
The Omicron XBB.1 Variant and Its Descendants: Genomic Mutations, Rapid Dissemination and Notable Characteristics
 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
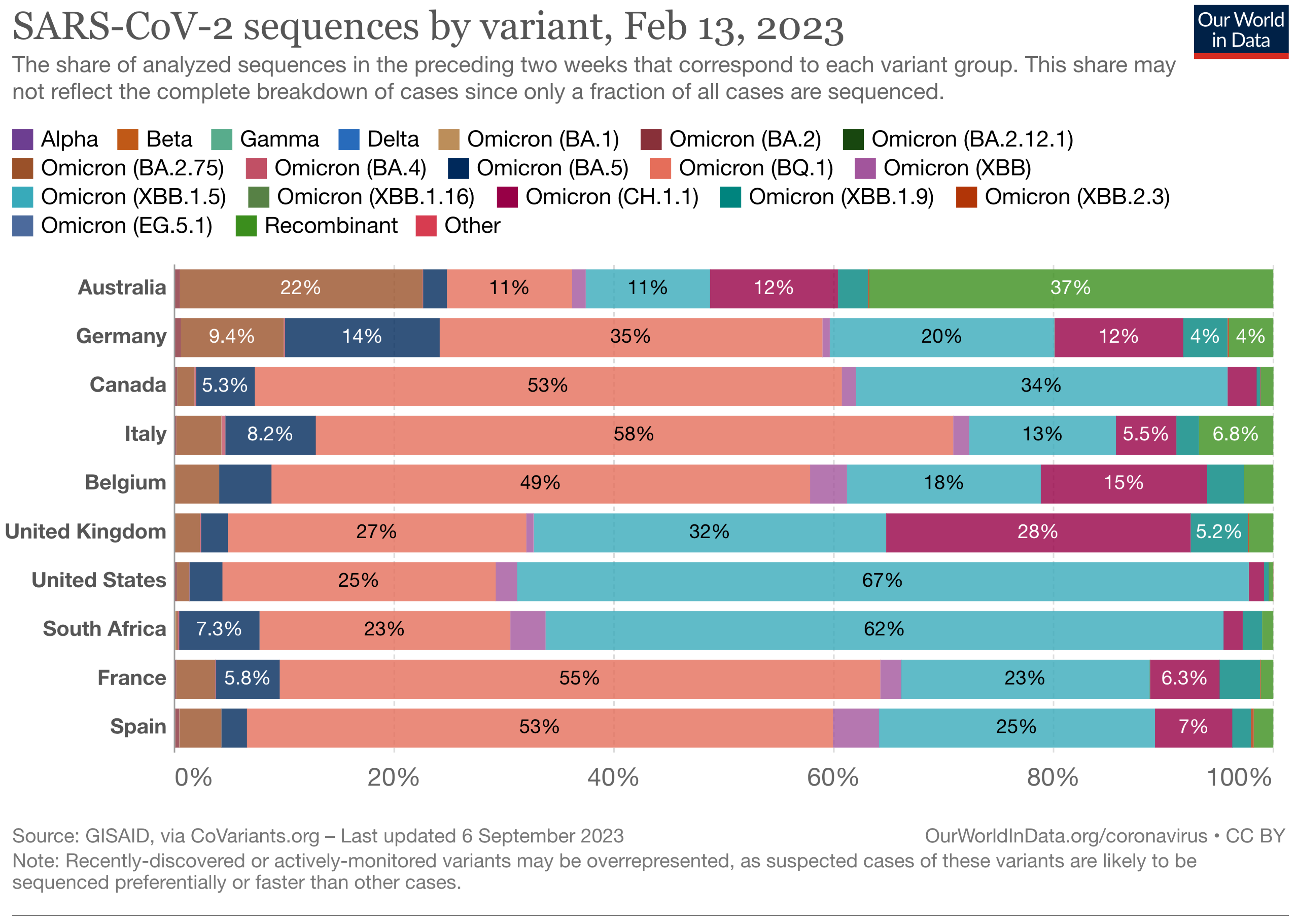
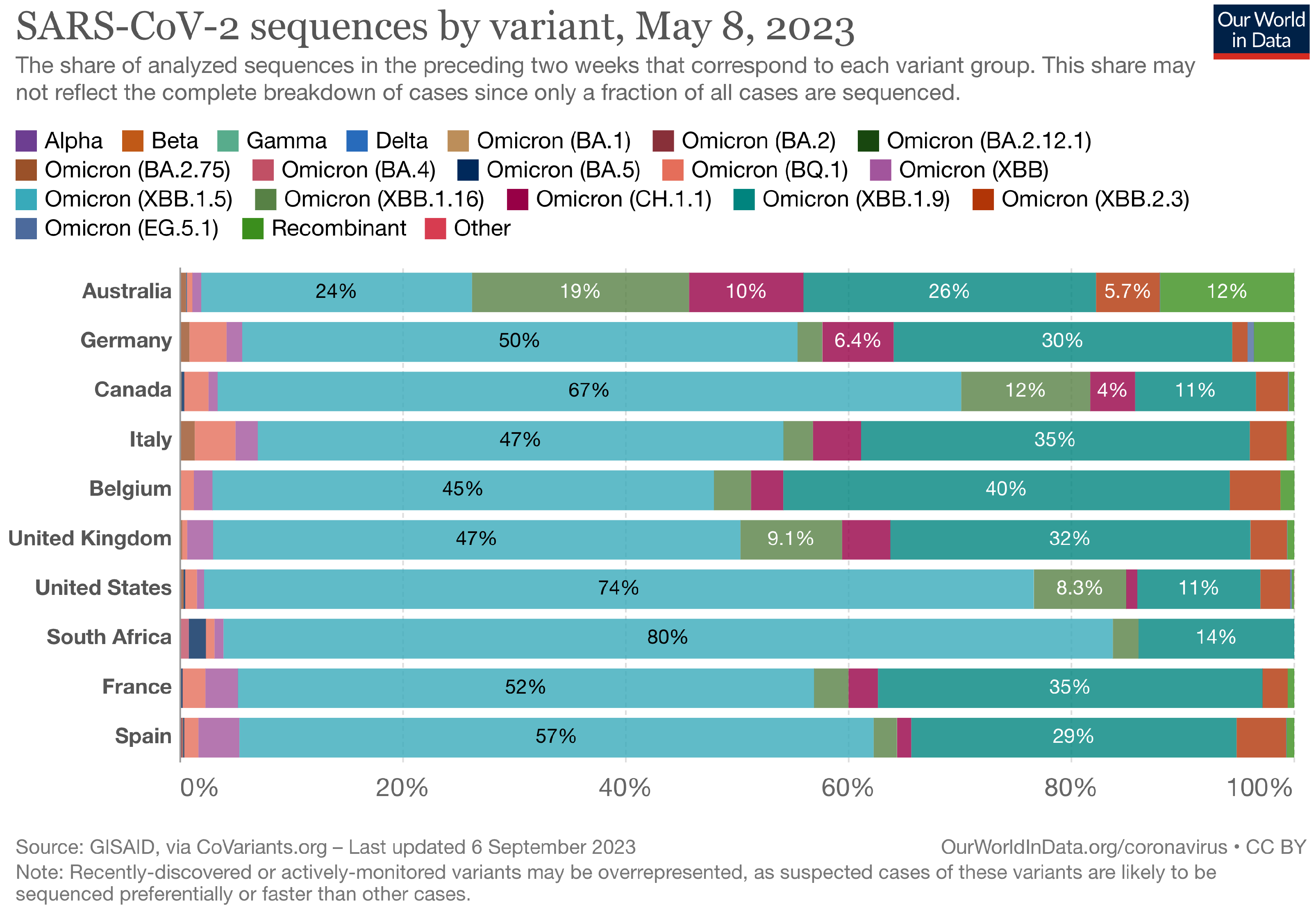
