
Drug Repurposing: Exploring Potential Anti-Cancer Strategies by Targeting Cancer Signalling Pathways

Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Signalling Pathways and Tumourigenesis
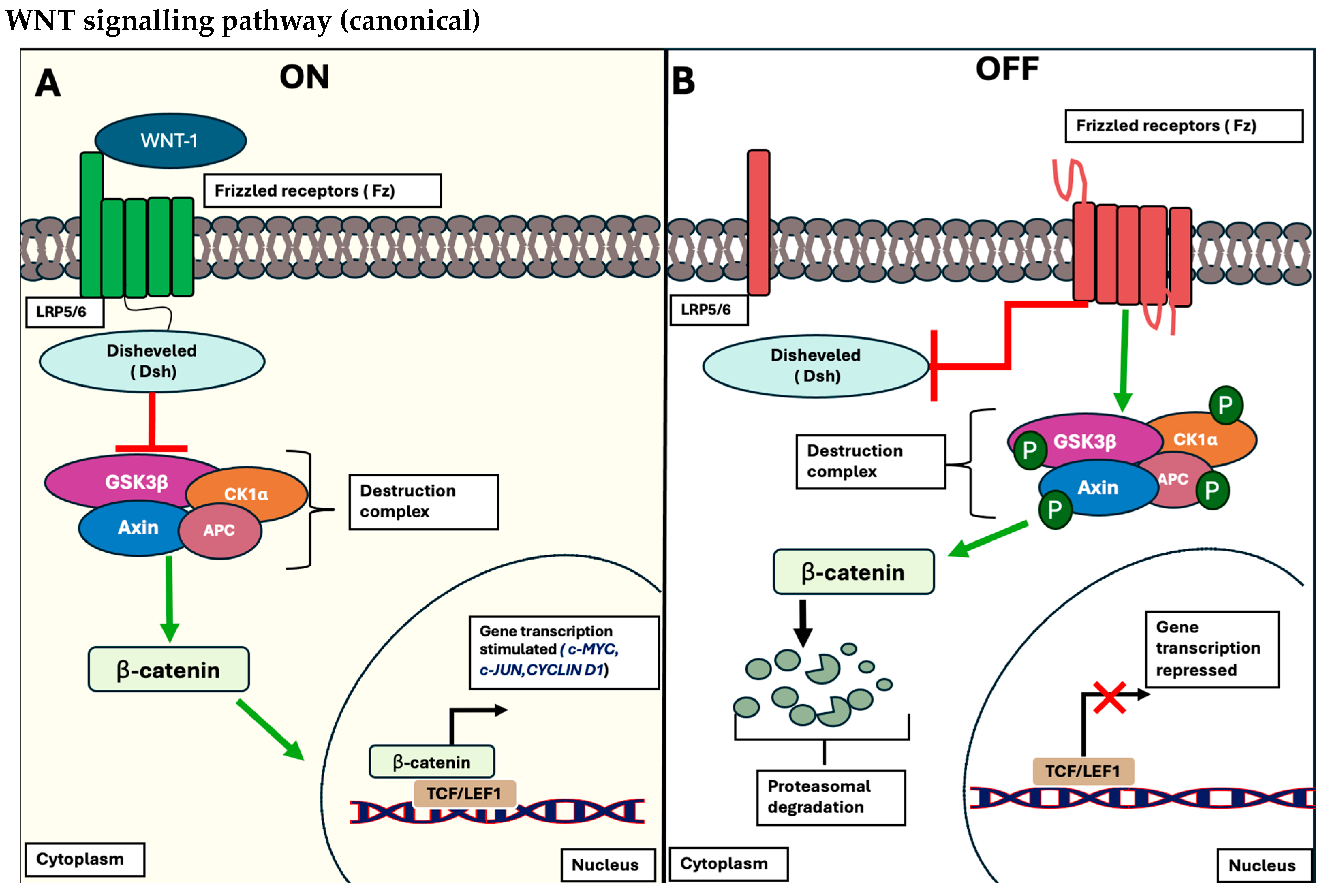
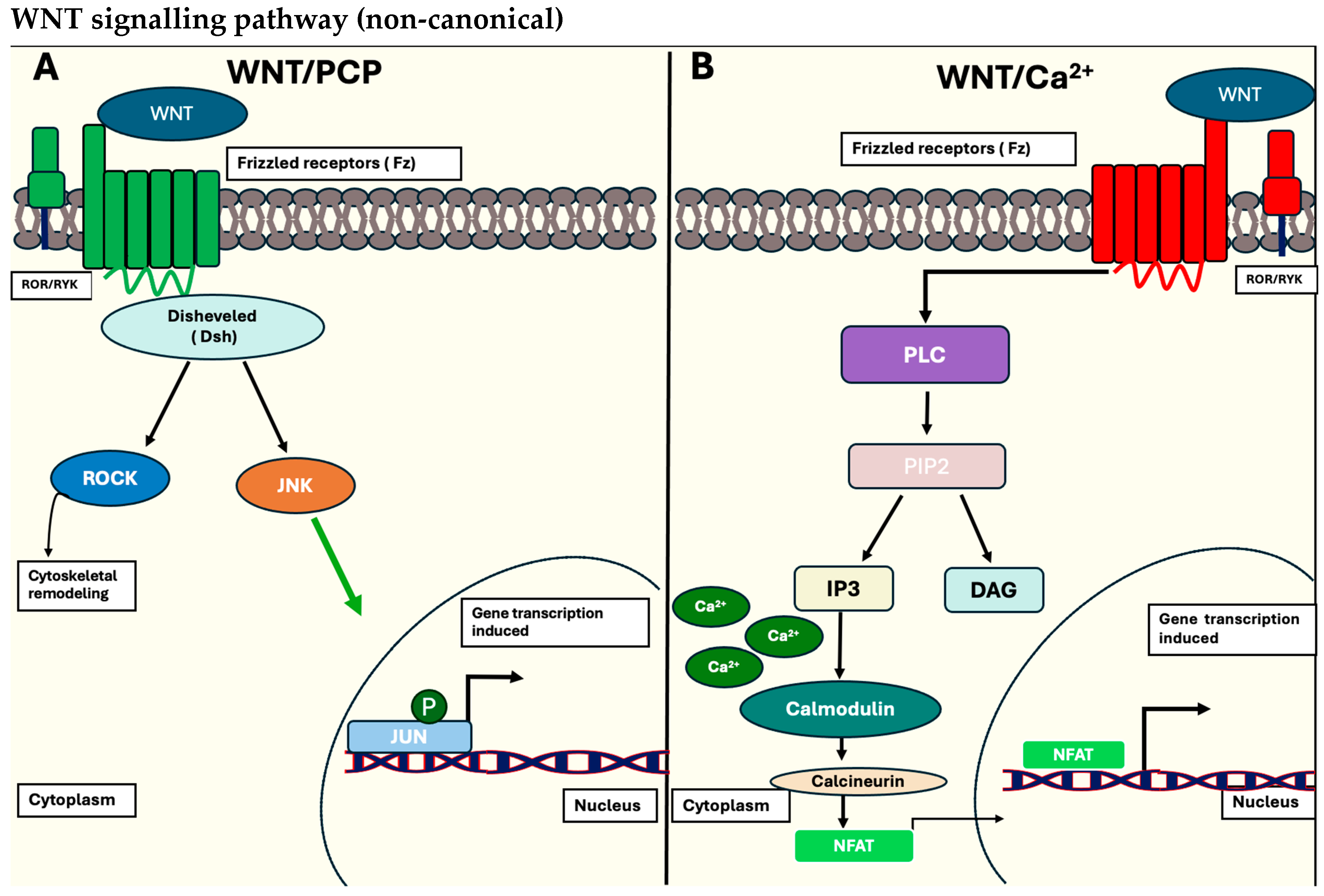
1.1.1. The Wnt/β-Catenin Signalling Pathway
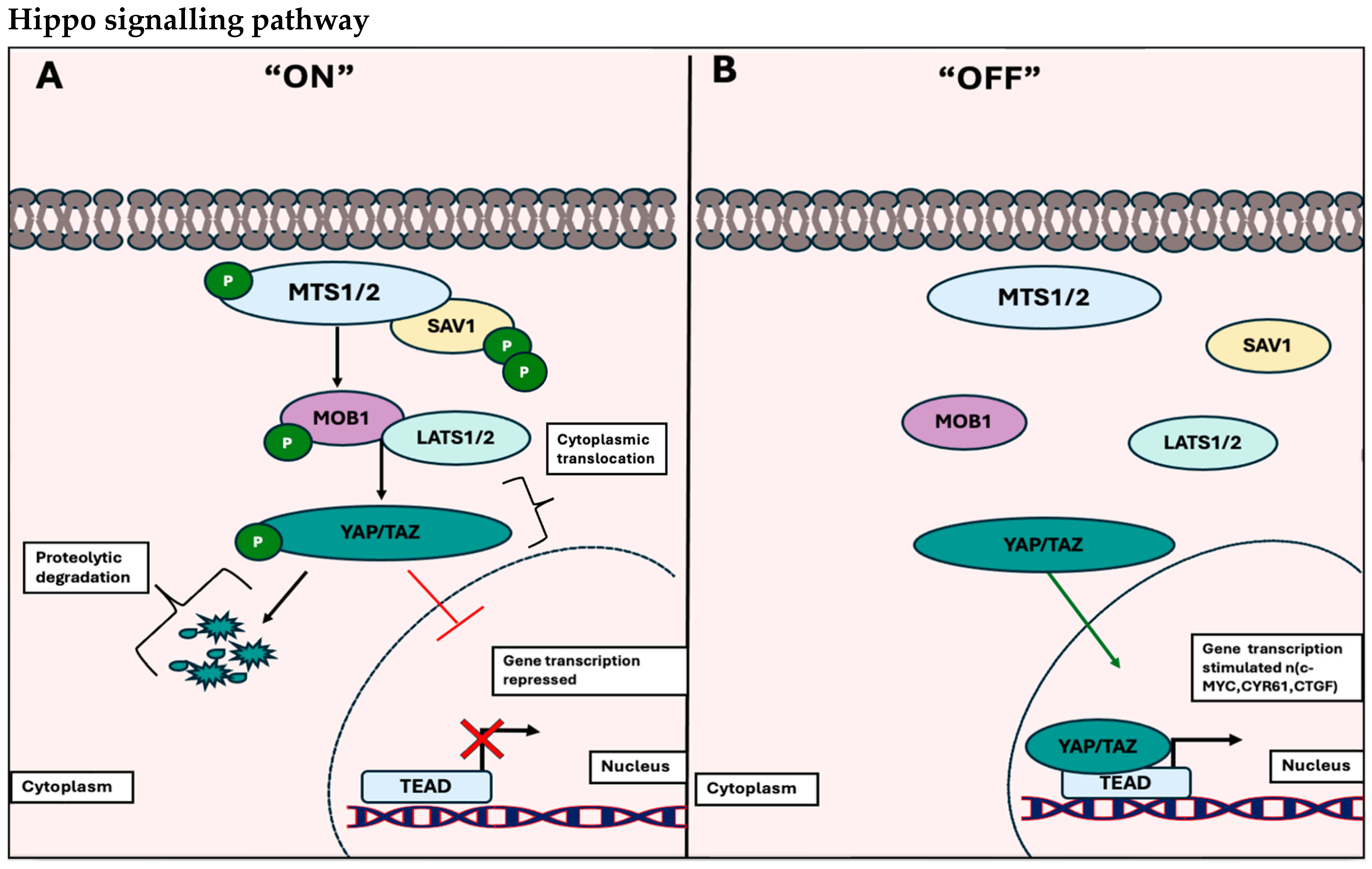
1.1.2. The Hippo Signalling Pathway
1.1.3. Deregulation of Cancer Signalling Pathways
1.2. Therapeutic Compounds Targeting the Hippo and WNT Signalling Cascades
1.3. Challenges of Targeted Therapy in Cancer
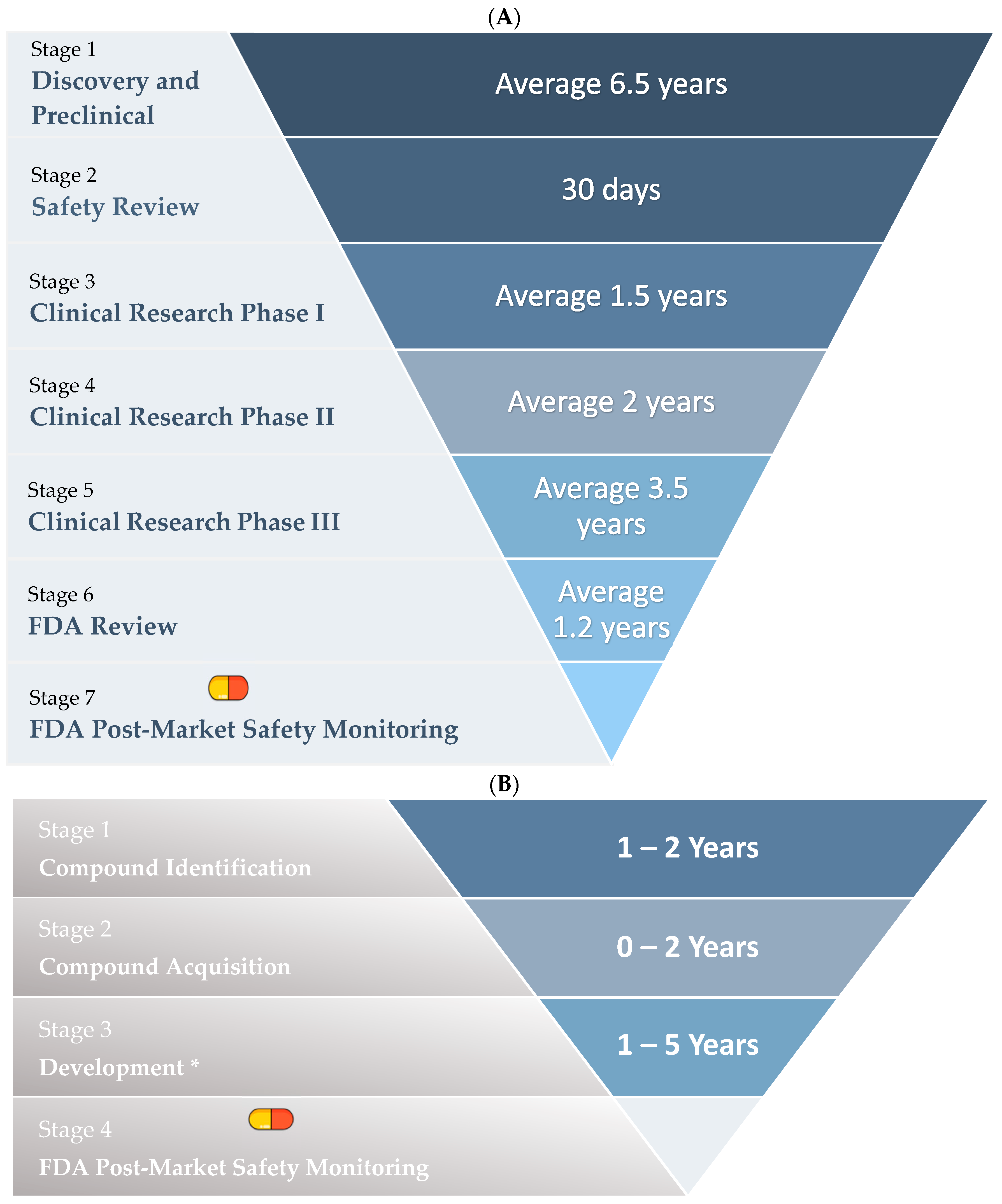
1.4. Drug Repurposing
Stages of Drug Repurposing
1.5. Examples of Repurposed Drugs
2. Repurposing Psychotropic Medications as Potential Therapeutic Drugs for Cancer
2.1. Sertraline
2.2. Fluoxetine
2.3. Repurposing Antimalarials as Potential Therapeutic Drugs for Cancer
Artemisinin/Artesunate
3. Drug Repurposing in Targeting Alternative Cancer-Associated Signalling Pathways
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 10 March 2024).
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–677. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Parsa, N. Environmental factors inducing human cancers. Iran. J. Public Health 2012, 41, 1–9. [Google Scholar]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Hu, Z.; Xu, X.; Dai, X.; Liu, Z. Key signal transduction pathways and crosstalk in cancer: Biological and therapeutic opportunities. Transl. Oncol. 2022, 26, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, T.; Zhang, S.; Qang, J.; Chen, Y.; Zhao, H.; Yang, Y.; Shi, S.; Chen, Q.; Liu, K. The Wnt signaling pathway in tumorigenesis, pharmacological targets, and drug development for cancer therapy. Biomark. Res. 2021, 9, 3–9. [Google Scholar] [CrossRef]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Prior, J.; Piwinca-Worms, D.; Bu, B. LRP6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 5136–5140. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.; Duarte, D.; Vale, N. Drug Repurposing in Cancer Therapy: Influence of Patient’s Genetic Background in Breast Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 4280. [Google Scholar] [CrossRef]
