
The Advancement and Application of the Single-Cell Transcriptome in Biological and Medical Research

 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
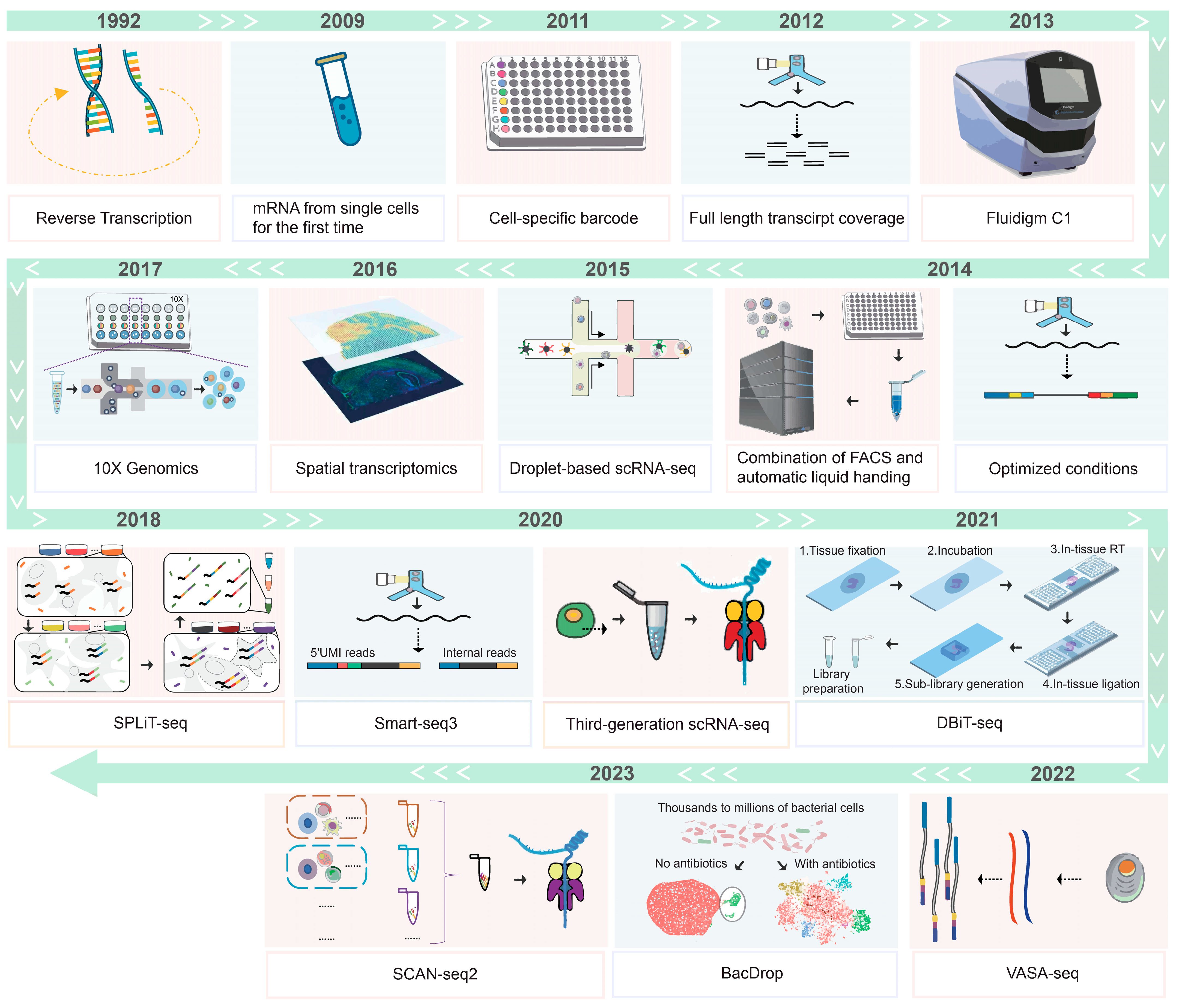
2. Development of the Single Cell Transcriptome
3. Single Cell Isolation and Transcriptome Analysis Process
3.1. Single Cell Isolation
3.2. Data Quality Control
3.3. Construction of Seurat Objects and Information Extraction
3.4. Cluster Analysis
3.5. Annotation of Cellular Taxa
3.5.1. Automatic Annotation
3.5.2. Manual Annotation
3.5.3. Validation
3.6. Calculation of Cell Ratios
3.7. Analysis of Cellular Interactions
3.8. Gene Differential Expression Analysis
3.9. Pseudotime Analysis
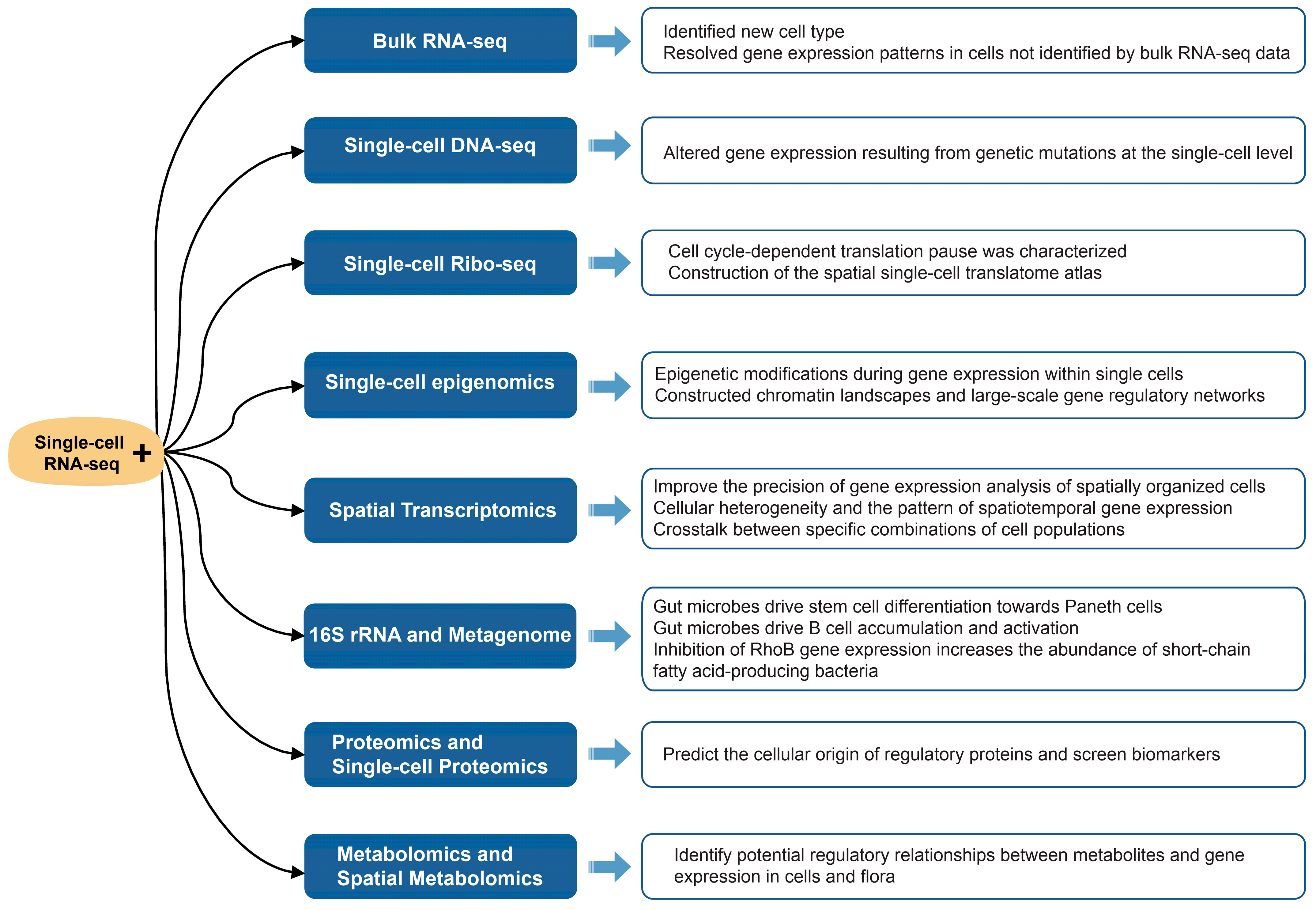
4. Integrated Analysis of scRNA-Seq and Multi-Omics
4.1. Bulk RNA-Seq
4.2. Single-Cell DNA-Seq
4.3. Single-Cell Ribo-Seq
4.4. Single-Cell Epigenomics
4.5. Spatial Transcriptomics
4.6. 16S rRNA and Metagenome
4.7. Proteomics and Single-Cell Proteomics
4.8. Metabolomics and Spatial Metabolomics
5. Application of Single-Cell Transcriptome Sequencing Technology
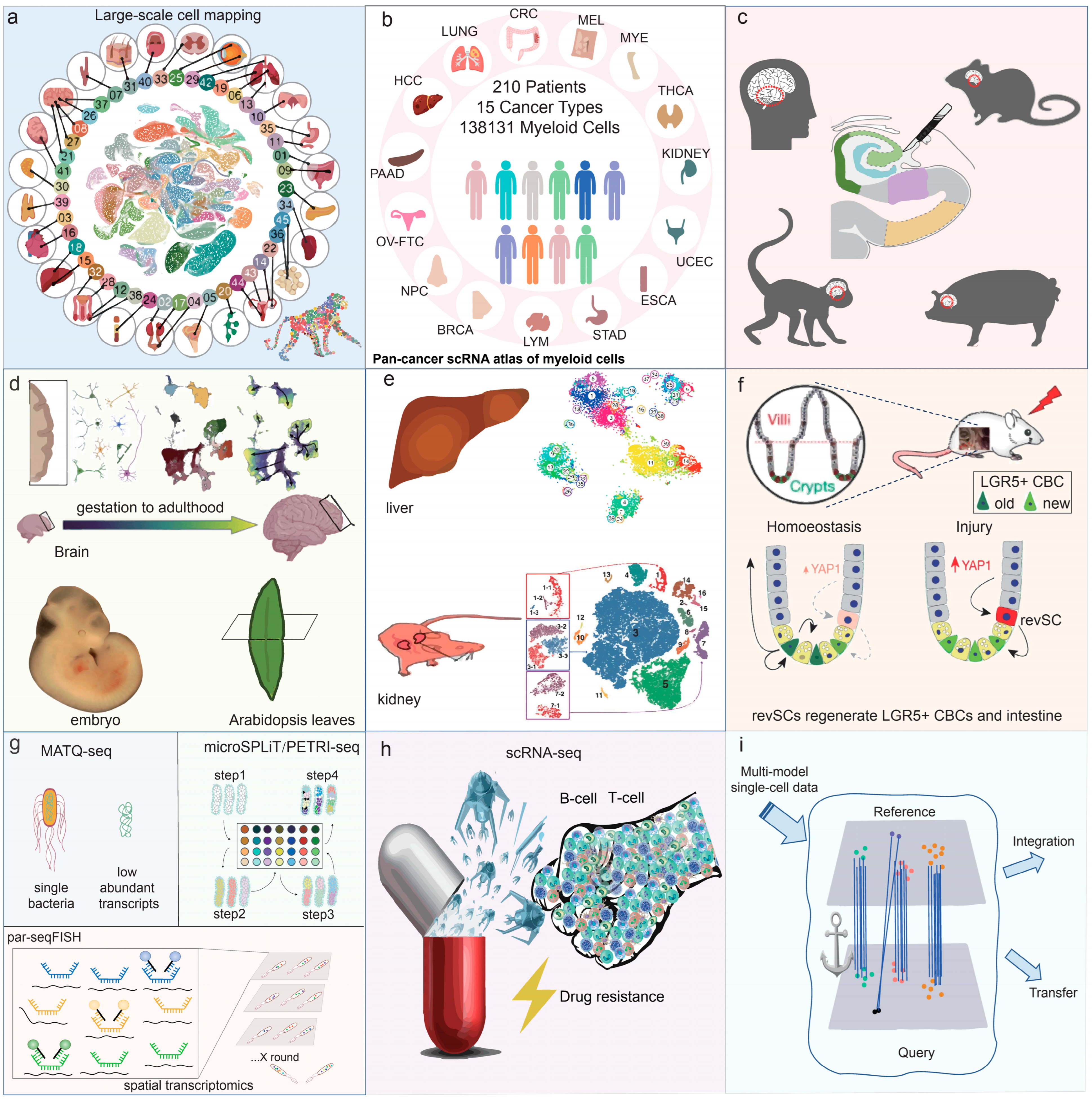
5.1. Large-Scale Cell Mapping Construction
5.2. Oncology Research
5.3. Neuroscience Research
5.4. Developmental Biology
5.5. Cell Subpopulation Refinement and Rare Cell Type Identification
5.6. Stem Cells Research
5.7. Applications in Microbiology
5.8. Drug Resistance Research
5.9. Integration and Utilization of Single-Cell Datasets
6. Summary and Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pennisi, E. Science’s 2018 Breakthrough of the Year: Tracking development cell by cell. Science 2018. Available online: https://vis.sciencemag.org/breakthrough2018/finalists/#cell-development (accessed on 10 April 2024).
- Method of the Year 2019: Single-cell multimodal omics. Nat. Methods 2020, 17, 1. [CrossRef]
- Method of the Year 2020: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 1. [CrossRef]
- Dong, F.; Hao, S.; Zhang, S.; Zhu, C.Y.; Cheng, H.; Yang, Z.N.; Hamey, F.K.; Wang, X.F.; Gao, A.; Wang, F.J.; et al. Differentiation of transplanted haematopoietic stem cells tracked by single-cell transcriptomic analysis. Nat. Cell Biol. 2020, 22, 630–639. [Google Scholar] [CrossRef]
- Xie, X.W.; Liu, M.Y.; Zhang, Y.W.; Wang, B.R.; Zhu, C.Y.; Wang, C.C.; Li, Q.; Huo, Y.Y.; Guo, J.J.; Xu, C.L.; et al. Single-cell transcriptomic landscape of human blood cells. Natl. Sci. Rev. 2021, 8, nwaa180. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.M.; Ren, J.; Sun, S.H.; Ma, S.; Zhang, W.Q.; Yu, Y.; Cai, Y.S.; Yan, K.W.; Li, W.; et al. Single-nucleus transcriptomic landscape of primate hippocampal aging. Protein Cell 2021, 12, 695–716. [Google Scholar] [CrossRef]
- Han, L.; Wei, X.Y.; Liu, C.Y.; Volpe, G.; Zhuang, Z.K.; Zou, X.X.; Wang, Z.F.; Pan, T.T.; Yuan, Y.; Zhang, X.; et al. Cell transcriptomic atlas of the non-human primate Macaca fascicularis. Nature 2022, 604, 723–731. [Google Scholar] [CrossRef]