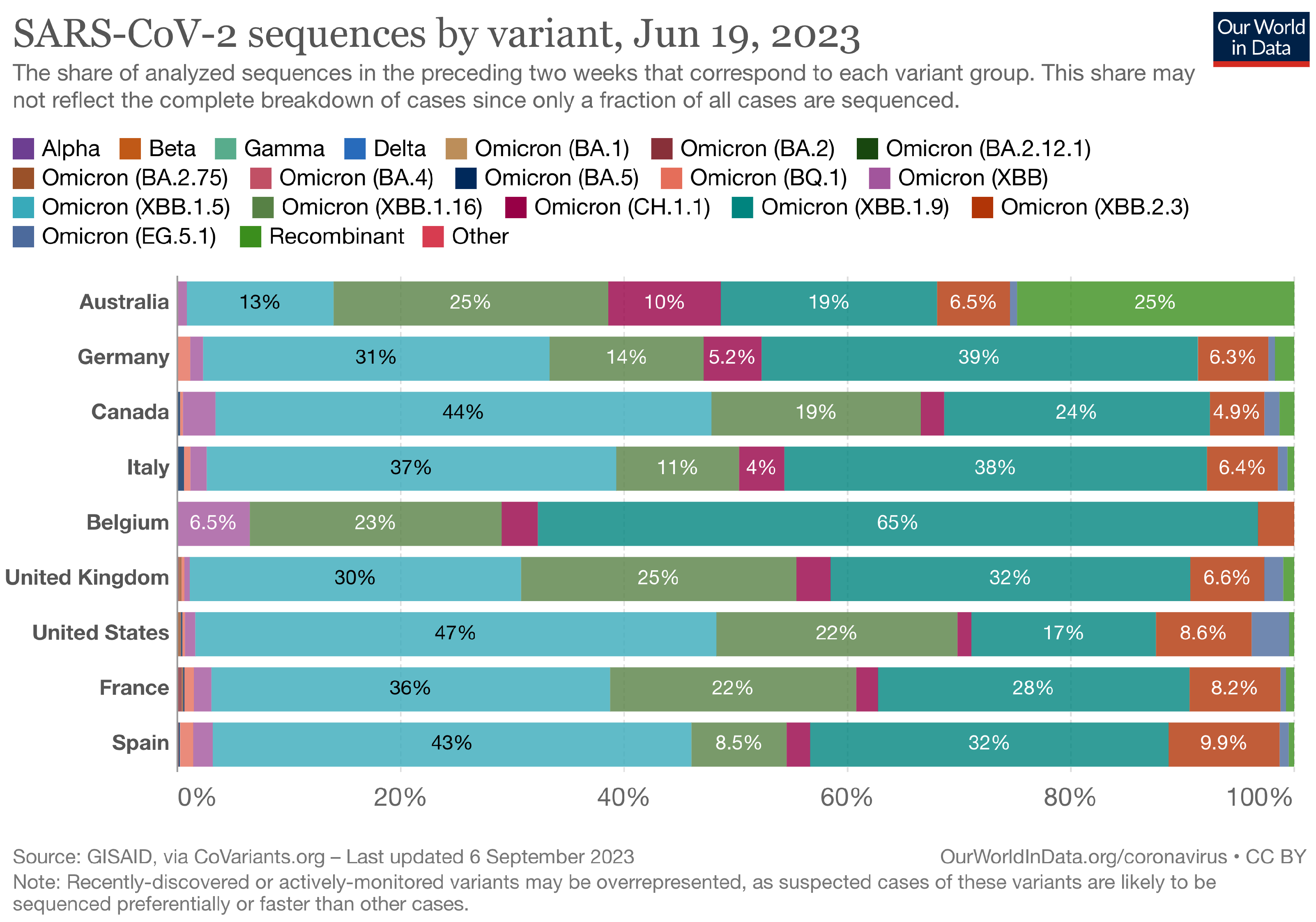
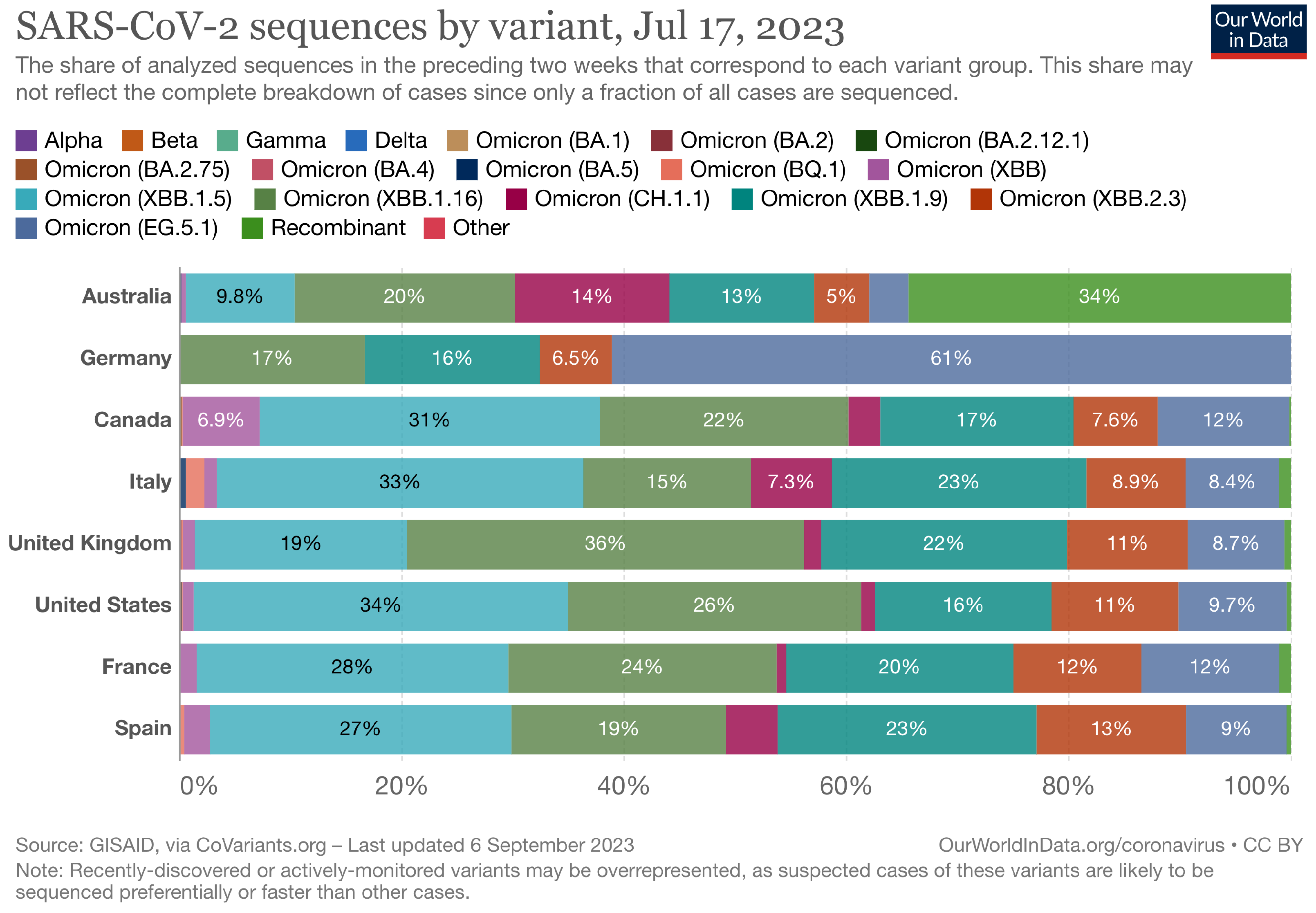
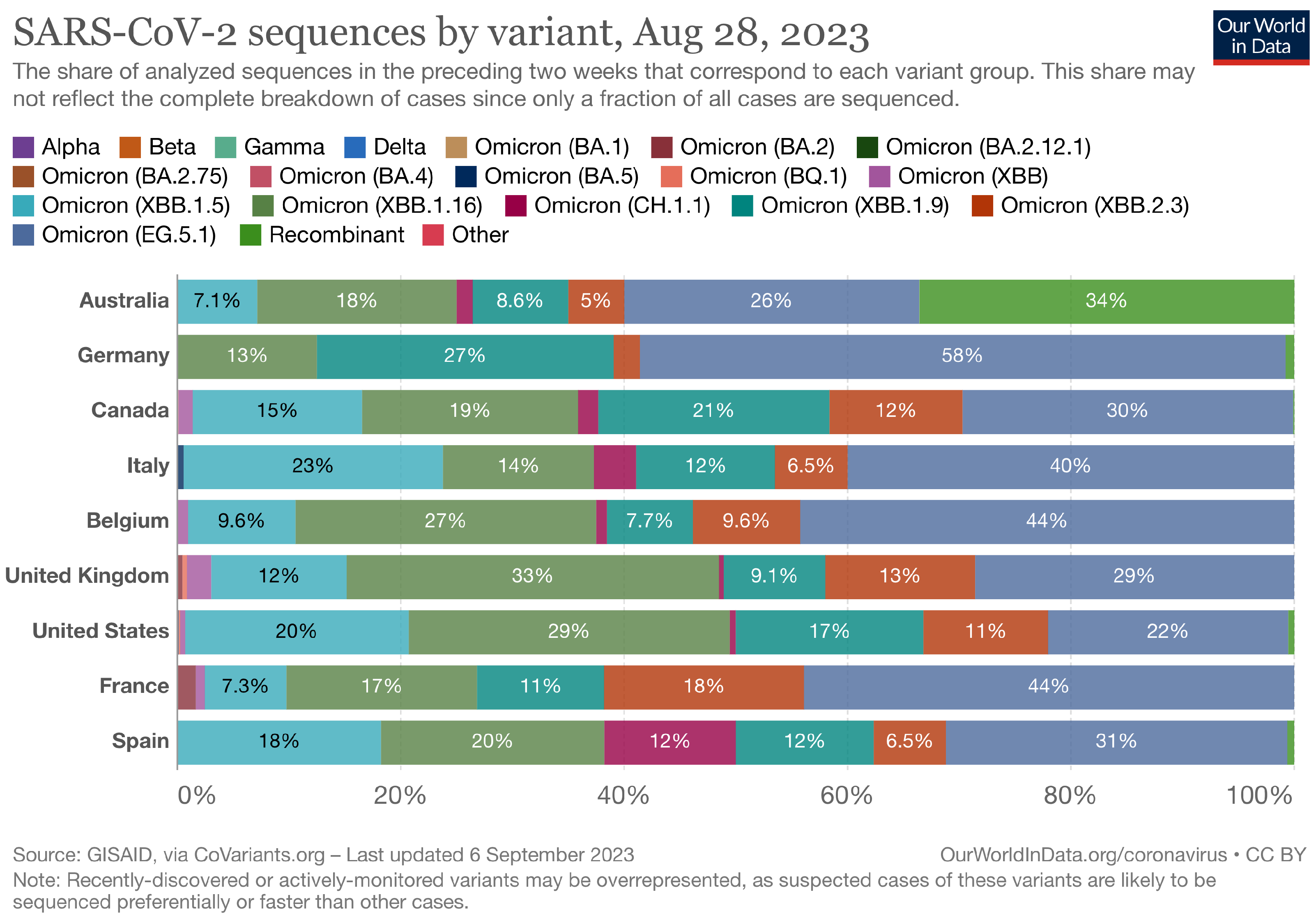
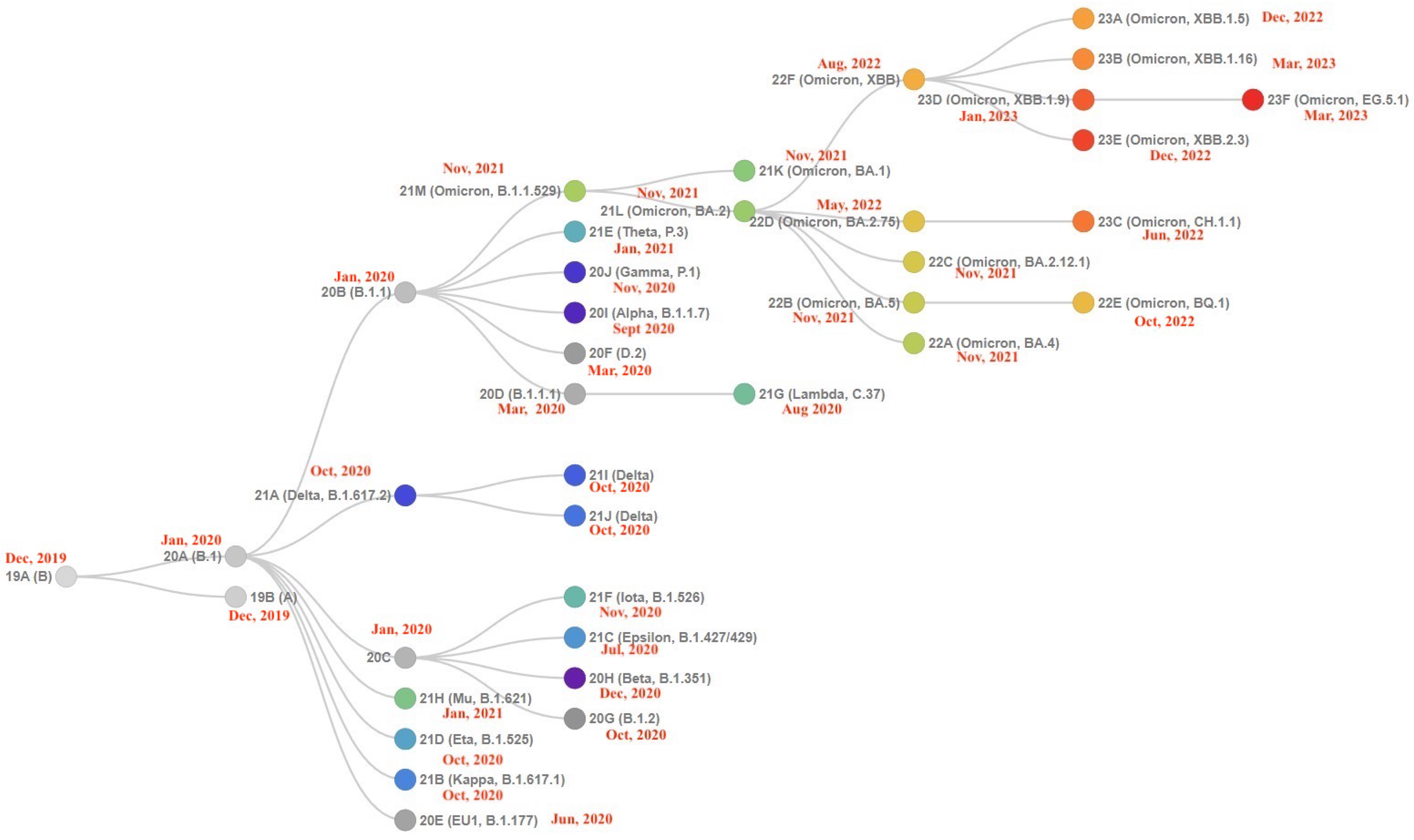
The Landscape of the Omicron Variants