- Unterleuthner, D.; Neuhold, P.; Schwarz, K.; Janker, L.; Neuditschko, B.; Nivarthi, H.; Crncec, I.; Kramer, N.; Unger, C.; Hengstschläger, M.; et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis 2020, 23, 163–167. [Google Scholar] [CrossRef]
- Kurnit, K.C.; Kim, G.N.; Fellman, B.M.; Urbauer, D.L.; Mills, G.B.; Zhang, W.; Broaddus, R.R. CTNNB1 (beta-catenin) mutation identifies low grade, early-stage endometrial cancer patients at increased risk of recurrence. Mod. Pathol. 2017, 30, 1032–1041. [Google Scholar] [CrossRef]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Res. 2018, 78, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Kaur, P.; Bunnag, N.; Suresh, J.; Sung, I.C.H.; Tan, Q.H.; Gruber, J.; Tolwinski, N.S. Wnt signalling in disease. Cells 2019, 8, 826. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Dannappel, M.; Wan, C.; Firestein, R. Transcriptional Regulation of Wnt/B-Catenin Pathway in Colorectal Cancer. Cells 2020, 9, 2125. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Hingole, S.; Chaudhary, V. The emerging mechanisms of Wnt secretion and signaling in development. Front. Cell Dev. Biol. 2021, 9, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Neiheisel, A.; Kaur, M.; Ma, N.; Havard, P.; Shenoy, A.K. Wnt pathway modulators in cancer therapeutics: An update on completed and ongoing clinical trials. Int. J. Cancer 2021, 150, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Rajamannan, N. Diseases of Wnt signaling. Rev. Endocr. Metab. Disord. 2006, 7, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Dong, J. The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective. Cancers 2021, 13, 6214. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 578–582. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, X.; Maglic, D.; Dill, M.T.; Mojumdar, K.; Ng, P.K.S.; Jeong, K.J.; Tsang, Y.H.; Moreno, D.; Bhavana, V.H.; et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018, 25, 3–10. [Google Scholar] [CrossRef]
- Díaz-Martín, J.; López-García, M.Á.; Romero-Pérez, L.; Atienza-Amores, M.R.; Pecero, M.L.; Castilla, M.Á.; Biscuola, M.; Santón, A.; Palacios, J. Nuclear TAZ expression associates with the triple-negative phenotype in breast cancer. Endocr.-Relat. Cancer 2015, 22, 443–454. [Google Scholar] [CrossRef]
