
Genetic Polymorphisms in Exon 5 and Intron 5 and 7 of AIRE Are Associated with Rheumatoid Arthritis Risk in a Hungarian Population

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Inclusion and Exclusion Criteria of Variations
2.2. Ethical Approval
2.3. Study Subjects
2.4. Assessment of Clinical Parameters
2.5. DNA Extraction
2.6. Genotyping AIRE Variants
2.7. Statistical Analyses
2.8. Analysing the Effects of Variants on Regulatory Binding Motifs
3. Results
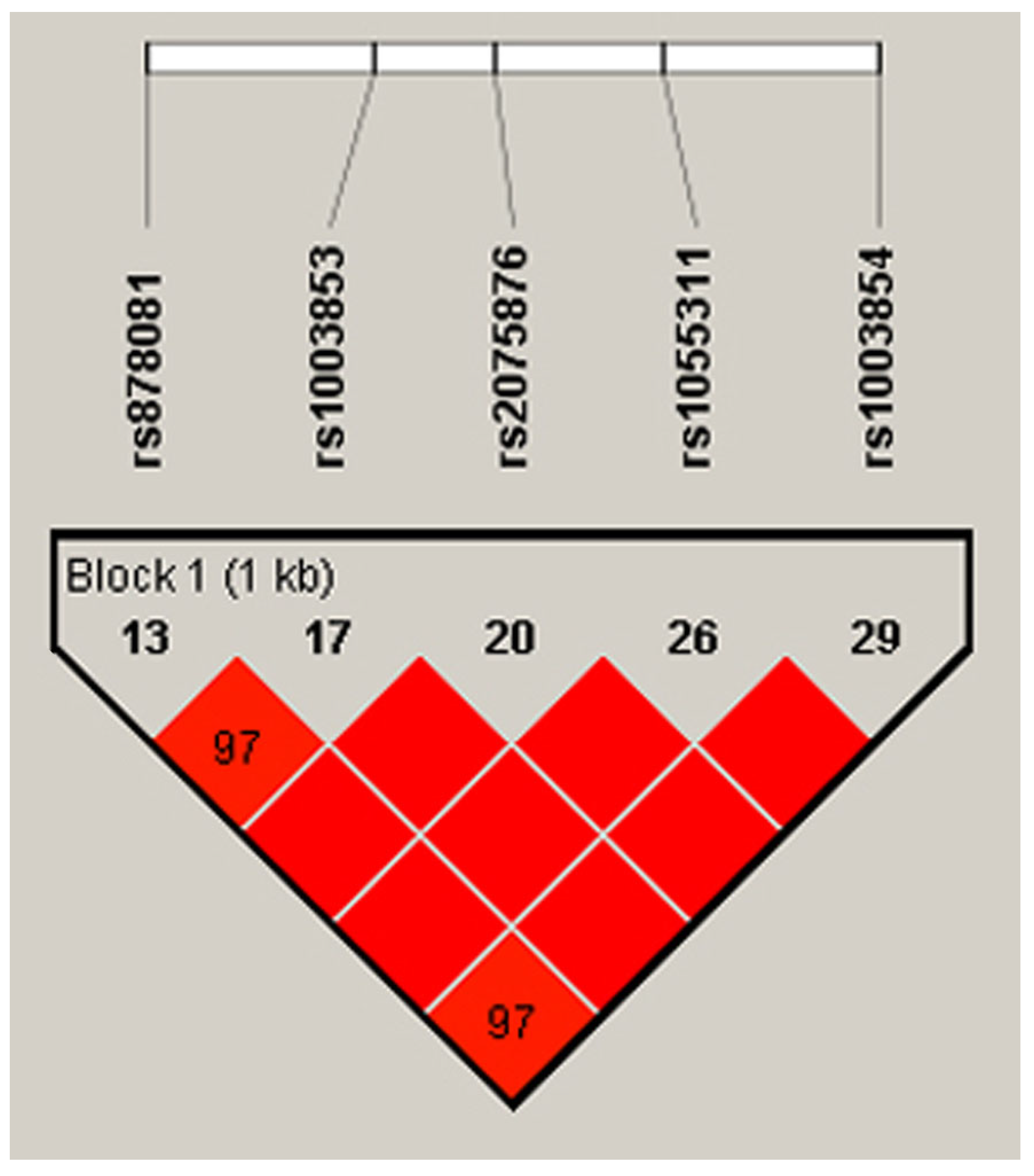
3.1. Preliminarily Included Variations
3.2. Characteristics of Study Populations
3.3. Results of HWE Analysis
3.4. Allelic Polymorphisms in Exon 5 and Intron 5 and 7 Are Associated with RA Risk
3.5. Bivariate Correlation and Association of Clinical Parameters with Genotypic Subgroups of Different Genetic Models in Patients with RA and Control Subjects
3.6. Allele-Specific Affinity to Transcriptional Factor Binding Motifs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finckh, A.; Gilbert, B.; Hodkinson, B.; Bae, S.C.; Thomas, R.; Deane, K.D.; Alpizar-Rodriguez, D.; Lauper, K. Global epidemiology of rheumatoid arthritis. Nat. Rev. Rheumatol. 2022, 18, 591–602. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, L.I.; Bogdanos, D.P.; Katsiari, C.; Platsoucas, C.D. Anti-citrullinated peptides as autoantigens in rheumatoid arthritis-relevance to treatment. Autoimmun. Rev. 2014, 13, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, J.; Wu, Z.; Mi, L.; Li, N.; Wang, Y.; Peng, X.; Xu, K.; Wu, F.; Zhang, L. Anti-citrullinated Protein Antibody Generation, Pathogenesis, Clinical Application, and Prospects. Front. Med. 2022, 8, 802934. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Martínez-Muñoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodríguez-Frade, J.M. T Cell Migration in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Bromberg, R.; Deane, K.D.; Lahey, L.J.; Derber, L.A.; Chandra, P.E.; Edison, J.D.; Gilliland, W.R.; Tibshirani, R.J.; Norris, J.M.; et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS ONE 2012, 7, e35296. [Google Scholar] [CrossRef] [PubMed]
- Amariuta, T.; Luo, Y.; Knevel, R.; Okada, Y.; Raychaudhuri, S. Advances in genetics toward identifying pathogenic cell states of rheumatoid arthritis. Immunol. Rev. 2020, 294, 188–204. [Google Scholar] [CrossRef] [PubMed]