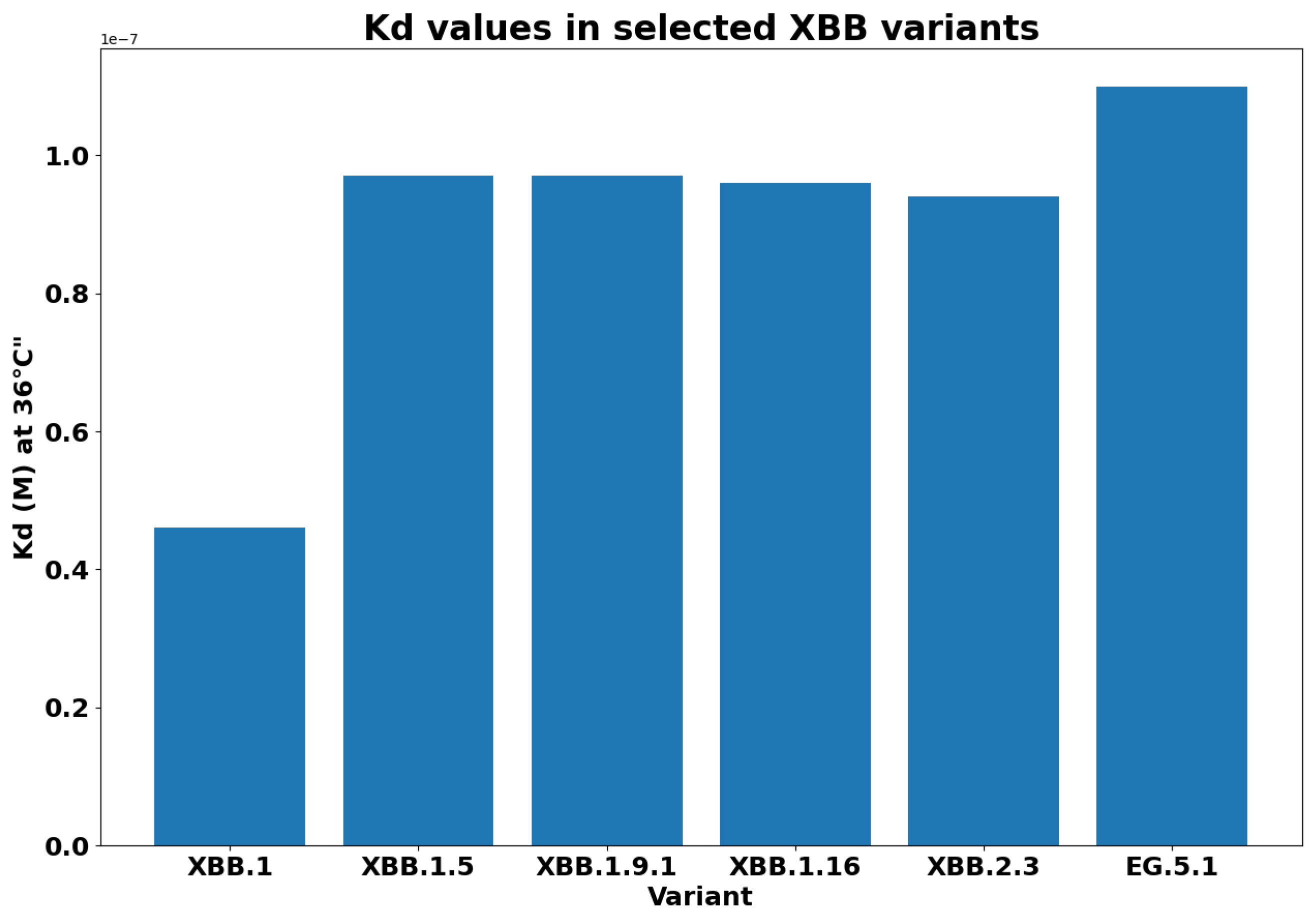
- XBB.1.5 (Kraken) emerged due to a genetic recombination between two BA.2 sublineages (see ancestors of XBB nodes in the tree) combined with S486P mutations at a significant point in its evolutionary history.