- Barbosa, I.A.M.; Gopalakrishnan, R.; Mercan, S.; Mourikis, T.P.; Martin, T.; Wengert, S.; Sheng, C.; Ji, F.; Lopes, R.; Knehr, J.; et al. Cancer lineage-specific regulation of YAP responsive elements revealed through large-scale functional epigenomic screens. Nat. Commun. 2023, 14, 2–8. [Google Scholar] [CrossRef]
- Kim, M.; Ly, S.H.; Xie, Y.; Duronio, G.N.; Ford-Roshon, D.; Hwang, J.H.; Sulahian, R.; Rennhack, J.P.; So, J.; Gjoerup, O.; et al. YAP1 and PRDM14 converge to promote cell survival and tumorigenesis. Dev. Cell 2022, 57, 213–216. [Google Scholar] [CrossRef]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat. Cancer 2021, 2, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Bata, N.; Chaikuad, A.; Bakas, N.A.; Limpert, A.S.; Lambert, L.J.; Sheffler, D.J.; Berger, L.M.; Liu, G.; Yuan, C.; Wang, L.; et al. Inhibitors of the Hippo Pathway Kinases STK3/MST2 and STK4/MST1 Have Utility for the Treatment of Acute Myeloid Leukemia. J. Med. Chem. 2022, 65, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; Hacken, E.T.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat. Med. 2014, 20, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, A.U.; Driver, L.M.; Zhao, M.T.; Wells, C.I.; Pickett, J.E.; O’Bryne, S.N.; Eduful, B.J.; Yang, X.; Howard, L.; You, S.; et al. Non-canonical role of Hippo tumor suppressor serine/threonine kinase 3 STK3 in prostate cancer. Mol. Ther. 2022, 30, 485–500. [Google Scholar] [CrossRef]
- Wei, L.; Ma, X.; Hou, Y.; Zhao, T.; Sun, R.; Qiu, C.; Liu, Y.; Qiu, Z.; Liu, Z.; Jiang, J. Verteporfin reverses progestin resistance through YAP/TAZ-PI3K-Akt pathway in endometrial carcinoma. Cell Death Discov. 2023, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Zhou, X. Targeting Hippo signaling in cancer: Novel perspectives and therapeutic potential. MedComm 2023, 4, 2–12. [Google Scholar] [CrossRef]
- Jiao, S.; Li, C.; Hao, Q.; Miao, H.; Zhang, L.; Li, L.; Zhou, Z. VGLL4 targets a TCF4-TEAD4 complex to coregulate Wnt and Hippo signalling in colorectal cancer. Nat. Commun. 2017, 8, 2–11. [Google Scholar] [CrossRef]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed]