- Hatscher, L.; Lehmann, C.H.K.; Purbojo, A.; Onderka, C.; Liang, C.; Hartmann, A.; Cesnjevar, R.; Bruns, H.; Gross, O.; Nimmerjahn, F.; et al. Select hyperactivating NLRP3 ligands enhance the T(H)1- and T(H)17-inducing potential of human type 2 conventional dendritic cells. Sci. Signal. 2021, 14, eabe1757. [Google Scholar] [CrossRef]
- Wahis, J.; Baudon, A.; Althammer, F.; Kerspern, D.; Goyon, S.; Hagiwara, D.; Lefevre, A.; Barteczko, L.; Boury-Jamot, B.; Bellanger, B.; et al. Astrocytes mediate the effect of oxytocin in the central amygdala on neuronal activity and affective states in rodents. Nat. Neurosci. 2021, 24, 529–541. [Google Scholar] [CrossRef]
- Aizarani, N.; Saviano, A.; Sagar, N.; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grun, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- Park, J.; Shrestha, R.; Qiu, C.; Kondo, A.; Huang, S.; Werth, M.; Li, M.; Barasch, J.; Susztak, K. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 2018, 360, 758–763. [Google Scholar] [CrossRef]
- Franjic, D.; Skarica, M.; Ma, S.J.; Arellano, J.I.; Tebbenkamp, A.T.N.; Choi, J.; Xu, C.; Li, Q.; Morozov, Y.M.; Andrijevic, D.; et al. Transcriptomic taxonomy and neurogenic trajectories of adult human, macaque, and pig hippocampal and entorhinal cells. Neuron 2022, 110, 452–469.e14. [Google Scholar] [CrossRef]
- Wang, K.K.; Wang, S.S.; Chen, Y.; Wu, D.; Hu, X.Y.; Lu, Y.J.; Wang, L.P.; Bao, L.; Li, C.L.; Zhang, X. Single-cell transcriptomic analysis of somatosensory neurons uncovers temporal development of neuropathic pain. Cell Res 2021, 31, 939–940. [Google Scholar] [CrossRef]
- Zhi, M.L.; Zhang, J.Y.; Tang, Q.Z.; Yu, D.W.; Gao, S.; Gao, D.F.; Liu, P.L.; Guo, J.X.; Hai, T.; Gao, J.; et al. Generation and characterization of stable pig pregastrulation epiblast stem cell lines. Cell Res. 2022, 32, 383–400. [Google Scholar] [CrossRef]
- Ayyaz, A.; Kumar, S.; Sangiorgi, B.; Ghoshal, B.; Gosio, J.; Ouladan, S.; Fink, M.; Barutcu, S.; Trcka, D.; Shen, J.; et al. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 2019, 569, 121–125. [Google Scholar] [CrossRef]
- Kuchina, A.; Brettner, L.M.; Paleologu, L.; Roco, C.M.; Rosenberg, A.B.; Carignano, A.; Kibler, R.; Hirano, M.; DePaolo, R.W.; Seelig, G. Microbial single-cell RNA sequencing by split-pool barcoding. Science 2021, 371, eaba5257. [Google Scholar] [CrossRef]
- Eberwine, J.; Yeh, H.; Miyashiro, K.; Cao, Y.; Nair, S.; Finnell, R.; Zettel, M.; Coleman, P. Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. USA 1992, 89, 3010–3014. [Google Scholar] [CrossRef]
- Lambolez, B.; Audinat, E.; Bochet, P.; Crepel, F.; Rossier, J. AMPA receptor subunits expressed by single Purkinje cells. Neuron 1992, 9, 247–258. [Google Scholar] [CrossRef]
- Peixoto, A.; Monteiro, M.; Rocha, B.; Veiga-Fernandes, H. Quantification of multiple gene expression in individual cells. Genome Res. 2004, 14, 1938–1947. [Google Scholar] [CrossRef]
- Sheng, H.Z.; Lin, P.X.; Nelson, P.G. Analysis of multiple heterogeneous mRNAs in single cells. Anal. Biochem. 1994, 222, 123–130. [Google Scholar] [CrossRef]
- Kurimoto, K.; Yabuta, Y.; Ohinata, Y.; Ono, Y.; Uno, K.D.; Yamada, R.G.; Ueda, H.R.; Saitou, M. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res. 2006, 34, e42. [Google Scholar] [CrossRef]