- MacKay, K.; Eyre, S.; Myerscough, A.; Milicic, A.; Barton, A.; Laval, S.; Barrett, J.; Lee, D.; White, S.; John, S.; et al. Whole-genome linkage analysis of rheumatoid arthritis susceptibility loci in 252 affected sibling pairs in the United Kingdom. Arthritis Rheum. 2002, 46, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Kurkó, J.; Besenyei, T.; Laki, J.; Glant, T.T.; Mikecz, K.; Szekanecz, Z. Genetics of rheumatoid arthritis—A comprehensive review. Clin. Rev. Allergy Immunol. 2013, 45, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Kawaguchi, T.; Stahl, E.A.; Kur-reeman, F.A.; Nishida, N.; et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat. Genet. 2012, 44, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, K.; Sakaue, S.; Terao, C.; Luo, Y.; Sonehara, K.; Yamaguchi, K.; Amariuta, T.; Too, C.L.; Laufer, V.A.; Scott, I.C.; et al. Multi-ancestry genome-wide association analyses identify novel genetic mechanisms in rheumatoid arthritis. Nat. Genet. 2022, 54, 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D.; Benoist, C. Aire. Annu. Rev. Immunol. 2009, 27, 287–312. [Google Scholar] [CrossRef] [PubMed]
- Perniola, R. Twenty Years of AIRE. Front. Immunol. 2018, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Kyewski, B.; Klein, L. A central role for central tolerance. Annu. Rev. Immunol. 2006, 24, 571–606. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef]
- Derbinski, J.; Schulte, A.; Kyewski, B.; Klein, L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2001, 2, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, R.; Biemelt, A.; Cordshagen, A.; Johl, A.; Kuthning, D.; Müller-Hilke, B. The Prerequisites for Central Tolerance Induction against Citrullinated Proteins in the Mouse. PLoS ONE 2016, 11, e0158773. [Google Scholar] [CrossRef] [PubMed]
- Constantine, G.M.; Lionakis, M.S. Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol. Rev. 2019, 287, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.N.; Waterfield, M.R.; Gardner, J.M.; Anderson, M.S. Aire in Autoimmunity. Annu. Rev. Immunol. 2024. epub ahead of print. [Google Scholar] [CrossRef]
- Walser-Kuntz, D.R.; Weyand, C.M.; Weaver, A.J.; O’Fallon, W.M.; Goronzy, J.J. Mechanisms under-lying the formation of the T cell receptor repertoire in rheumatoid arthritis. Immunity 1995, 2, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Weyand, C.M. Thymic function and peripheral T-cell homeostasis in rheumatoid arthritis. Trends Immunol. 2001, 22, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Lovewell, T.R.; McDonagh, A.J.; Messenger, A.G.; Azzouz, M.; Tazi-Ahnini, R. The AIRE -230Y Polymorphism Affects AIRE Transcriptional Activity: Potential Influence on AIRE Function in the Thymus. PLoS ONE 2015, 10, e0127476. [Google Scholar] [CrossRef] [PubMed]