- Le, P.N.; McDermott, J.D.; Jimeno, A. Targeting the Wnt pathway in human cancers: Therapeutic targeting with a focus on OMP-54F28. Pharmacol. Ther. 2015, 146, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Janku, F. Tumor heterogeneity in the clinic: Is it a real problem? Ther. Adv. Med. Oncol. 2014, 6, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Concepcion, J.; Uprety, D.; Adjei, A.A. Challenges in the Use of Targeted Therapies in Non-Small Cell Lung Cancer. Cancer Res. Treat. 2022, 54, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Ahamad, S.; Qazi, S.; Raza, K. Translational bioinformatics methods for drug discovery and drug repurposing. Adv. Ubiquitous Sens. Appl. Healthc. 2021, 13, 127–139. [Google Scholar]
- Jourdan, J.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.S.; Alagarsamy, V.; Solomon, V.R.; Jose, P.A.; Murugesan, S. Drug Repurposing: An Effective Tool in Modern Drug Discovery. Russ. J. Bioorganic Chem. 2023, 49, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Khairnar, S.J.; Jadhav, A.G. Drug Repurposing (DR): An Emergin Approach in Drug Disocvery. In Drug Repurposing—Hyposthesis, Molecular Aspects and Therapeutic Applications; Badria, F.A., Ed.; IntechOpen: London, UK, 2020. [Google Scholar]
- Xue, H.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef]
- Kaushik, I.; Ramachandran, S.; Prasad, S.; Srivastava, S.K. Drug rechannelling: A novel paradigm for cancer treatment. Semin. Cancer Biol. 2020, 68, 279–290. [Google Scholar] [CrossRef]
- Dhir, N.; Jain, A.; Mahendru, D.; Prakash, A.; Medhi, B. Drug Repurposing and Orphan Disease Therapeutics. Drug Repurposing—Hypothesis, Molecular Aspects and Therapeutic Applications; Badria, F.A., Ed.; IntechOpen: London, UK, 2020. [Google Scholar]
- Wang, Y.; Aldahdooh, J.; Hu, Y.; Yang, H.; Vähä-Koskela, M.; Tang, J.; Tanoli, Z. DrugRepo: A novel approach to repurposing drugs based on chemical and genomic features. Sci. Rep. 2022, 12, 4–8. [Google Scholar] [CrossRef]
- Hijazi, M.A.; Gessner, A.; El-Najjar, N. Repurposing of Chronically Used Drugs in Cancer Therapy: A Chance to Grasp. Cancers 2023, 15, 3199. [Google Scholar] [CrossRef] [PubMed]