- Tietjen, I.; Rihel, J.M.; Cao, Y.; Koentges, G.; Zakhary, L.; Dulac, C. Single-cell transcriptional analysis of neuronal progenitors. Neuron 2003, 38, 161–175. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Bao, S.; Lee, C.; Nordman, E.; Wang, X.; Lao, K.; Surani, M.A. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell 2010, 6, 468–478. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Ranaivo, H.; Thirion, F.; Bera-Maillet, C.; Guilly, S.; Simon, C.; Sothier, M.; Van den Berghe, L.; Feugier-Favier, N.; Lambert-Porcheron, S.; Dussouse, I.; et al. Increasing the diversity of dietary fibers in a daily-consumed bread modifies gut microbiota and metabolic profile in subjects at cardiometabolic risk. Gut Microbes 2022, 14, 2044722. [Google Scholar] [CrossRef]
- Islam, S.; Kjallquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lonnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef]
- Ramskold, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef]
- Goetz, J.J.; Trimarchi, J.M. Transcriptome sequencing of single cells with Smart-Seq. Nat. Biotechnol. 2012, 30, 763–765. [Google Scholar] [CrossRef]
- Tan, S.J.; Phan, H.; Gerry, B.M.; Kuhn, A.; Hong, L.Z.; Min Ong, Y.; Poon, P.S.; Unger, M.A.; Jones, R.C.; Quake, S.R.; et al. A microfluidic device for preparing next generation DNA sequencing libraries and for automating other laboratory protocols that require one or more column chromatography steps. PLoS ONE 2013, 8, e64084. [Google Scholar] [CrossRef]
- Guo, G.; Huss, M.; Tong, G.Q.; Wang, C.; Li Sun, L.; Clarke, N.D.; Robson, P. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev. Cell 2010, 18, 675–685. [Google Scholar] [CrossRef]
- Picelli, S.; Bjorklund, A.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef]
- Sheng, K.; Cao, W.; Niu, Y.; Deng, Q.; Zong, C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat. Methods 2017, 14, 267–270. [Google Scholar] [CrossRef]
- Homberger, C.; Saliba, A.E.; Vogel, J. A MATQ-seq-Based Protocol for Single-Cell RNA-seq in Bacteria. Methods Mol. Biol. 2023, 2584, 105–121. [Google Scholar]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W.; et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef]
- Salmen, F.; De Jonghe, J.; Kaminski, T.S.; Alemany, A.; Parada, G.E.; Verity-Legg, J.; Yanagida, A.; Kohler, T.N.; Battich, N.; van den Brekel, F.; et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat. Biotechnol. 2022, 40, 1780–1793. [Google Scholar] [CrossRef]
- Ma, P.; Amemiya, H.M.; He, L.L.; Gandhi, S.J.; Nicol, R.; Bhattacharyya, R.P.; Smillie, C.S.; Hung, D.T. Bacterial droplet-based single-cell RNA-seq reveals antibiotic-associated heterogeneous cellular states. Cell 2023, 186, 877–891.e814. [Google Scholar] [CrossRef]
- Stahl, P.L.; Salmen, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramskold, D.; Hendriks, G.J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef]
- Fan, X.; Tang, D.; Liao, Y.; Li, P.; Zhang, Y.; Wang, M.; Liang, F.; Wang, X.; Gao, Y.; Wen, L.; et al. Single-cell RNA-seq analysis of mouse preimplantation embryos by third-generation sequencing. PLoS Biol. 2020, 18, e3001017. [Google Scholar] [CrossRef]