- Tazi-Ahnini, R.; McDonagh, A.J.; Wengraf, D.A.; Lovewell, T.R.; Vasilopoulos, Y.; Messenger, A.G.; Cork, M.J.; Gawkrodger, D.J. The autoimmune regulator gene (AIRE) is strongly associated with vitiligo. Br. J. Dermatol. 2008, 159, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Wengraf, D.A.; McDonagh, A.J.; Lovewell, T.R.; Vasilopoulos, Y.; Macdonald-Hull, S.P.; Cork, M.J.; Messenger, A.G.; Tazi-Ahnini, R. Genetic analysis of autoimmune regulator haplotypes in alopecia areata. Tissue Antigens 2008, 71, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Bianchi, M.; Landegren, N.; Dalin, F.; Skov, J.; Hultin-Rosenberg, L.; Mathioudaki, A.; Nordin, J.; Hallgren, Å.; Andersson, G.; et al. Common genetic variation in the autoimmune regulator (AIRE) locus is associated with autoimmune Addison’s disease in Sweden. Sci. Rep. 2018, 8, 8395. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, F.; Rizzi, M.; Sprecacenere, B.; Balestra, P.; Sessarego, M.; Di Carlo, A.; Filaci, G.; Gabrielli, A.; Ravazzolo, R.; Indiveri, F. AIRE gene polymorphisms in systemic sclerosis associated with autoimmune thyroiditis. Clin. Immunol. 2007, 122, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, G.; Ferrera, F.; Pastorino, L.; Fenoglio, D.; Negrini, S.; Sormani, M.P.; Indiveri, F.; Scarrà, G.B.; Filaci, G. The role of AIRE polymorphisms in melanoma. Clin. Immunol. 2010, 136, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Colobran, R.; Giménez-Barcons, M.; Marín-Sánchez, A.; Porta-Pardo, E.; Pujol-Borrell, R. AIRE genetic variants and predisposition to polygenic autoimmune disease: The case of Graves’ disease and a systematic literature review. Hum. Immunol. 2016, 77, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Bérczi, B.; Gerencsér, G.; Farkas, N.; Hegyi, P.; Veres, G.; Bajor, J.; Czopf, L.; Alizadeh, H.; Ra-konczay, Z.; Vigh, É.; et al. Association between AIRE gene polymorphism and rheumatoid arthritis: A systematic review and meta-analysis of case-control studies. Sci. Rep. 2017, 7, 14096. [Google Scholar] [CrossRef] [PubMed]
