M1 CTR | 24.99 | 28.02 | 9.76 | 188.88 | 121.12 | 35.97 | 36.14 | 113.41 |
| M1 0.1 µg/mL sTREM2 | 24.99 | 33.67 | 9.17 | 135.57 | 86.16 | 29.71 | 31.58 | 109.87 |
| M1 1 µg/mL sTREM2 | 24.99 | 35.28 | 9.46 | 151.9 | 98.05 | 43.81 | 32.09 | 107.21 |
| M2 CTR | 24.99 | 36.9 | 10.64 | 110.74 | 87.64 | 43.8 | 125.28 | 117.84 |
| M2 0.1 µg/mL sTREM2 | 26.16 | 38.52 | 9.76 | 109.32 | 77.97 | 53.98 | 125.29 | 124.04 |
| M2 1 µg/mL sTREM2 | 34.28 | 41.75 | 10.94 | 128.48 | 119.63 | 79.83 | 133.38 | 120.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arsenault, R.; Marshall, S.; Salois, P.; Li, Q.; Zhang, W. sTREM2 Differentially Affects Cytokine Expression in Myeloid-Derived Cell Models via MAPK–JNK Signaling Pathway. Biology 2024, 13, 87. https://doi.org/10.3390/biology13020087
Arsenault R, Marshall S, Salois P, Li Q, Zhang W. sTREM2 Differentially Affects Cytokine Expression in Myeloid-Derived Cell Models via MAPK–JNK Signaling Pathway. Biology. 2024; 13(2):87. https://doi.org/10.3390/biology13020087
Chicago/Turabian StyleArsenault, Ryan, Steven Marshall, Patrick Salois, Qiao Li, and Wandong Zhang. 2024. "sTREM2 Differentially Affects Cytokine Expression in Myeloid-Derived Cell Models via MAPK–JNK Signaling Pathway" Biology 13, no. 2: 87. https://doi.org/10.3390/biology13020087
APA StyleArsenault, R., Marshall, S., Salois, P., Li, Q., & Zhang, W. (2024). sTREM2 Differentially Affects Cytokine Expression in Myeloid-Derived Cell Models via MAPK–JNK Signaling Pathway. Biology, 13(2), 87. https://doi.org/10.3390/biology13020087