- Su, G.; Qin, X.; Enninful, A.; Bai, Z.; Deng, Y.; Liu, Y.; Fan, R. Spatial multi-omics sequencing for fixed tissue via DBiT-seq. STAR Protoc. 2021, 2, 100532. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, Z.; Zhang, Y.; Lu, P.; Wen, L.; Tang, F. High-throughput and high-sensitivity full-length single-cell RNA-seq analysis on third-generation sequencing platform. Cell Discov. 2023, 9, 5. [Google Scholar] [CrossRef]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef]
- Kim, D.W.; Yao, Z.; Graybuck, L.T.; Kim, T.K.; Nguyen, T.N.; Smith, K.A.; Fong, O.; Yi, L.; Koulena, N.; Pierson, N.; et al. Multimodal Analysis of Cell Types in a Hypothalamic Node Controlling Social Behavior. Cell 2019, 179, 713–728.e717. [Google Scholar] [CrossRef]
- Coskun, A.F.; Cai, L. Dense transcript profiling in single cells by image correlation decoding. Nat. Methods 2016, 13, 657–660. [Google Scholar] [CrossRef]
- Eng, C.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Wei, X.; Fu, S.; Li, H.; Liu, Y.; Wang, S.; Feng, W.; Yang, Y.; Liu, X.; Zeng, Y.Y.; Cheng, M.; et al. Single-cell Stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science 2022, 377, eabp9444. [Google Scholar] [CrossRef]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792.e1721. [Google Scholar] [CrossRef]
- Xia, C.; Fan, J.; Emanuel, G.; Hao, J.; Zhuang, X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. USA 2019, 116, 19490–19499. [Google Scholar] [CrossRef]
- Wen, L.; Tang, F. Recent advances in single-cell sequencing technologies. Precis. Clin. Med. 2022, 5, pbac002. [Google Scholar] [CrossRef]
- Slovin, S.; Carissimo, A.; Panariello, F.; Grimaldi, A.; Bouche, V.; Gambardella, G.; Cacchiarelli, D. Single-Cell RNA Sequencing Analysis: A Step-by-Step Overview. Methods Mol. Biol. 2021, 2284, 343–365. [Google Scholar]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Lun, A.T.L.; Riesenfeld, S.; Andrews, T.; Dao, T.P.; Gomes, T.; participants in the 1st Human Cell Atlas, J.; Marioni, J.C. EmptyDrops: Distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 2019, 20, 63. [Google Scholar] [CrossRef]
- Xi, J.; Park, S.R.; Lee, J.H.; Kang, H.M. SiftCell: A robust framework to detect and isolate cell-containing droplets from single-cell RNA sequence reads. Cell Syst. 2023, 14, 620–628.e623. [Google Scholar] [CrossRef]
- Heiser, C.N.; Wang, V.M.; Chen, B.; Hughey, J.J.; Lau, K.S. Automated quality control and cell identification of droplet-based single-cell data using dropkick. Genome Res. 2021, 31, 1742–1752. [Google Scholar] [CrossRef]
- Muskovic, W.; Powell, J.E. DropletQC: Improved identification of empty droplets and damaged cells in single-cell RNA-seq data. Genome Biol. 2021, 22, 329. [Google Scholar] [CrossRef]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 2019, 8, 281–291.e289. [Google Scholar] [CrossRef]