- XBB.1.9.1 (Hyperion) is XBB.1.5’s sibling.
- XBB.1.16 (Arcturus) was initially identified in India with a single mutation (K478R) in the RBD of XBB.1.5. Earlier studies demonstrated that mutations K417N, Q498R, and N501Y in the RBD region increase the ability of the variant to bind to the human ACE2 receptor. Mutations in residue 484 in the loop area have been associated with the virus’s ability to evade the immune system.
- XBB.2.3 (Acrux) first appeared in late December 2022 in India, even though it did not begin to spread until March 2023. It presents a highly evasive mutation, S:T478K.
- EG.5.1 (Eris) is a direct descendent of XBB.1.9.2, which has the same spike amino acid profile as XBB.1.5. EG.5.1 was first reported in February 2023 and designated as a variant under monitoring (VUM) on 19th July 2023 [23].
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ORF | Open reading frame |
| S | Spike |
| E | Envelope |
| M | Membrane |
| N | Nucleocapsid |
| WHO | World Health Organization |
| VBM | Variant Being Monitored |
| VOF | Variant of Concern |
| VOF | Variant of Concern |
| VOI | Variant of Interest |
| RBD | Receptor binding domain |
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
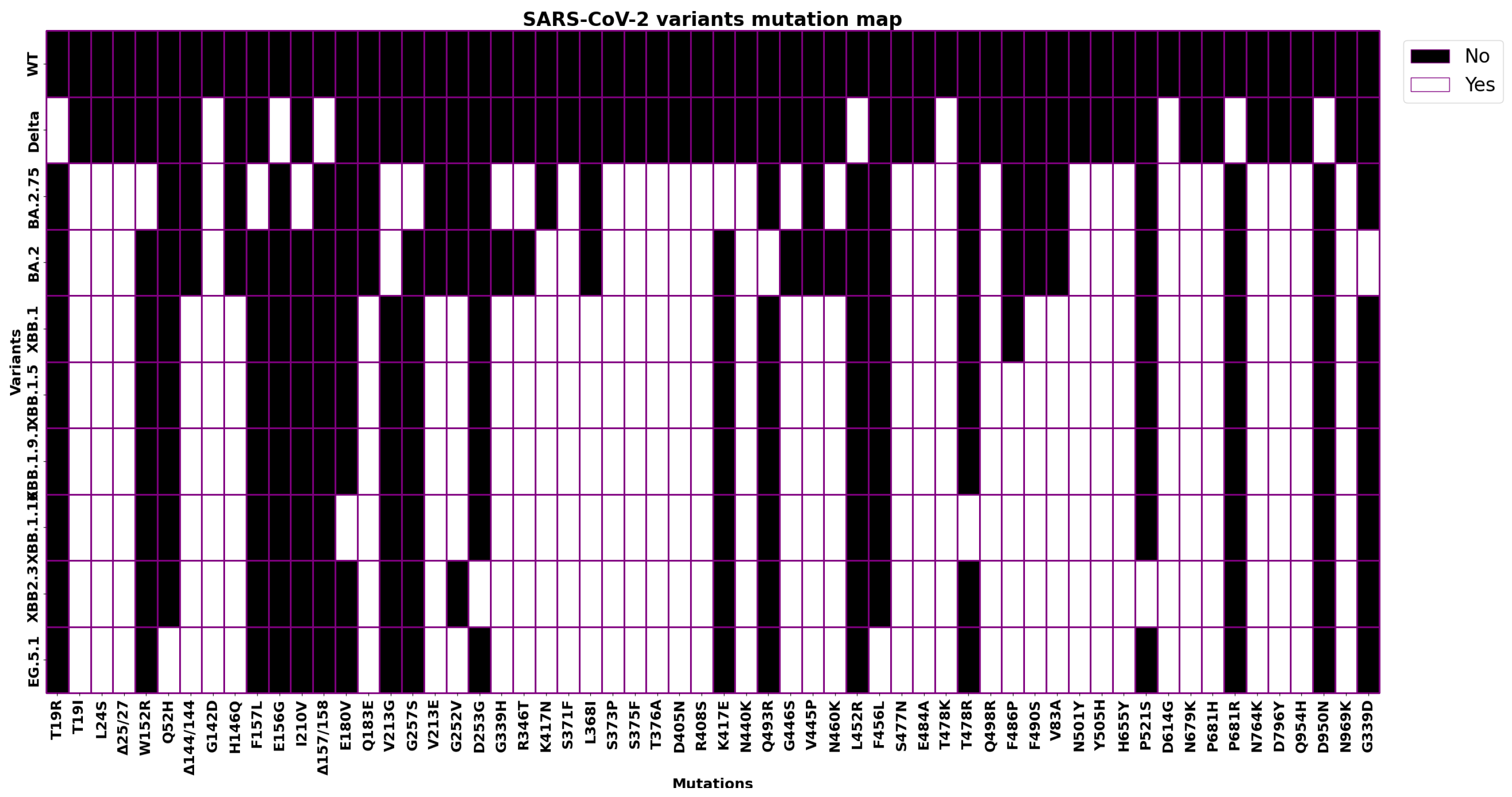
| Mutation | Alpha B1.1.7 | Gamma P.1 | Beta B.1.351 | Delta B.1.167.2 | Omicron BA.2 | XBB.1.0 | EG.5.1 |
|---|---|---|---|---|---|---|---|
| L18F | X | X | |||||
| T19R | X | ||||||
| T19I | X | X | X | ||||
| T20N | X | ||||||
| L24S | X | X | X | ||||
| P26S | X | ||||||
| DEL25/27 | X | X | X | ||||
| Q52H | X | ||||||
| DEL 69/70 | X | ||||||
| D80A | X | ||||||
| V83A | X | X | |||||
| D138Y | X | ||||||
| G142D | X | X | X | ||||
| DEL144/144 | X | X | X | ||||
| H146Q | X | X | |||||
| E156G | X | ||||||
| DEL157/158 | X | ||||||
| Q183E | X | X | |||||
| R190S | X | ||||||
| V213G | X | ||||||
| V213E | X | X | |||||
| D215G | X | ||||||
| DEL 242/244 | X | ||||||
| R346I | X | ||||||
| G252V | X | X | |||||
| G339D | X | ||||||
| G339H | X | X | |||||
| R346T | X | X | |||||
| L381I | X | X | |||||
| S371F | X | X | X | ||||
| S373P | X | X | X | ||||
| S375F | X | X | X | ||||
| T376A | X | X | X | ||||
| D405N | X | X | X | ||||
| R408S | X | X | X | ||||
| K417N | X | X | X | X | |||
| N440K | X | X | X | ||||
| V445P | X | X | |||||
| G446S | X | X | |||||
| L452R | X | ||||||
| N460K | X | X | |||||
| K417T | X | ||||||
| S477N | X | X | X | ||||
| T478K | X | X | X | X | |||
| F456L | X | ||||||
| E484A | X | X | X | ||||
| E484K | X | X | |||||
| F486P | X | ||||||
| F490S | X | X | |||||
| Q493R | X | ||||||
| Q498R | X | X | X | ||||
| N501Y | X | X | X | X | X | X | |