- García-Lozano, J.R.; Torres-Agrela, B.; Montes-Cano, M.A.; Ortiz-Fernández, L.; Conde-Jaldón, M.; Teruel, M.; García, A.; Núñez-Roldán, A.; Martín, J.; González-Escribano, M.F. Association of the AIRE gene with susceptibility to rheumatoid arthritis in a European population: A case control study. Arthritis Res. Ther. 2013, 15, R11. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, R.; Lu, Y.; Zou, X.; Yang, W. Aire and Fezf2, two regulators in medullary thymic epithelial cells, control autoimmune diseases by regulating TSAs: Partner or complementer? Front. Immunol. 2022, 13, 948259. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Cavalli-Sforza, L.L.; Bodmer, W.F. The Genetics of Human Populations, 1st ed.; Dover Publications: San Francisco, CA, USA, 1971; pp. 41–118. [Google Scholar]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020, 48, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, M.H.; Qian, H.; Hou, Z.; Rosen, J.D.; Tapia, A.L.; Shan, Y.; Jain, D.; Argos, M.; Arnett, D.K.; Avery, C.; et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019, 15, e1008500. [Google Scholar] [CrossRef] [PubMed]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S.; et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Ingegnoli, F.; Castelli, R.; Gualtierotti, R. Rheumatoid factors: Clinical applications. Dis. Markers 2013, 35, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Niewold, T.B.; Harrison, M.J.; Paget, S.A. Anti-CCP antibody testing as a diagnostic and prognostic tool in rheumatoid arthritis. QJM 2007, 100, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Steiner, G.; Smolen, J. Autoantibodies in rheumatoid arthritis and their clinical significance. Arthritis Res. 2002, 4 (Suppl. 2), S1–S5. [Google Scholar] [CrossRef]
- Graham, J.E.; Granger, C.V.; Karmarkar, A.M.; Deutsch, A.; Niewczyk, P.; Divita, M.A.; Ottenbacher, K.J. The Uniform Data System for Medical Rehabilitation: Report of follow-up information on patients discharged from inpatient rehabilitation programs in 2002–2010. Am. J. Phys. Med. Rehabil. 2014, 93, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Jou, J.M.; Lewis, S.M.; Briggs, C.; Lee, S.H.; De La Salle, B.; McFadden, S.; International Council for Standardization in Haematology. ICSH review of the measurement of the erythocyte sedimentation rate. Int. J. Lab. Hematol. 2011, 33, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Lewontin, R.C.; Kojima, K. The Evolutionary Dynamics of Complex Polymorphisms. Evolution 1960, 14, 458–472. [Google Scholar]
- HaploReg v4.2. Available online: https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php (accessed on 25 March 2024).