- DePasquale, E.A.K.; Schnell, D.J.; Van Camp, P.J.; Valiente-Alandi, I.; Blaxall, B.C.; Grimes, H.L.; Singh, H.; Salomonis, N. DoubletDecon: Deconvoluting Doublets from Single-Cell RNA-Sequencing Data. Cell Rep. 2019, 29, 1718–1727.e1718. [Google Scholar] [CrossRef]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e324. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Belkina, A.C.; Ciccolella, C.O.; Anno, R.; Halpert, R.; Spidlen, J.; Snyder-Cappione, J.E. Automated optimized parameters for T-distributed stochastic neighbor embedding improve visualization and analysis of large datasets. Nat. Commun. 2019, 10, 5415. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef]
- Clarke, Z.A.; Andrews, T.S.; Atif, J.; Pouyabahar, D.; Innes, B.T.; MacParland, S.A.; Bader, G.D. Tutorial: Guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nat. Protoc. 2021, 16, 2749–2764. [Google Scholar] [CrossRef]
- Franzén, O.; Gan, L.M.; Björkegren, J.L.M. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef]
- Zhang, X.X.; Lan, Y.J.; Xu, J.Y.; Quan, F.; Zhao, E.J.; Deng, C.Y.; Luo, T.; Xu, L.W.; Liao, G.M.; Yan, M.; et al. CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Snyder, M.P.; Lin, S.; Posgai, A.; Atkinson, M.; Regev, A.; Rood, J.; Rozenblatt-Rosen, O.; Gaffney, L.; Hupalowska, A.; Satija, R.; et al. The human body at cellular resolution: The NIH Human Biomolecular Atlas Program. Nature 2019, 574, 187–192. [Google Scholar]
- Rosati, E.; Dowds, C.M.; Liaskou, E.; Henriksen, E.K.K.; Karlsen, T.H.; Franke, A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol. 2017, 17, 61. [Google Scholar] [CrossRef]
- Setliff, I.; Shiakolas, A.R.; Pilewski, K.A.; Murji, A.A.; Mapengo, R.E.; Janowska, K.; Richardson, S.; Oosthuysen, C.; Raju, N.; Ronsard, L.; et al. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell 2019, 179, 1636–1646.e15. [Google Scholar] [CrossRef]
- Liu, F.L.; Zhang, Y.Y.; Zhang, L.; Li, Z.Y.; Fang, Q.; Gao, R.R.; Zhang, Z.M. Systematic comparative analysis of single-nucleotide variant detection methods from single-cell RNA sequencing data. Genome Biol. 2019, 20, 242. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e813. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e1821. [Google Scholar] [CrossRef]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B.; et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2016, 7, 7866. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ma, Y.; Sun, S.; Shang, X.; Keller, E.T.; Chen, M.; Zhou, X. Integrative differential expression and gene set enrichment analysis using summary statistics for scRNA-seq studies. Nat. Commun. 2020, 11, 1585. [Google Scholar] [CrossRef]
- Haghverdi, L.; Büttner, M.; Wolf, F.A.; Buettner, F.; Theis, F.J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 2016, 13, 845–848. [Google Scholar] [CrossRef]
- Qiu, X.J.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallee, V.P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 2019, 179, 846–863.e824. [Google Scholar] [CrossRef]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef]
- Xu, Z.; You, W.; Chen, W.; Zhou, Y.; Nong, Q.; Valencak, T.G.; Wang, Y.; Shan, T. Single-cell RNA sequencing and lipidomics reveal cell and lipid dynamics of fat infiltration in skeletal muscle. J. Cachexia Sarcopenia Muscle 2021, 12, 109–129. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M.; et al. G&T-seq: Parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar]