| Y505H | X | X | X | ||||
| A570D | X | ||||||
| D614G | X | X | X | X | X | X | X |
| H655Y | X | X | X | X | |||
| N679K | X | X | X | ||||
| P681H | X | X | X | X | |||
| P681R | X | ||||||
| A701V | X | ||||||
| T716I | X | ||||||
| N764K | X | X | X | ||||
| D796Y | X | X | X | ||||
| D950N | X | ||||||
| Q954H | X | X | X | ||||
| N969K | X | X | X | ||||
| S982A | X | ||||||
| T1027Y | X | ||||||
| V1176F | X | ||||||
| D1118H | X |
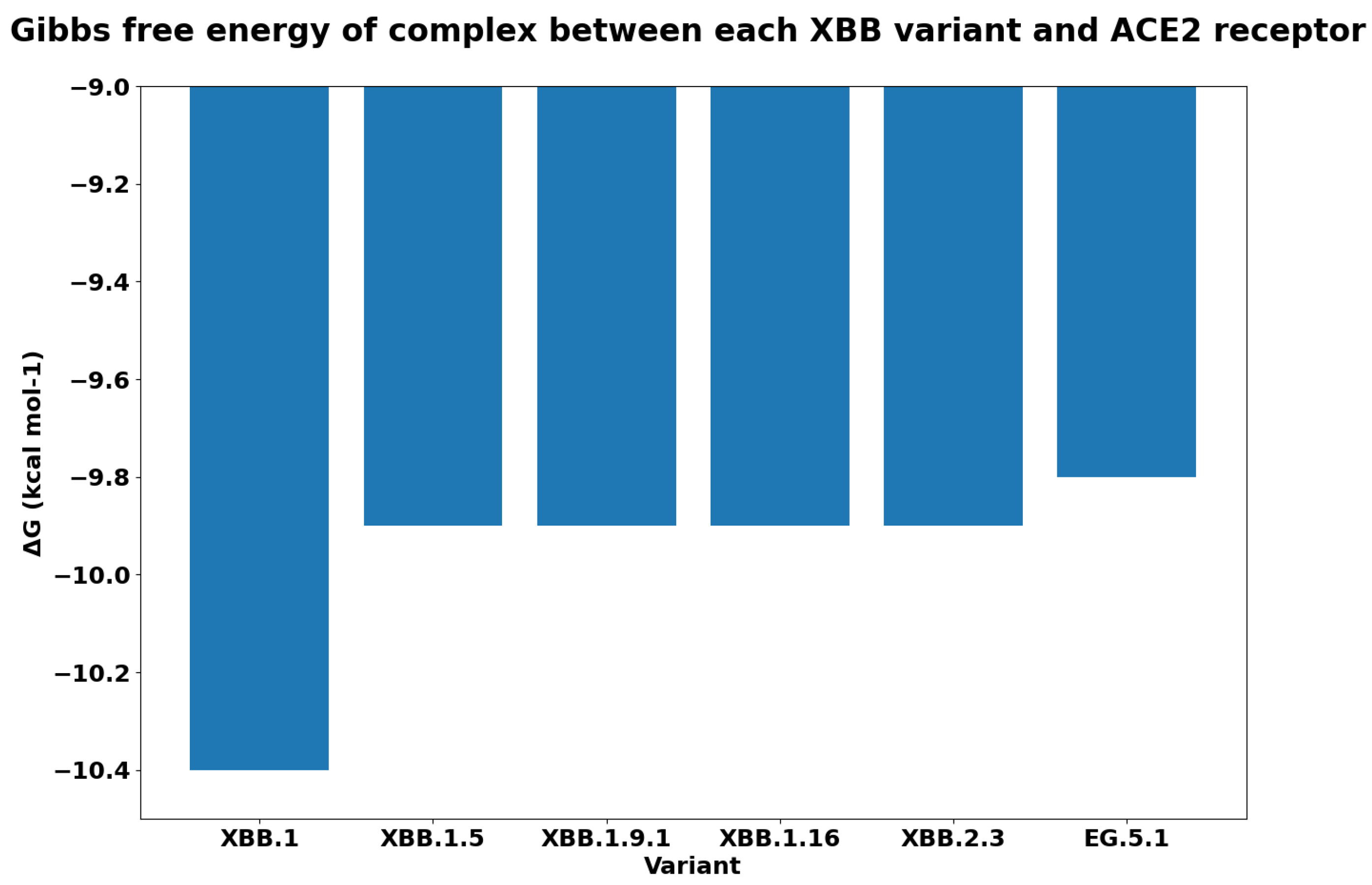
| Mutation | G | Variants |
|---|---|---|
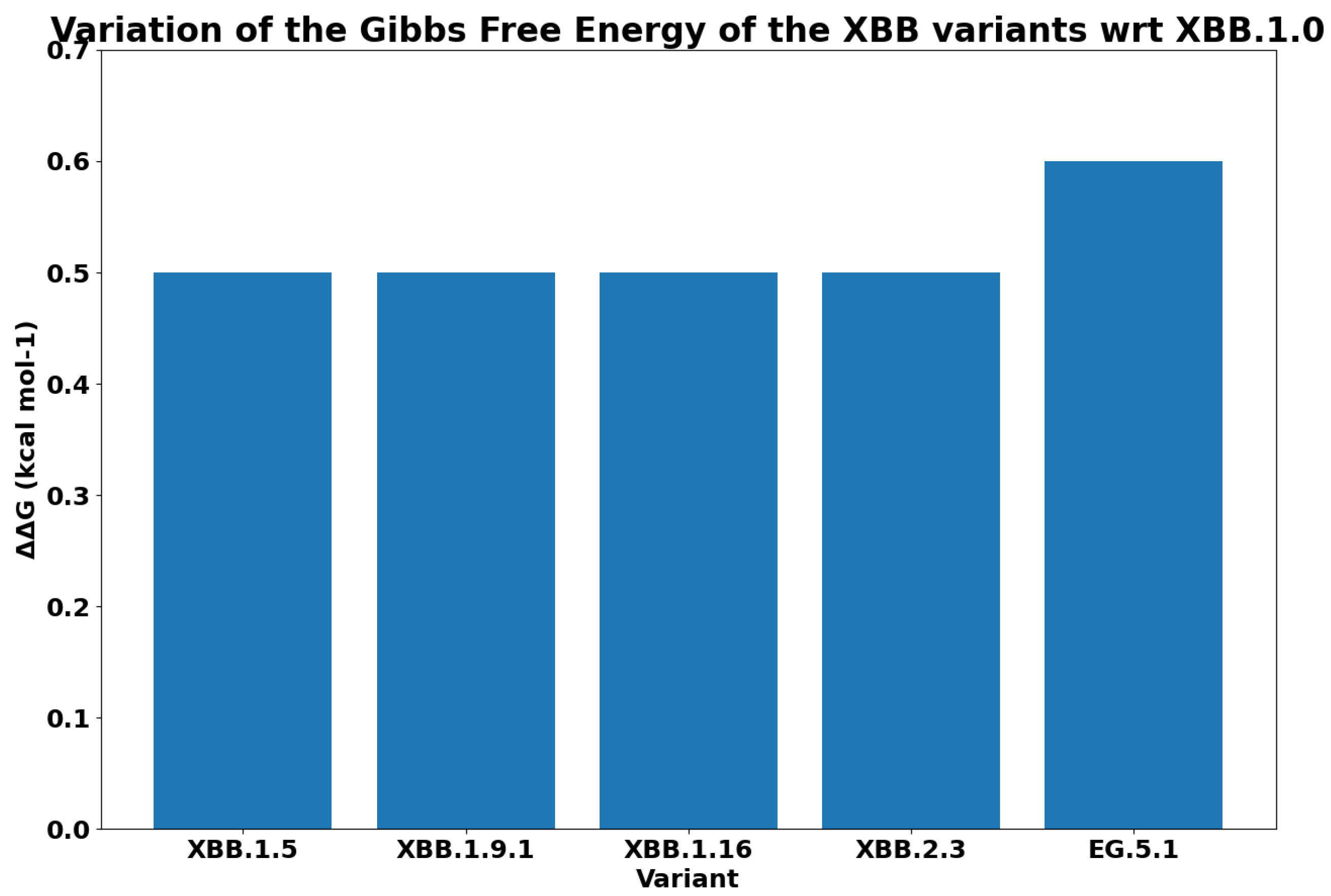
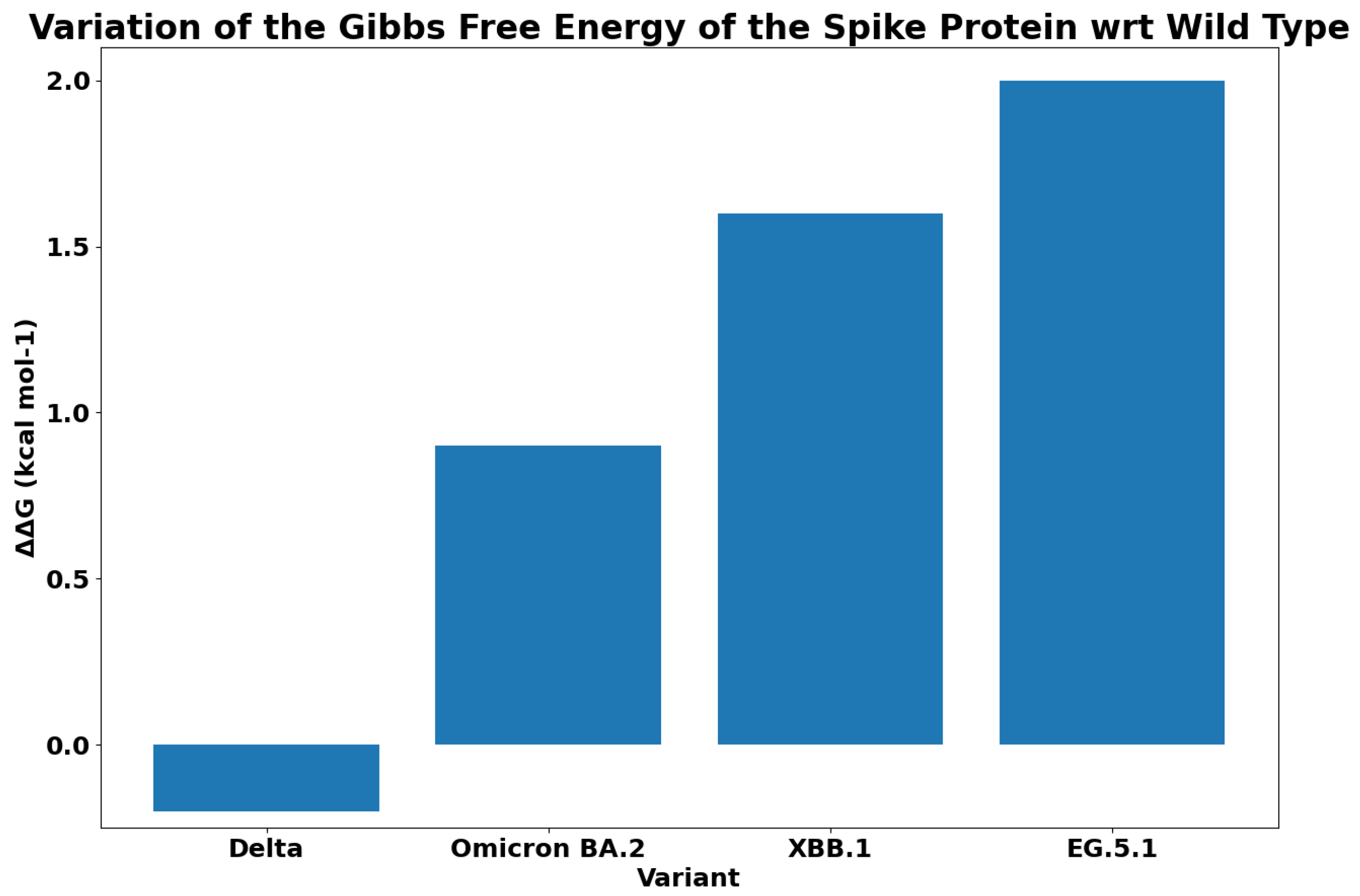
| WT (PDB: 7DF4) | −13.6 | |
| D405N | −13.9 | Omicron (BA.2), XBB.1.0, |
| EG.5.1 | ||
| D614G | −13.6 | Alpha (B1.1.7), Gamma (P.1), Beta (B.1.351), Delta (B.1.167.2), Omicron (BA.2), XBB.1.0, |
| EG.5.1 | ||
| F456L | −13.5 | EG.5.1 |
| F486P | −13.4 | EG.5.1 |
| H655Y | −13.6 | Gamma (P.1), Omicron (BA.2), XBB.1.0 |
| EG.5.1 | ||
| K417N | −13.7 | Beta (B.1.351), Omicron (BA.2), XBB.1.0 |
| EG.5.1 | ||
| N501Y | −13.3 | Alpha (B1.1.7), Gamma (P.1), Beta (B.1.351), Omicron (BA.2), XBB.1.0, |
| EG.5.1 | ||
| P681H | −13.6 | Alpha (B1.1.7), Omicron (BA.2), XBB.1.0 |
| EG.5.1 | ||
| Q52H | −13.6 | EG.5.1 |
| R408S | −13.7 | Omicron (BA.2), XBB.1.0, |
| EG.5.1 | ||
| T376A | −13.6 | Omicron (BA.2), XBB.1.0, |
| EG.5.1 | ||
| T478K | −13.6 | Delta (B.1.167.2), Omicron (BA.2), XBB.1.0, |
| EG.5.1 |
References
- Guzzi, P.H.; Mercatelli, D.; Ceraolo, C.; Giorgi, F.M. Master regulator analysis of the SARS-CoV-2/human interactome. J. Clin. Med. 2020, 9, 982. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef]
- Kumar Das, J.; Tradigo, G.; Veltri, P.; Guzzi, P.H.; Roy, S. Data science in unveiling COVID-19 pathogenesis and diagnosis: Evolutionary origin to drug repurposing. Briefings Bioinform. 2021, 22, 855–872. [Google Scholar] [CrossRef]
- Domingo, E.; Holland, J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151. [Google Scholar] [CrossRef]
- Madhi, S.A.; Kwatra, G.; Myers, J.E.; Jassat, W.; Dhar, N.; Mukendi, C.K.; Nana, A.J.; Blumberg, L.; Welch, R.; Ngorima-Mabhena, N.; et al. Population immunity and COVID-19 severity with Omicron variant in South Africa. N. Engl. J. Med. 2022, 386, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Frydman, J.; Andino, R. The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 2013, 11, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tian, C.; Liu, P.; Guo, D.; Zheng, W.; Huang, X.; Zhang, Y.; Liu, L. Effects of SARS-CoV-2 mutations on protein structures and intraviral protein–protein interactions. J. Med. Virol. 2021, 93, 2132–2140. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Tournier, J.D.; Mori, S.; Leemans, A. Diffusion tensor imaging and beyond. Magn. Reson. Med. 2011, 65, 1532–1556. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Worp, N.; Nieuwenhuijse, D.F.; Sikkema, R.S.; Haagmans, B.; Fouchier, R.A.; Koopmans, M. The next phase of SARS-CoV-2 surveillance: Real-time molecular epidemiology. Nat. Med. 2021, 27, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Hiram Guzzi, P.; Petrizzelli, F.; Mazza, T. Disease spreading modeling and analysis: A survey. Briefings Bioinform. 2022, 23, bbac230. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, P.H.; Di Paola, L.; Puccio, B.; Lomoio, U.; Giuliani, A.; Veltri, P. Computational analysis of the sequence-structure relation in SARS-CoV-2 spike protein using protein contact networks. Sci. Rep. 2023, 13, 2837. [Google Scholar] [CrossRef] [PubMed]
- Burki, T.K. Omicron variant and booster COVID-19 vaccines. Lancet Respir. Med. 2022, 10, e17. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Hong, H.; Wang, S.; Ma, L.; Liu, C.; Bai, Y.; Adam, D.C.; Tian, L.; Wang, L.; Lau, E.H.; et al. Reproduction number of the omicron variant triples that of the delta variant. Viruses 2022, 14, 821. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, F.; Tang, J.; Nussinov, R.; Cheng, F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Health 2020, 2, e667–e676. [Google Scholar] [CrossRef]
- Yamasoba, D.; Uriu, K.; Plianchaisuk, A.; Kosugi, Y.; Pan, L.; Zahradnik, J.; Ito, J.; Sato, K. Virological characteristics of the SARS-CoV-2 Omicron XBB. 1.16 variant. Lancet Infect. Dis. 2023, 23, 655–656. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Azzena, I.; Casu, M.; Cossu, P.; Fiori, P.L.; Benvenuto, D.; Imperia, E.; Giovanetti, M.; Ceccarelli, G.; et al. Genome-based comparison between the recombinant SARS-CoV-2 XBB and its parental lineages. J. Med. Virol. 2023, 95, e28625. [Google Scholar] [CrossRef]
- Uriu, K.; Ito, J.; Zahradnik, J.; Fujita, S.; Kosugi, Y.; Schreiber, G.; Sato, K. Enhanced transmissibility, infectivity, and immune resistance of the SARS-CoV-2 omicron XBB. 1.5 variant. Lancet Infect. Dis. 2023, 23, 280–281. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, D.; Lee, C.C.D.; Wu, N.C.; Jackson, A.M.; Zhu, X.; Liu, H.; Peng, L.; Van Gils, M.J.; Sanders, R.W.; et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science 2021, 373, 818–823. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.H.; Friis, N.U.; Bager, P.; Stegger, M.; Fonager, J.; Fomsgaard, A.; Gram, M.A.; Christiansen, L.E.; Ethelberg, S.; Legarth, R.; et al. Risk of reinfection, vaccine protection, and severity of infection with the BA. 5 omicron subvariant: A nation-wide population-based study in Denmark. Lancet Infect. Dis. 2023, 23, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shine, M.; Pyle, A.M.; Zhang, Y. US-align: Universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods 2022, 19, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2003, 1, 2.3.1–2.3.22. [Google Scholar] [CrossRef] [PubMed]