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J.; Ramu, C.; Gemünd, C.; Aasland, R. The APECED polyglandular autoimmune syndrome protein, AIRE-1, contains the SAND domain and is probably a transcription factor. Trends Biochem. Sci. 1998, 23, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Berczi, B.; Nusser, N.; Peter, I.; Nemeth, B.; Gyongyi, Z. Association Between AIRE Polymorphisms rs870881(C>T), rs1003854(T>C) and Rheumatoid Arthritis Risk: A Hungarian Case-control Study. In Vivo 2024, 38, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Horita, N.; Kaneko, T. Genetic model selection for a case-control study and a meta-analysis. Meta Gene 2015, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Di Matteo, A.; Bathon, J.M.; Emery, P. Rheumatoid arthritis. Lancet 2023, 402, 2019–2033. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.C.; Rudensky, A.Y. Conceiving the Inconceivable: The Function of Aire in Immune Tolerance to Peripheral Tissue-Restricted Antigens in the Thymus. J. Immunol. 2021, 206, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.G.; Laloraya, M.; Wang, C.Y.; Ruan, Q.G.; Davoodi-Semiromi, A.; Kao, K.J.; She, J.X. The autoimmune regulator (AIRE) is a DNA-binding protein. J. Biol. Chem. 2001, 276, 41357–41364. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, M.J.; Collard, M.W.; Huggenvik, J.I.; Liu, Z.; Gibson, T.J.; Sattler, M. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat. Struct. Biol. 2001, 8, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.; Hanzu, F.A.; Pradas-Juni, M.; Aranda, G.B.; Halperin, I.; Puig-Domingo, M.; Aguiló, S.; Fernández-Rebollo, E. New splice site acceptor mutation in AIRE gene in autoimmune polyendocrine syndrome type 1. PLoS ONE 2014, 9, e101616. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Modica, R.; Allotta, M.L.; Guarnotta, V.; Cervato, S.; Masiero, S.; Giordano, R.; Garelli, S.; Betterle, C. Autoimmune polyendocrinopathy-candidiasis-ectodermal-dystrophy (APECED) in Sicily: Confirmation that R203X is the peculiar AIRE gene mutation. J. Endocrinol. Investig. 2012, 35, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Aytac, G.; Guven, B.; Aydin, I.; Topyildiz, E.; Aykut, A.; Durmaz, A.; Edeer Karaca, N.; Aksu, G.; Kutukculer, N. An Extraordinary Case of Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED) Syndrome Misdiagnosed as Juvenile Idiopathic Arthritis on Admission. Case Rep. Immunol. 2023, 2023, 2363760. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Yamada, R.; Ohmura, K.; Takahashi, M.; Kawaguchi, T.; Kochi, Y.; Human Disease Genomics Working Group; RA Clinical and Genetic Study Consortium; Okada, Y.; Nakamura, Y.; et al. The human AIRE gene at chromosome 21q22 is a genetic determinant for the predisposition to rheumatoid arthritis in Japanese population. Hum. Mol. Genet. 2011, 20, 2680–2685. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Li, X.R.; Cen, H.; Yin, Z.S. Association of AIRE polymorphisms with genetic susceptibility to rheumatoid arthritis in a Chinese population. Inflammation 2014, 37, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.J.; Zhang, S.L.; Wen, H.F.; Liang, Y. Association of rs2075876 polymorphism of AIRE gene with rheumatoid arthritis risk. Hum. Immunol. 