
A Cyclic Permutation Approach to Removing Spatial Dependency between Clustered Gene Ontology Terms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. SAGO Pipeline
2.2. Hypergeometric Test for GO Term Enrichment
2.3. Cyclic and Random Permutations
2.4. Linear Regression Analysis
2.5. Random Intervals Analysis
2.6. Data Sources and Processing
3. Results
3.1. Spatial Dependencies Affect Enrichment Analyses
3.2. Developing the Spatial Adjusted Gene Ontology (SAGO) Analysis Tool
3.3. Multiple Hypothesis Corrections
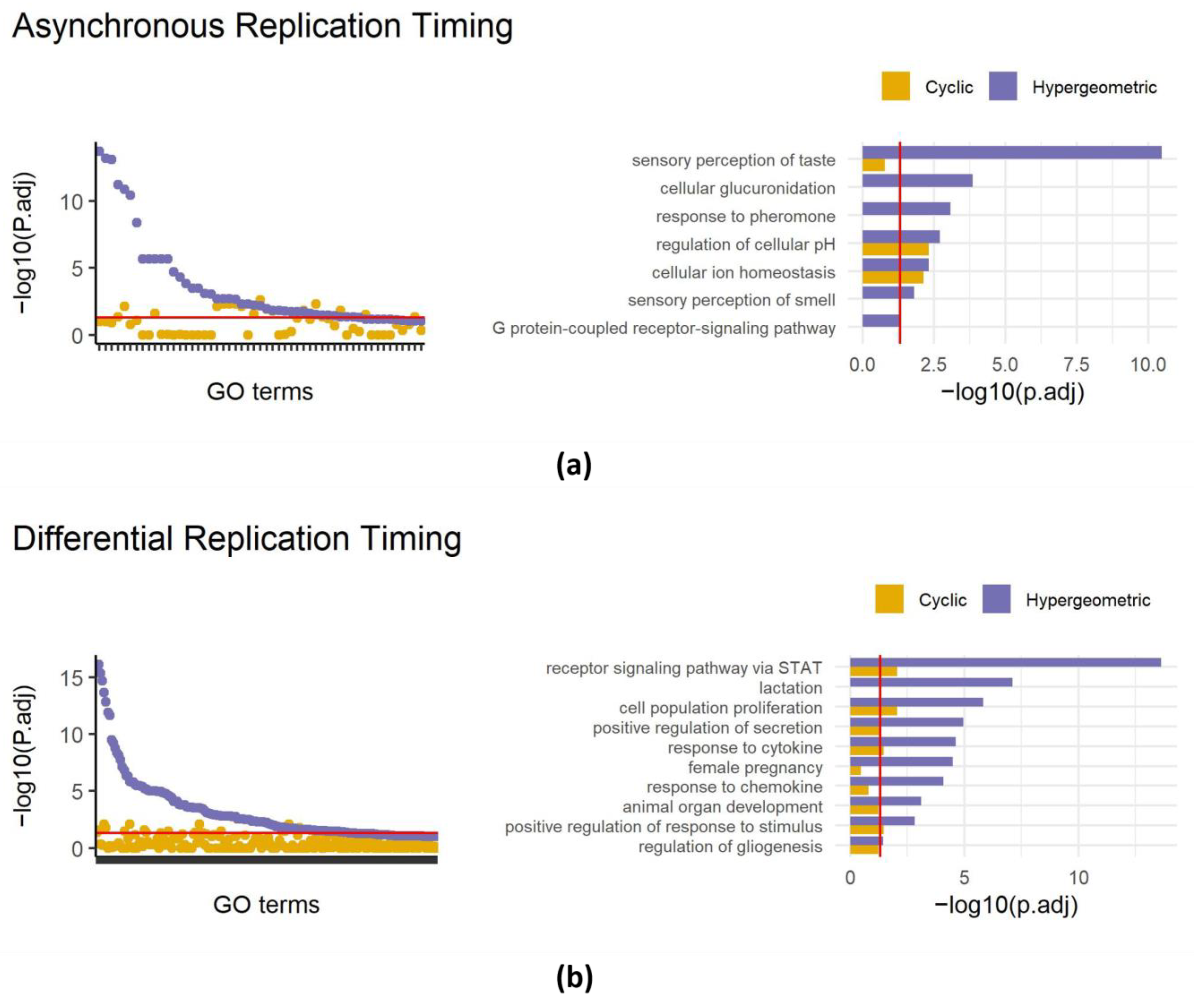
3.4. Applying SAGO to Replication Timing Data
3.5. Expanding the Use of SAGO to Additional Types of Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Mooney, M.A.; Wilmot, B. Gene set analysis: A step-by-step guide. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015, 168, 517–527. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]
- Rivals, I.; Personnaz, L.; Taing, L.; Potier, M.C. Enrichment or depletion of a GO category within a class of genes: Which test? Bioinformatics 2007, 23, 401–407. [Google Scholar] [CrossRef]
- Li, W.; Shih, A.; Freudenberg-Hua, Y.; Fury, W.; Yang, Y. Beyond standard pipeline and p < 0.05 in pathway enrichment analyses. Comput. Biol. Chem. 2021, 92, 107455. [Google Scholar]
- Takebayashi, S.I.; Ogata, M.; Okumura, K. Anatomy of Mammalian Replication Domains. Genes 2017, 8, 110. [Google Scholar] [CrossRef]
- Poulet, A.; Li, B.; Dubos, T.; Rivera-Mulia, J.C.; Gilbert, D.M.; Qin, Z.S. RT States: Systematic annotation of the human genome using cell type-specific replication timing programs. Bioinformatics 2019, 35, 2167–2176. [Google Scholar] [CrossRef]
- Du, Q.; Bert, S.A.; Armstrong, N.J.; Caldon, C.E.; Song, J.Z.; Nair, S.S.; Gould, C.M.; Luu, P.L.; Peters, T.; Khoury, A.; et al. Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nat. Commun. 2019, 10, 416. [Google Scholar] [CrossRef]
- Kosak, S.T.; Groudine, M. Gene order and dynamic domains. Science 2004, 306, 644–647. [Google Scholar] [CrossRef]
- Hurst, L.D.; Pal, C.; Lercher, M.J. The evolutionary dynamics of eukaryotic gene order. Nat. Rev. Genet. 2004, 5, 299–310. [Google Scholar] [CrossRef]
- Michalak, P. Coexpression, coregulation, and cofunctionality of neighboring genes in eukaryotic genomes. Genomics 2008, 91, 243–248. [Google Scholar] [CrossRef]
- Ben-Elazar, S.; Yakhini, Z.; Yanai, I. Spatial localization of co-regulated genes exceeds genomic gene clustering in the Saccharomyces cerevisiae genome. Nucleic Acids Res. 2013, 41, 2191–2201. [Google Scholar] [CrossRef][Green Version]