- Guo, L.; Yi, X.; Chen, L.; Zhang, T.; Guo, H.; Chen, Z.; Cheng, J.; Cao, Q.; Liu, H.; Hou, C.; et al. Single-Cell DNA Sequencing Reveals Punctuated and Gradual Clonal Evolution in Hepatocellular Carcinoma. Gastroenterology 2022, 162, 238–252. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, M.; Shi, J.; Zhu, Y.; Wang, X.; Zhang, S.; Wang, F. Cracking the pattern of tumor evolution based on single-cell copy number alterations. Brief. Bioinform. 2023, 24, bbad341. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Pastor, V.B.; Goodings, C.; Voss, R.K.; Kozyra, E.J.; Szvetnik, A.; Noellke, P.; Dworzak, M.; Stary, J.; Locatelli, F.; et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat. Med. 2021, 27, 1806–1817. [Google Scholar] [CrossRef]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- VanInsberghe, M.; van den Berg, J.; Andersson-Rolf, A.; Clevers, H.; van Oudenaarden, A. Single-cell Ribo-seq reveals cell cycle-dependent translational pausing. Nature 2021, 597, 561–565. [Google Scholar] [CrossRef]
- Zeng, H.; Huang, J.; Ren, J.; Wang, C.K.; Tang, Z.; Zhou, H.; Zhou, Y.; Shi, H.; Aditham, A.; Sui, X.; et al. Spatially resolved single-cell translatomics at molecular resolution. Science 2023, 380, eadd3067. [Google Scholar] [CrossRef]
- Bao, Y.; Zhai, J.; Chen, H.; Wong, C.C.; Liang, C.; Ding, Y.; Huang, D.; Gou, H.; Chen, D.; Pan, Y.; et al. Targeting m(6)A reader YTHDF1 augments antitumour immunity and boosts anti-PD-1 efficacy in colorectal cancer. Gut 2023, 72, 1497–1509. [Google Scholar] [CrossRef]
- Gao, W.; Gallardo-Dodd, C.J.; Kutter, C. Cell type-specific analysis by single-cell profiling identifies a stable mammalian tRNA-mRNA interface and increased translation efficiency in neurons. Genome Res. 2022, 32, 97–110. [Google Scholar] [CrossRef]
- Lai, Y.; Ramirez-Pardo, I.; Isern, J.; An, J.; Perdiguero, E.; Serrano, A.L.; Li, J.; Garcia-Dominguez, E.; Segales, J.; Guo, P.; et al. Multimodal cell atlas of the ageing human skeletal muscle. Nature 2024, 629, 154–164. [Google Scholar] [CrossRef]
- Zhang, G.; Fu, Y.; Yang, L.; Ye, F.; Zhang, P.; Zhang, S.; Ma, L.; Li, J.; Wu, H.; Han, X.; et al. Construction of single-cell cross-species chromatin accessibility landscapes with combinatorial-hybridization-based ATAC-seq. Dev. Cell. 2024, 59, 793–811.e798. [Google Scholar] [CrossRef]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 2018, 361, 1380–1385. [Google Scholar] [CrossRef]
- Chen, S.; Lake, B.B.; Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 2019, 37, 1452–1457. [Google Scholar] [CrossRef]
- Zhu, C.; Yu, M.; Huang, H.; Juric, I.; Abnousi, A.; Hu, R.; Lucero, J.; Behrens, M.M.; Hu, M.; Ren, B. An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat. Struct. Mol. Biol. 2019, 26, 1063–1070. [Google Scholar] [CrossRef]