2015, 76, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, T.; Chen, M.; Chai, Y. Association of AIRE gene polymorphisms with susceptibility to rheumatoid arthritis among ethnic Han Chinese from Shaanxi. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2016, 33, 373–377. (In Chinese) [Google Scholar] [CrossRef]
- Yang, H.; Li, J.; Jiang, L.; Jiang, X.; Zhou, X.; Xu, N. The rs878081 polymorphism of AIRE gene increases the risk of rheumatoid arthritis in a Chinese Han population: A case-control study. Braz. J. Med. Biol. Res. 2018, 51, e7944. [Google Scholar] [CrossRef] [PubMed]
- Yap, H.Y.; Tee, S.Z.; Wong, M.M.; Chow, S.K.; Peh, S.C.; Teow, S.Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells 2018, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- van Delft, M.A.; Huitema, L.F.; Tas, S.W. The contribution of NF-κB signalling to immune regulation and tolerance. Eur. J. Clin. Investig. 2015, 45, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Haljasorg, U.; Bichele, R.; Saare, M.; Guha, M.; Maslovskaja, J.; Kõnd, K.; Remm, A.; Pihlap, M.; Tomson, L.; Kisand, K.; et al. A highly conserved NF-κB-responsive enhancer is critical for thymic expression of Aire in mice. Eur. J. Immunol. 2015, 45, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Barnabei, L.; Laplantine, E.; Mbongo, W.; Rieux-Laucat, F.; Weil, R. NF-κB: At the Borders of Autoimmunity and Inflammation. Front. Immunol. 2021, 12, 716469. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y. GATA3: A master of many trades in immune regulation. Trends Immunol. 2014, 35, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.B. Intron-mediated regulation of gene expression. Curr. Top. Microbiol. Immunol. 2008, 326, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Osaki, D.; Hiramatsu, H. Citrullination and deamidation affect aggregation properties of amyloid β-proteins. Amyloid 2016, 23, 234–241. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Group 1 | Group 2 | |||
|---|---|---|---|---|
| Characteristics | Patients with RA (n = 270) | Control Subjects (n = 322) | Patients with RA (n = 170) | Control Subjects (n = 222) |
| Age (years), mean ± SD | 65.47 ± 9.85 a | 78.66 ± 5.86 | 65.53 ± 9.89 d | 77.53 ± 6.03 |
| Sex | ||||
| Female, no. (%) | 227 (84.0) b | 252 (78.2) c | 142 (83.5) e | 184 (82.8) f |
| Male, no. (%) | 43 (15.9) | 70 (21.7) | 28 (16.4) | 38 (17.1) |
| ESR (mm/h), mean ± SD (normal range: 3–13 mm/h) | 39.52 ± 25.21 a | 17.87 ± 12.81 | 39.09 ± 26.04 d | 19.02 ± 13.82 |
| CRP (mg/dL), mean ± SD (normal range: 0–10 mg/dL) | 29.79 ± 29.88 a | 7.10 ± 6.80 | 30.36 ± 30.34 d | 7.53 ± 8.03 |
| Rf (IgG) (IU/mL), mean ± SD (normal range: 0–15 IU/mL) | 118.80 ± 126.63 a | 13.49 ± 5.55 | 104.71 ± 113.31 d | 14.41 ± 13.75 |
| aCCP (U/mL), mean ± SD (normal range: <20.0 U/mL; weak positivity: ≥20.0–39.0 U/mL; moderate positivity: ≥39.0–59.0 U/mL; strong positivity: ≥59.0 U/mL) | 590.69 ± 947.96 a | 4.56 ± 2.29 | 596.27 ± 1003.71 d | 4.55 ± 1.66 |