- Elizondo, L.I.; Jafar-Nejad, P.; Clewing, J.M.; Boerkoel, C.F. Gene clusters, molecular evolution and disease: A speculation. Curr. Genom. 2009, 10, 64–75. [Google Scholar] [CrossRef]
- Singer, G.A.; Lloyd, A.T.; Huminiecki, L.B.; Wolfe, K.H. Clusters of co-expressed genes in mammalian genomes are conserved by natural selection. Mol. Biol. Evol. 2005, 22, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Lercher, M.J.; Urrutia, A.O.; Hurst, L.D. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat. Genet. 2002, 31, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Sonnhammer, E.L. Genomic gene clustering analysis of pathways in eukaryotes. Genome Res. 2003, 13, 875–882. [Google Scholar] [CrossRef]
- Tiirikka, T.; Siermala, M.; Vihinen, M. Clustering of gene ontology terms in genomes. Gene 2014, 550, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, C.P.; Navarro, P.; Huffman, J.E.; Wright, A.F.; Hayward, C.; Campbell, H.; Wilson, J.F.; Rudan, I.; Hastie, N.D.; Vitart, V.; et al. Uncovering networks from genome-wide association studies via circular genomic permutation. G3 Genes Genomes Genet. 2012, 2, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Gel, B.; Diez-Villanueva, A.; Serra, E.; Buschbeck, M.; Peinado, M.A.; Malinverni, R. regioneR: An R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 2016, 32, 289–291. [Google Scholar] [CrossRef]
- Zang, C.; Wang, Y.; Peng, W. RECOGNICER: A coarse-graining approach for identifying broad domains from ChIP-seq data. Quant. Biol. 2020, 8, 359–368. [Google Scholar] [CrossRef]
- Chakraborty, A.; Wang, J.G.; Ay, F. dcHiC detects differential compartments across multiple Hi-C datasets. Nat. Commun. 2022, 13, 6827. [Google Scholar] [CrossRef]
- Yehuda, Y.; Blumenfeld, B.; Mayorek, N.; Makedonski, K.; Vardi, O.; Cohen-Daniel, L.; Mansour, Y.; Baror-Sebban, S.; Masika, H.; Farago, M.; et al. Germline DNA replication timing shapes mammalian genome composition. Nucleic Acids Res. 2018, 46, 8299–8310. [Google Scholar] [CrossRef]
- Malnic, B.; Godfrey, P.A.; Buck, L.B. The human olfactory receptor gene family. Proc. Natl. Acad. Sci. USA 2004, 101, 2584–2589. [Google Scholar] [CrossRef]
- Wen, S.H.; Tzeng, J.Y.; Kao, J.T.; Hsiao, C.K. A two-stage design for multiple testing in large-scale association studies. J. Hum. Genet. 2006, 51, 523–532. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Luo, Y.; Hitz, B.C.; Gabdank, I.; Hilton, J.A.; Kagda, M.S.; Lam, B.; Myers, Z.; Sud, P.; Jou, J.; Lin, K.; et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 2020, 48, D882–D889. [Google Scholar] [CrossRef]
- Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; Lee, B.T.; et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016, 44, D726–D732. [Google Scholar] [CrossRef]
- Bonev, B.; Mendelson Cohen, N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.P.; Tanay, A.; et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Keough, K.C.; Gjoni, K.; Santini, G.T.; Abdill, R.J.; Wickramasinghe, N.M.; Dundes, C.E.; Karnay, A.; Chen, A.; Salomon, R.E.A.; et al. An atlas of lamina-associated chromatin across twelve human cell types reveals an intermediate chromatin subtype. Genome Biol. 2023, 24, 16. [Google Scholar] [CrossRef] [PubMed]
- Labani, M.; Afrasiabi, A.; Beheshti, A.; Lovell, N.H.; Alinejad-Rokny, H. PeakCNV: A multi-feature ranking algorithm-based tool for genome-wide copy number variation-association study. Comput. Struct. Biotechnol. J. 2022, 20, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Lipson, D.; Yogev, S.; Yakhini, Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol. 2007, 3, e39. [Google Scholar] [CrossRef] [PubMed]
- Lazar, N.H.; Celik, S.; Chen, L.; Fay, M.; Irish, J.C.; Jensen, J.; Tillinghast, C.A.; Urbanik, J.; Bone, W.P.; Roberts, G.H.L.; et al. High-resolution genome-wide mapping of chromosome-arm-scale truncations induced by CRISPR-Cas9 editing. bioRxiv 2023. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapoport, R.; Greenberg, A.; Yakhini, Z.; Simon, I. A Cyclic Permutation Approach to Removing Spatial Dependency between Clustered Gene Ontology Terms. Biology 2024, 13, 175. https://doi.org/10.3390/biology13030175
Rapoport R, Greenberg A, Yakhini Z, Simon I. A Cyclic Permutation Approach to Removing Spatial Dependency between Clustered Gene Ontology Terms. Biology. 2024; 13(3):175. https://doi.org/10.3390/biology13030175
Chicago/Turabian StyleRapoport, Rachel, Avraham Greenberg, Zohar Yakhini, and Itamar Simon. 2024. "A Cyclic Permutation Approach to Removing Spatial Dependency between Clustered Gene Ontology Terms" Biology 13, no. 3: 175. https://doi.org/10.3390/biology13030175
APA StyleRapoport, R., Greenberg, A., Yakhini, Z., & Simon, I. (2024). A Cyclic Permutation Approach to Removing Spatial Dependency between Clustered Gene Ontology Terms. Biology, 13(3), 175. https://doi.org/10.3390/biology13030175