- Bittrich, S.; Rose, Y.; Segura, J.; Lowe, R.; Westbrook, J.D.; Duarte, J.M.; Burley, S.K. RCSB Protein Data Bank: Improved annotation, search and visualization of membrane protein structures archived in the PDB. Bioinformatics 2022, 38, 1452–1454. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Markosian, C.; Staquicini, D.I.; Dogra, P.; Dodero-Rojas, E.; Lubin, J.H.; Tang, F.H.; Smith, T.L.; Contessoto, V.G.; Libutti, S.K.; Wang, Z.; et al. Genetic and Structural Analysis of SARS-CoV-2 Spike Protein for Universal Epitope Selection. Mol. Biol. Evol. 2022, 39, msac091. [Google Scholar] [CrossRef]
- Benjamini, Y. Discovering the False Discovery Rate. J. R. Stat. Soc. Ser. Stat. Methodol. 2010, 72, 405–416. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual Comparisons by Ranking Methods. In Breakthroughs in Statistics: Methodology and Distribution; Springer: New York, NY, USA, 1998. [Google Scholar]
- Xia, S.; Wang, L.; Jiao, F.; Yu, X.; Xu, W.; Huang, Z.; Li, X.; Wang, Q.; Zhu, Y.; Man, Q.; et al. SARS-CoV-2 Omicron subvariants exhibit distinct fusogenicity, but similar sensitivity, to pan-CoV fusion inhibitors. Emerg. Microbes Infect. 2023, 12, 2178241. [Google Scholar] [CrossRef]
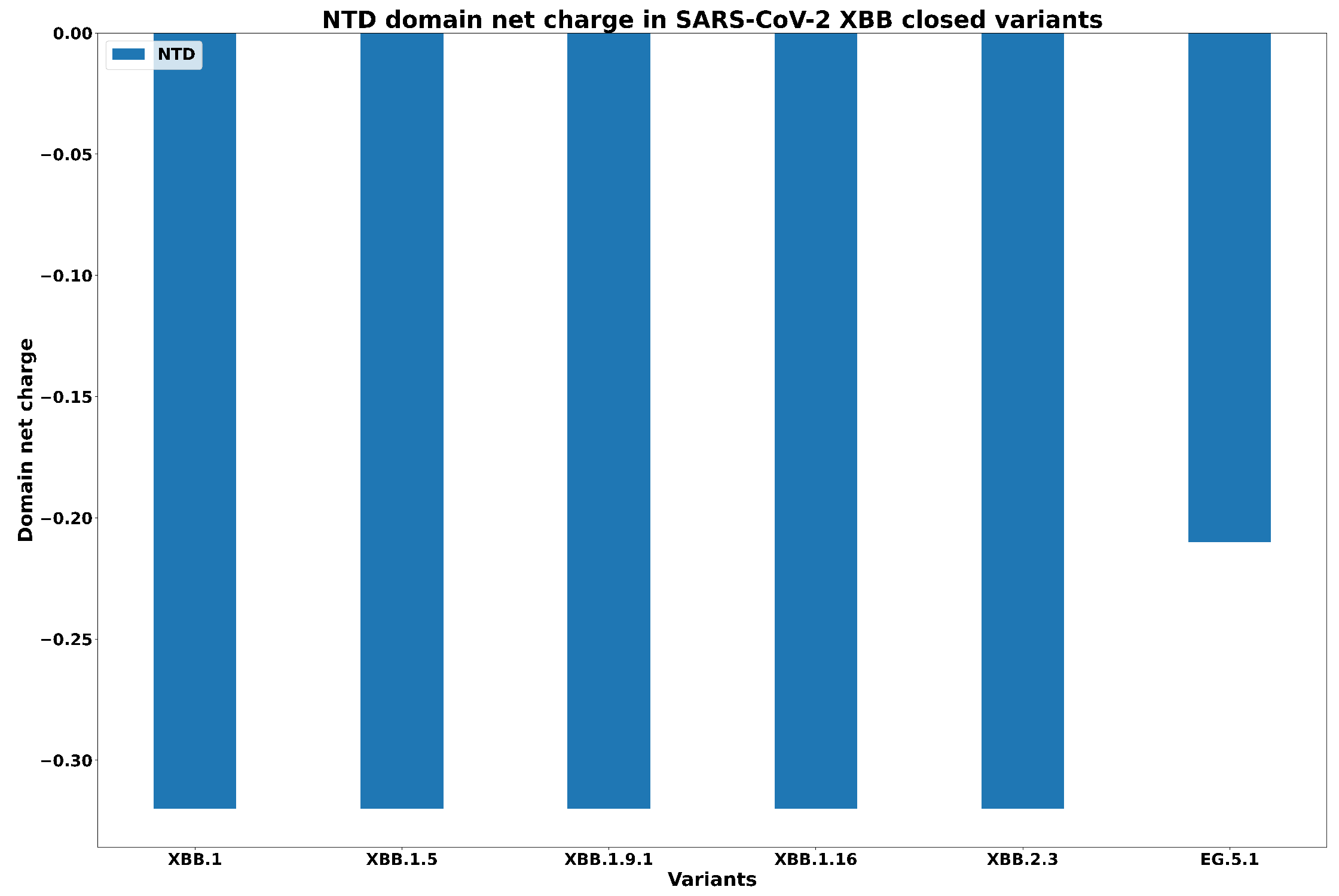
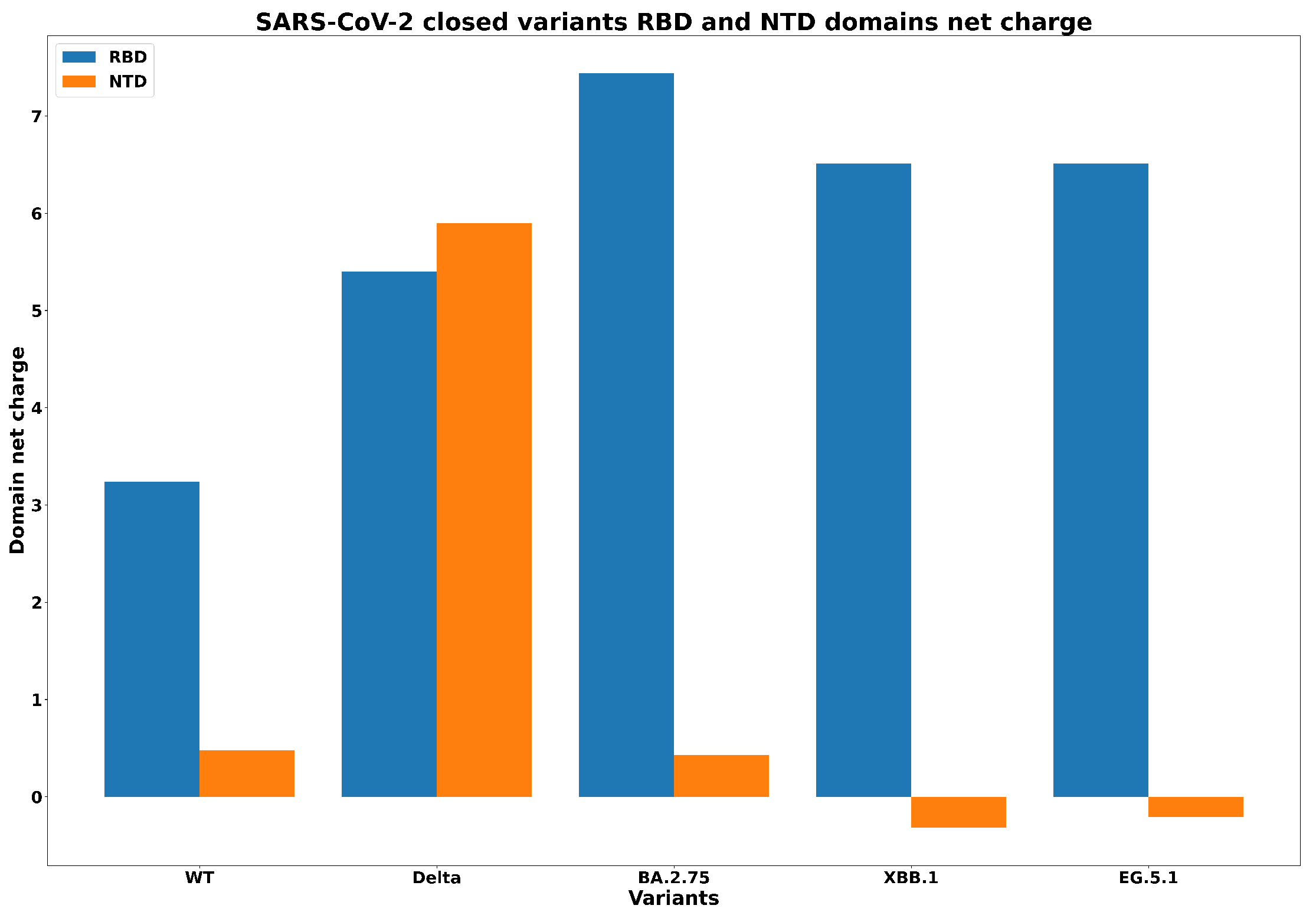
- Ciccozzi, M.; Pascarella, S. Two sides of the same coin: The N-terminal and the receptor-binding domains of SARS-CoV-2 Spike. Future Virol. 2023, 18, 75–78. [Google Scholar] [CrossRef]
- Jalali, N.; Brustad, H.K.; Frigessi, A.; MacDonald, E.A.; Meijerink, H.; Feruglio, S.L.; Nygård, K.M.; Rø, G.; Madslien, E.H.; De Blasio, B.F. Increased household transmission and immune escape of the SARS-CoV-2 Omicron compared to Delta variants. Nat. Commun. 2022, 13, 5706. [Google Scholar] [CrossRef] [PubMed]
- Uraki, R.; Ito, M.; Kiso, M.; Yamayoshi, S.; Iwatsuki-Horimoto, K.; Furusawa, Y.; Sakai-Tagawa, Y.; Imai, M.; Koga, M.; Yamamoto, S.; et al. Antiviral and bivalent vaccine efficacy against an omicron XBB. 1.5 isolate. Lancet Infect. Dis. 2023, 23, 402–403. [Google Scholar] [CrossRef] [PubMed]