| Seropositive, no. (%) | 164 (60.7) | 121 (71.1) | ||
| Seronegative, no. (%) | 23 (8.5) a | 290 (90.0) | 33 (19.4) d | 187 (84.2) |
| Without serology (%) | 83 (30.8) | 32 (10.0) | 16 (9.5) | 35 (15.8) |
| DAS28, mean ± SD (disease remission: <2.6; low: ≥2.6–3.1; moderate: ≥3.1–5.1; high: ≥5.1) | 5.35 ± 1.20 | - | 5.44 ± 1.12 | - |
| AIRE rs878081 (Exon 5) | RA n = 170 (%) | Control Subjects n = 222 (%) | OR (95% CI) | p a |
|---|---|---|---|---|
| Alleles | ||||
| T | 66 (19.4) | 117 (26.4) | ||
| C | 274 (80.6) | 327 (73.6) | 1.48 (1.05–2.09) | 0.023 * |
| Genotypes | ||||
| TT | 7 (4.1) | 13 (5.9) | ||
| CT | 53 (31.2) | 92 (41.4) | 1.07 (0.40–2.84) | 0.892 |
| CC | 110 (64.7) | 117 (52.7) | 1.74 (0.67–4.53) | 0.253 |
| Dominant model | ||||
| TT | 7 (4.1) | 13 (5.9) | ||
| CC + CT | 163 (95.9) | 209 (94.1) | 1.44 (0.56–3.71) | 0.441 |
| Recessive model | ||||
| CT + TT | 60 (35.3) | 105 (47.3) | ||
| CC | 110 (64.7) | 117 (52.7) | 1.64 (1.09–2.48) | 0.017 * |
| Overdominant model | ||||
| CC + TT | 117 (68.8) | 130 (58.6) | ||
| CT | 53 (31.2) | 92 (41.4) | 0.64 (0.42–0.97) | 0.037 * |
| AIRE rs1003853 (Intron 5) | RA n = 270 (%) | Control subjects n = 322 (%) | OR (95% CI) | p a |
| Alleles | ||||
| T | 116 (21.5) | 172 (26.7) | ||
| C | 424 (78.5) | 472 (73.3) | 1.33 (1.01–1.74) | 0.037 * |
| Genotypes | ||||
| TT | 15 (5.6) | 16 (5.0) | ||
| CT | 85 (31.5) | 141 (43.8) | 0.64 (0.30–1.36) | 0.251 |
| CC | 170 (63.0) | 165 (51.2) | 1.09 (0.52–2.29) | 0.802 |
| Dominant model | ||||
| TT | 15 (5.6) | 16 (5.0) | ||
| CC + CT | 255 (94.4) | 306 (95.0) | 0.88 (0.43–1.83) | 0.750 |
| Recessive model | ||||
| CT + TT | 100 (37.0) | 157 (48.8) | ||
| CC | 170 (63.0) | 165 (51.2) | 1.618 (1.16–2.25) | 0.004 * |
| Overdominant model | ||||
| CC + TT | 185 (68.5) | 181 (56.2) | ||
| CT | 85 (31.5) | 141 (43.8) | 0.59 (0.42–0.82) | 0.002 * |
| AIRE rs1003854 (Intron 7) | RA n = 170 (%) | Control subjects n = 222 (%) | OR (95% CI) | p a |
| Alleles | ||||
| C | 69 (20.3) | 124 (27.9) | ||
| T | 271 (79.7) | 320 (72.1) | 1.52 (1.08–2.12) | 0.014 * |
| Genotypes | ||||
| CC | 6 (3.5) | 13 (5.9) | ||
| TC | 57 (33.5) | 99 (44.6) | 1.34 (0.48–3.69) | 0.567 |
| TT | 106 (62.4) | 109 (41.1) | 2.10 (0.77–5.74) | 0.146 |
| Dominant model | ||||
| CC | 6 (3.5) | 14 (6.3) | ||
| TC + TT | 164 (96.5) | 208 (93.7) | 1.84 (0.69–4.89) | 0.222 |
| Recessive model | ||||
| TC + CC | 63 (37.1) | 112 (50.5) | ||
| TT | 107 (62.9) | 110 (49.5) | 1.72 (1.15–1.44) | 0.008 * |
| Overdominant model | ||||
| CC + TT | 113 (66.5) | 124 (55.9) | ||
| TC | 57 (33.5) | 98 (44.1) | 0.63 (0.42–0.96) | 0.034 * |
| AIRE rs878081 (Exon 5) | Patients with RA | Control Subjects | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ESR | p a | Pearson Correlation | p b | OR (95% CI) | p c | ESR | p d | Pearson Correlation | p e | OR (95% CI) | p f | |
| Genotype | ||||||||||||
| TT | 24.25 ± 13.50 | 16.69 ± 13.11 | ||||||||||