- Johnson, K.C.; Anderson, K.J.; Courtois, E.T.; Gujar, A.D.; Barthel, F.P.; Varn, F.S.; Luo, D.; Seignon, M.; Yi, E.; Kim, H.; et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat. Genet. 2021, 53, 1456–1468. [Google Scholar] [CrossRef]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Furth, D.; Qian, X.; Wardell, E.; Custodio, J.; Reimegard, J.; Salmen, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e1619. [Google Scholar] [CrossRef]
- Farah, E.N.; Hu, R.K.; Kern, C.; Zhang, Q.; Lu, T.Y.; Ma, Q.; Tran, S.; Zhang, B.; Carlin, D.; Monell, A.; et al. Spatially organized cellular communities form the developing human heart. Nature 2024, 627, 854–864. [Google Scholar] [CrossRef]
- Chen, A.; Sun, Y.; Lei, Y.; Li, C.; Liao, S.; Meng, J.; Bai, Y.; Liu, Z.; Liang, Z.; Zhu, Z.; et al. Single-cell spatial transcriptome reveals cell-type organization in the macaque cortex. Cell 2023, 186, 3726–3743.e3724. [Google Scholar] [CrossRef]
- Allen, W.E.; Blosser, T.R.; Sullivan, Z.A.; Dulac, C.; Zhuang, X. Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell 2023, 186, 194–208.e118. [Google Scholar] [CrossRef]
- Cao, S.; Feng, H.; Yi, H.; Pan, M.; Lin, L.; Zhang, Y.S.; Feng, Z.; Liang, W.; Cai, B.; Li, Q.; et al. Single-cell RNA sequencing reveals the developmental program underlying proximal-distal patterning of the human lung at the embryonic stage. Cell Res. 2023, 33, 421–433. [Google Scholar] [CrossRef]
- Kim, J.E.; Li, B.; Fei, L.; Horne, R.; Lee, D.; Loe, A.K.; Miyake, H.; Ayar, E.; Kim, D.K.; Surette, M.G.; et al. Gut microbiota promotes stem cell differentiation through macrophage and mesenchymal niches in early postnatal development. Immunity 2022, 55, 2300–2317.e2306. [Google Scholar] [CrossRef]
- Yang, J.; Pei, G.; Sun, X.; Xiao, Y.; Miao, C.; Zhou, L.; Wang, B.; Yang, L.; Yu, M.; Zhang, Z.S.; et al. RhoB affects colitis through modulating cell signaling and intestinal microbiome. Microbiome 2022, 10, 149. [Google Scholar] [CrossRef]
- Overacre-Delgoffe, A.E.; Bumgarner, H.J.; Cillo, A.R.; Burr, A.H.P.; Tometich, J.T.; Bhattacharjee, A.; Bruno, T.C.; Vignali, D.A.A.; Hand, T.W. Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity 2021, 54, 2812–2824.e2814. [Google Scholar] [CrossRef]
- Barrow, F.; Khan, S.; Fredrickson, G.; Wang, H.; Dietsche, K.; Parthiban, P.; Robert, S.; Kaiser, T.; Winer, S.; Herman, A.; et al. Microbiota-Driven Activation of Intrahepatic B Cells Aggravates NASH Through Innate and Adaptive Signaling. Hepatology 2021, 74, 704–722. [Google Scholar] [CrossRef]
- Lai, H.C.; Lin, T.L.; Chen, T.W.; Kuo, Y.L.; Chang, C.J.; Wu, T.R.; Shu, C.C.; Tsai, Y.H.; Swift, S.; Lu, C.C. Gut microbiota modulates COPD pathogenesis: Role of anti-inflammatory Parabacteroides goldsteinii lipopolysaccharide. Gut 2022, 71, 309–321. [Google Scholar] [CrossRef]
- Hezaveh, K.; Shinde, R.S.; Klotgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 2022, 55, 324–340.e328. [Google Scholar] [CrossRef]
- Xue, M.Y.; Wu, J.J.; Xie, Y.Y.; Zhu, S.L.; Zhong, Y.F.; Liu, J.X.; Sun, H.Z. Investigation of fiber utilization in the rumen of dairy cows based on metagenome-assembled genomes and single-cell RNA sequencing. Microbiome 2022, 10, 11. [Google Scholar] [CrossRef]