- Mohi-ud-din, R.; Chawla, A.; Sharma, P.; Mir, A.H.; Potoo, H.F.; Reiner, Ž.; Atessahin, D.A.; Sharifi-Rad, J.; Mir, H.R.; Calina, D.; et al. Repurposing approved non-oncology drugs for cancer therapy: A comprehensive review of mechanisms, efficacy, and clinical prospects. Eur. J. Med. Res. 2023, 28, 345. [Google Scholar] [CrossRef] [PubMed]
- Varalda, M.; Antona, A.; Bettio, V.; Roy, K.; Vachamaram, A.; Yellenki, V.; Massarotti, A.; Baldanzi, G.; Capello, D. Psychotropic Drugs Show Anticancer Activity by Disrupting Mitochondrial and Lysosomal Function. Front. Oncol. 2020, 10, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Kanner, Y.B.; Teng, Q.; Ganoth, A.; Peer, D.; Wang, J.; Chen, Z.; Tsfadia, Y. Cytotoxicity and reversal effect of sertraline, fluoxetine, and citalopram on MRP1- and MRP7-mediated MDR. Front. Pharmacol. 2023, 14, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.M. SSRI Antidepressant Medications: Adverse Effects and Tolerability. Prim. Care Companion J. Clin. Psychiatry 2001, 3, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Edinoff, A.N.; Akuly, H.A.; Hanna, T.A.; Ochoa, C.O.; Patto, S.J.; Ghaffer, Y.A.; Kaye, A.D.; Viswanath, I.U.; Boyer, A.G.; Cornett, E.M.; et al. Selective Serotonin Reuptake Inhibitors and Adverse Effects: A Narrative Review. Neurol. Int. 2021, 13, 387–401. [Google Scholar] [CrossRef]
- Duarte, D.; Vale, N. Antidepressant Drug Sertraline against Human Cancer Cells. Biomolecules 2022, 12, 1513. [Google Scholar] [CrossRef] [PubMed]
- Baú-Carneiro, J.L.; Sumida, I.A.G.; Gallon, M.; Zaleski, T.; Bola-Ferreira, M.; Cavassin, F.B. Sertraline repositioning: An overview of its potential use as a chemotherapeutic agent after four decades of tumor reversal studies. Transl. Oncol. 2022, 16, 2–6. [Google Scholar] [CrossRef]
- Gil-Ad, I.; Zolokov, A.; Lomnitski, L.; Taler, M.; Bar, M.; Luria, D.; Ra, E.; Weizman, A. Evaluation of the potential anti-cancer activity of the antidepressant sertraline in human colon cancer cell lines and in colorectal cancer-xenografted mice. Int. J. Oncol. 2008, 33, 277–286. [Google Scholar] [CrossRef]