| CT | 33.16 ± 23.37 | 0.599 | 0.10 | 0.458 | 1.02 (0.96–1.08) | 0.454 | 21.73 ± 14.96 | 0.147 | 0.11 | 0.255 | 1.02 (0.97–1.08) | 0.259 |
| CC | 42.77 ± 27.11 | 0.196 | 0.13 | 0.179 | 1.03 (0.98–1.09) | 0.196 | 17.33 ± 12.79 | 0.708 | 0.01 | 0.86 | 1.004 (0.95–1.05) | 0.863 |
| Dominant | ||||||||||||
| TT | 24.25 ± 13.50 | 16.69 ± 13.11 | ||||||||||
| CC + CT | 39.52 ± 26.22 | 0.295 | 0.09 | 0.24 | 1.03 (0.97–1.09) | 0.262 | 19.18 ± 13.88 | 0.391 | 0.04 | 0.530 | 1.01 (0.96–1.06) | 0.530 |
| Recessive | ||||||||||||
| TT + CT | 32.46 ± 22.79 | 21.04 ± 14.76 | ||||||||||
| CC | 42.77 ± 27.11 | 0.027 * | 0.19 | 0.023 * | 1.01 (1.002–1.03) | 0.026 * | 17.33 ± 12.79 | 0.063 | −0.13 | 0.054 | 0.98 (0.96–1.001) | 0.980 |
| Overdominant | ||||||||||||
| TT + CC | 42.00 ± 26.90 | 17.26 ± 12.77 | ||||||||||
| CT | 33.16 ± 23.37 | 0.059 | −0.16 | 0.056 | 0.98 (0.97–1.001) | 0.059 | 21.73 ± 14.96 | 0.020 * | 0.15 | 0.023 * | 1.02 (1.003–1.04) | 0.025 * |
| AIRE rs1055311 (Exon 6) | Patients with RA | Control Subjects | ||||||
|---|---|---|---|---|---|---|---|---|
| aCCP | p a | Pearson Correlation | p b | aCCP | p c | Pearson Correlation | p d | |
| Genotype | ||||||||
| TT | 213.23 ± 258.53 | 4.44 ± 2.43 | ||||||
| CT | 725.33 ± 998.49 | 0.028 * | 0.25 | 0.036 * | 4.43 ± 2.32 | 0.950 | −0.001 | 0.997 |
| CC | 591.51 ± 999.63 | 0.093 | 0.16 | 0.116 | 4.83 ± 2.26 | 0.779 | 0.06 | 0.679 |
| Dominant | ||||||||
| TT | 213.23 ± 258.53 | 4.44 ± 2.43 | ||||||
| CC + CT | 644.19 ± 997.37 | 0.044 * | 0.15 | 0.071 | 4.62 ± 2.29 | 0.918 | 0.02 | 0.843 |
| Recessive | ||||||||
| TT + CT | 589.77 ± 893.25 | 4.43 ± 2.31 | ||||||
| CC | 591.51 ± 999.63 | 0.982 | 0.001 | 0.991 | 4.83 ± 2.26 | 0.187 | 0.085 | 0.403 |
| Overdominant | ||||||||
| TT + CC | 519.84 ± 917.72 | 4.77 ± 2.26 | ||||||
| CT | 725.33 ± 998.49 | 0.157 | 0.103 | 0.216 | 4.43 ± 2.32 | 0.209 | −0.07 | 0.467 |
| Locus | Ref | Alt | Position Weight Matrix ID | Strand | LOD Value of Ref | LOD Value of Alt |
|---|---|---|---|---|---|---|
| rs878081 | C | T | NF-kappaB_known4 | + | 12.5 | 14.5 |
| rs1003854 | T | C | GATA_disc3 | + | 12.2 | 13.2 |
| rs1055311 | C | T | NF-kappaB_disc2 | + | 1.7 | 12.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bérczi, B.; Nusser, N.; Péter, I.; Németh, B.; Kulisch, Á.; Kiss, Z.; Gyöngyi, Z. Genetic Polymorphisms in Exon 5 and Intron 5 and 7 of AIRE Are Associated with Rheumatoid Arthritis Risk in a Hungarian Population. Biology 2024, 13, 439. https://doi.org/10.3390/biology13060439
Bérczi B, Nusser N, Péter I, Németh B, Kulisch Á, Kiss Z, Gyöngyi Z. Genetic Polymorphisms in Exon 5 and Intron 5 and 7 of AIRE Are Associated with Rheumatoid Arthritis Risk in a Hungarian Population. Biology. 2024; 13(6):439. https://doi.org/10.3390/biology13060439
Chicago/Turabian StyleBérczi, Bálint, Nóra Nusser, Iván Péter, Balázs Németh, Ágota Kulisch, Zsuzsanna Kiss, and Zoltán Gyöngyi. 2024. "Genetic Polymorphisms in Exon 5 and Intron 5 and 7 of AIRE Are Associated with Rheumatoid Arthritis Risk in a Hungarian Population" Biology 13, no. 6: 439. https://doi.org/10.3390/biology13060439