- Conforti, C.; Dianzani, C.; Agozzino, M.; Giuffrida, R.; Marangi, G.F.; di Meo, N.; Morariu, S.H.; Persichetti, P.; Segreto, F.; Zalaudek, I.; et al. Cutaneous manifestations in confirmed COVID-19 patients: A systematic review. Biology 2020, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Fayad, N.; Abi Habib, W.; Kandeil, A.; El-Shesheny, R.; Kamel, M.N.; Mourad, Y.; Mokhbat, J.; Kayali, G.; Goldstein, J.; Abdallah, J. SARS-CoV-2 variants in Lebanon: Evolution and current situation. Biology 2021, 10, 531. [Google Scholar] [CrossRef] [PubMed]
| Spike Variant | Mutations |
|---|---|
| Wild Type | No Mutations |
| Delta B.1.617.2 | T19R + G142D + E156G + DEL157/158 + L452R + T478K + D614G + P681R + D950N |
| Omicron BA.2 | T19I + L24S + DEL25/27 + G142D + V213G + G339D + S371F + S373P + S375F + T376A + D405N + R408S + K417N + N440K + S477N + T478K + E484A + Q493R + Q498R + N501Y + Y505H + D614G + H655Y + N679K + P681H + N764K + D796Y + Q954H + N969K |
| Omicron BA.2.75 | T19I + L24S + DEL25/27 + G142D + W152R + F157L + I210V + V213G + G257S + G339H + R346T + S371F + S373P + S375F + T376A + D405N + R408S + K417E + N440K + G446S + N460K + S477N + T478K + E484A + Q498R + N501Y + Y505H + D614G + H655Y + N679K + P681H + N764K + D796Y + Q954H + N969K |
| XBB.1.0 | T19I + L24S + DEL25/27 + V83A + G142D + DEL144/144 + H146Q + Q183E + V213E + G252V + G339H + R346T + L368I + S371F + S373P + S375F + T376A + D405N + R408S + K417N + N440K + V445P + G446S + N460K + S477N + T478K + E484A + F490S + Q498R + N501Y + Y505H + D614G + H655Y + N679K + P681H + N764K + D796Y + Q954H + N969K |
| XBB.1.9.1 | XBB.1.0 + F486P |
| XBB.2.3 | XBB.1.0 + V252G + D253G + F486P + P521S |
| XBB.1.5 | XBB.1.0 + F486P |
| XBB.1.16 | XBB.1.0 + E180V + T478R + F486P |
| EG.5.1 | XBB.1.0 + Q52H + F456L + F486P |
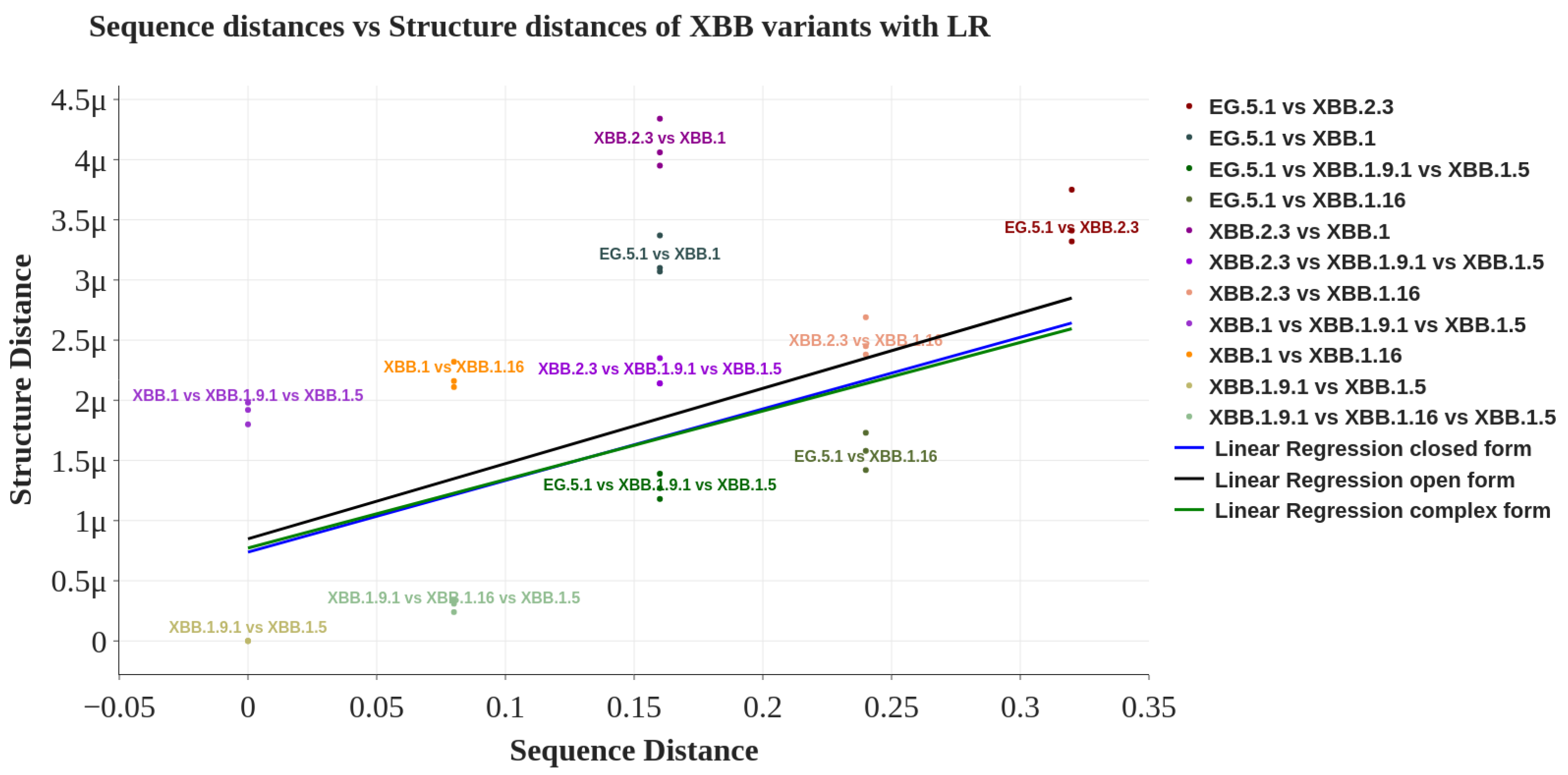
| Spike Structure | Pearson Coefficient | FDR Corrected p-Value |
|---|---|---|
| Closed | 0.462 | 0.042 |
| Open | 0.447 | 0.042 |
| Complex | 0.447 | 0.042 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giancotti, R.; Lomoio, U.; Puccio, B.; Tradigo, G.; Vizza, P.; Torti, C.; Veltri, P.; Guzzi, P.H. The Omicron XBB.1 Variant and Its Descendants: Genomic Mutations, Rapid Dissemination and Notable Characteristics. Biology 2024, 13, 90. https://doi.org/10.3390/biology13020090
Giancotti R, Lomoio U, Puccio B, Tradigo G, Vizza P, Torti C, Veltri P, Guzzi PH. The Omicron XBB.1 Variant and Its Descendants: Genomic Mutations, Rapid Dissemination and Notable Characteristics. Biology. 2024; 13(2):90. https://doi.org/10.3390/biology13020090
Chicago/Turabian StyleGiancotti, Raffaele, Ugo Lomoio, Barbara Puccio, Giuseppe Tradigo, Patrizia Vizza, Carlo Torti, Pierangelo Veltri, and Pietro Hiram Guzzi. 2024. "The Omicron XBB.1 Variant and Its Descendants: Genomic Mutations, Rapid Dissemination and Notable Characteristics" Biology 13, no. 2: 90. https://doi.org/10.3390/biology13020090