- Baldissera, A.B.; Boia-Ferreira, M.; Basílio, A.B.C.; Resende, J.S.S.; Castro, M.A.A.; Chaim, O.M.; Gremski, L.H.; Veiga, S.S.; Senff-Ribeiro, A. Sertraline as a potential cancer therapeutic approach: Biological relevance of TCTP in breast cancer cell lines and tumors. Adv. Med. Sci. 2023, 68, 227–237. [Google Scholar] [CrossRef]
- Malard, F.; Jacquet, E.; Nhiri, N.; Sizun, C.; Chabrier, A.; Messaoudi, S.; Dejeu, J.; Betzi, S.; Zhang, X.; Thureau, A.; et al. Revisiting the Molecular Interactions between the Tumor Protein TCTP and the Drugs Sertraline/Thioridazine. ChemMedChem 2022, 17, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Qu, T.; Yang, J.; Dai, Q. Serotonin acts through YAP to promote cell proliferation: Mechanism and implication in colorectal cancer progression. Cell Commun. Signal. 2023, 21, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Frick, L.R.; Palumbo, M.L.; Zappia, M.P.; Brocco, M.A.; Cremaschi, G.A. Inhibitory effect of fluoxetine on lymphoma growth through the modulation of antitumor T-cell response by serotonin-dependent and independent mechanisms. Biochem. Pharmacol. 2008, 75, 1817–1826. [Google Scholar] [CrossRef]
- Kannen, V.; Marini, T.; Turatti, A.; Carvalho, M.C.; Brandão, M.L.; Jabor, V.A.P.; Bonato, P.S.; Ferreira, F.R.; Zanette, D.L.; Silva, W.A., Jr.; et al. Fluoxetine induces preventive and complex effects against colon cancer development in epithelial and stromal areas in rats. Toxicol. Lett. 2011, 204, 134–140. [Google Scholar] [CrossRef]
- Warkus EL, L.; Marikawa, Y. Fluoxetine Inhibits Canonical Wnt Signaling to Impair Embryoid Body Morphogenesis: Potential Teratogenic Mechanisms of a Commonly Used Antidepressant. Toxicol. Sci. 2018, 165, 372–388. [Google Scholar] [CrossRef]
- Krishna, S.; Bustamente, L.; Haynes, R.K.; Staines, H.M. Artemisinins: Their growing importance in medicine. Trends Pharmacol. Sci. 2008, 29, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, S.; Schlegel, C.R.; Wang, G.; Belinson, J.L. Use of artemisinin and its derivatives to treat HPV-infected/transformed cells and cervical cancer: A review. Future Oncol. 2014, 10, 647–654. [Google Scholar] [CrossRef]
- Li, L.; Zhang, H.; Yuan, S.; Tian, Z.; Wang, L.; Sun, Z. Artesunate attenuates the growth of human colorectal carcinoma and inhibits hyperactive Wnt/beta-catenin pathway. Int. J. Cancer 2007, 121, 1360–1365. [Google Scholar] [CrossRef]
- Zhou, X.; Suo, F.; Haslinger, K.; Quax, W.J. Artemisinin—Type Drugs in Tumor Cell Death: Mechanisms, Combination Treatment with Biologics and Nanoparticle Delivery. Pharmaceutics 2022, 14, 395. [Google Scholar] [CrossRef]
- Gong, X.; Zhang, Q.; Torossian, A.; Cao, J. Selective Radiosensitization of Human Cervical Cancer Cells and Normal Cells by Artemisinin Through the Abrogation of Radiation-Induced G2Block. Int. J. Gynecol. Cancer 2019, 22, 718–724. [Google Scholar] [CrossRef]
- Luo, J.; Zhu, W.; Tang, Y.; Cao, H.; Zhou, Y.; Ji, R.; Zhou, X.; Lu, Z.; Yang, H.; Zhang, S.; et al. Artemisinin derivative artesunate induces radiosensitivity in cervical cancer cells in vitro and in vivo. Radiat. Oncol. 2014, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, J.; Chen, Q.; Han, S.; Shao, H.; Chen, P.; Kin, Q.; Yang, M.; Shangguan, F.; Fei, M.; et al. Artemisinin suppresses hepatocellular carcinoma cell growth, migration and invasion by targeting cellular bioenergetics and Hippo-YAP signaling. Arch. Toxicol. 2019, 93, 3367–3383. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Trigger, Mechanism, and Consequence. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Elsherbeny, A.; Pittalà, V.; Fallica, A.N.; Alghamdi, M.A.; Greish, K. The Potential Role of Sildenafil in Cancer Management through EPR Augmentation. J. Pers. Med. 2021, 11, 585. [Google Scholar] [CrossRef]
- Anindita, D.; Durrant, D.; Mitchell, C.; Dent, P.; Batra, S.K.; Kukreja, R.C. Sildenafil (Viagra) sensitizes prostate cancer cells to doxorubicin-mediated apoptosis through CD95. Oncotarget 2016, 7, 4399–4413. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Hippo Genes | Type of Alteration | Role in Tumourigenesis | References |
|---|---|---|---|---|
| Breast Cancer (triple- negative subtype) | YAP/TAZ | Amplification | TAZ expression induces metastatic and aggressive-like properties in breast cancer stem cells. | [22] |
| Uveal Melanoma | YAP | Amplification | YAP-TEAD activation leads to transcription of canonical target genes (CTGF, CYR61, and AMOTL2) and c-MYC hyperactivation. | [23] |
| Colon Cancer | YAP1/TAZ | Amplification | YAP1 expression is associated with enhanced transcription of target genes associated with colon cancer progression and poor prognosis. | [24] |
| Glioblastoma | YAP/TAZ | Overexpressed | YAP/TAZ prevents glioblastoma–stem cells from differentiating and mediates cell cycle progression in the gliomaspheres. | [25] |
| Cancer Type | Drug Name | Original Indication |
|---|---|---|
| General cancers | Gemcitabine | Antiviral |
| Breast cancer | Raloxifene | Osteoporosis |
| Breast, colorectal, endometrial, and prostate cancers | Metformin | Diabetes |
| General cancers | Orlistat | Obesity |
| General cancers | Itraconazole | Fungal infection |
| Multiple myeloma, neuroblastoma, and leukaemia | Flubendazole | Anthelmintic drug |
| Multiple myeloma | Thalidomide | Morning sickness |
| Colon cancer | Azithromycin | Antibacterial drug |
| Bone and prostate cancers | Doxycycline | |
| General cancers | Itraconazole | |
| Lymphoma and leukemic cells | Griseofulvin | Antifungal |
| Glioblastoma | Clotrimazole | |
| Breast and colorectal cancer | Ciclopirox | |
| Colorectal cancer | Aspirin | |
| General cancers | Etodolac | NSAIDs/Anti-inflammatory |
| General cancers | Etoricoxib | |
| General cancers | Celecoxib | |
| Breast cancer and glioblastoma | Hydroxychloroquine | Antimalarial drugs |
| Lung, breast, and colon cancers | Atovaquone |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haddad, N.; Gamaethige, S.M.; Wehida, N.; Elbediwy, A. Drug Repurposing: Exploring Potential Anti-Cancer Strategies by Targeting Cancer Signalling Pathways. Biology 2024, 13, 386. https://doi.org/10.3390/biology13060386
Haddad N, Gamaethige SM, Wehida N, Elbediwy A. Drug Repurposing: Exploring Potential Anti-Cancer Strategies by Targeting Cancer Signalling Pathways. Biology. 2024; 13(6):386. https://doi.org/10.3390/biology13060386
Chicago/Turabian StyleHaddad, Natalia, Sara Magura Gamaethige, Nadine Wehida, and Ahmed Elbediwy. 2024. "Drug Repurposing: Exploring Potential Anti-Cancer Strategies by Targeting Cancer Signalling Pathways" Biology 13, no. 6: 386. https://doi.org/10.3390/biology13060386