- Ghaddar, B.; Biswas, A.; Harris, C.; Omary, M.B.; Carpizo, D.R.; Blaser, M.J.; De, S. Tumor microbiome links cellular programs and immunity in pancreatic cancer. Cancer Cell 2022, 40, 1240–1253.e1245. [Google Scholar] [CrossRef]
- Chan, M.Y.; Efthymios, M.; Tan, S.H.; Pickering, J.W.; Troughton, R.; Pemberton, C.; Ho, H.H.; Prabath, J.F.; Drum, C.L.; Ling, L.H.; et al. Prioritizing Candidates of Post-Myocardial Infarction Heart Failure Using Plasma Proteomics and Single-Cell Transcriptomics. Circulation 2020, 142, 1408–1421. [Google Scholar] [CrossRef]
- Fava, A.; Buyon, J.; Mohan, C.; Zhang, T.; Belmont, H.M.; Izmirly, P.; Clancy, R.; Trujillo, J.M.; Fine, D.; Zhang, Y.; et al. Integrated urine proteomics and renal single-cell genomics identify an IFN-gamma response gradient in lupus nephritis. JCI Insight 2020, 5, e138345. [Google Scholar] [CrossRef]
- Cui Zhou, D.; Jayasinghe, R.G.; Chen, S.; Herndon, J.M.; Iglesia, M.D.; Navale, P.; Wendl, M.C.; Caravan, W.; Sato, K.; Storrs, E.; et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 2022, 54, 1390–1405. [Google Scholar] [CrossRef]
- Fu, Q.; Jiang, H.; Qian, Y.; Lv, H.; Dai, H.; Zhou, Y.; Chen, Y.; He, Y.; Gao, R.; Zheng, S.; et al. Single-cell RNA sequencing combined with single-cell proteomics identifies the metabolic adaptation of islet cell subpopulations to high-fat diet in mice. Diabetologia 2023, 66, 724–740. [Google Scholar] [CrossRef]
- Yao, L.; Wang, J.T.; Jayasinghe, R.G.; O’Neal, J.; Tsai, C.F.; Rettig, M.P.; Song, Y.; Liu, R.; Zhao, Y.; Ibrahim, O.M.; et al. Single-Cell Discovery and Multiomic Characterization of Therapeutic Targets in Multiple Myeloma. Cancer Res. 2023, 83, 1214–1233. [Google Scholar] [CrossRef]
- Mirji, G.; Worth, A.; Bhat, S.A.; El Sayed, M.; Kannan, T.; Goldman, A.R.; Tang, H.Y.; Liu, Q.; Auslander, N.; Dang, C.V.; et al. The microbiome-derived metabolite TMAO drives immune activation and boosts responses to immune checkpoint blockade in pancreatic cancer. Sci. Immunol. 2022, 7, eabn0704. [Google Scholar] [CrossRef]
- Chai, X.; Wang, J.; Li, H.; Gao, C.; Li, S.; Wei, C.; Huang, J.; Tian, Y.; Yuan, J.; Lu, J.; et al. Intratumor microbiome features reveal antitumor potentials of intrahepatic cholangiocarcinoma. Gut Microbes 2023, 15, 2156255. [Google Scholar] [CrossRef]
- Huang, L.J.; Mao, X.T.; Li, Y.Y.; Liu, D.D.; Fan, K.Q.; Liu, R.B.; Wu, T.T.; Wang, H.L.; Zhang, Y.; Yang, B.; et al. Multiomics analyses reveal a critical role of selenium in controlling T cell differentiation in Crohn’s disease. Immunity 2021, 54, 1728–1744.e1727. [Google Scholar] [CrossRef]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; Garcia-Canaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e1826. [Google Scholar] [CrossRef]
- Zhang, J.; Song, J.; Tang, S.; Zhao, Y.; Wang, L.; Luo, Y.; Tang, J.; Ji, Y.; Wang, X.; Li, T.; et al. Multi-omics analysis reveals the chemoresistance mechanism of proliferating tissue-resident macrophages in PDAC via metabolic adaptation. Cell Rep. 2023, 42, 112620. [Google Scholar] [CrossRef]
- Wang, G.; Heijs, B.; Kostidis, S.; Rietjens, R.G.J.; Koning, M.; Yuan, L.; Tiemeier, G.L.; Mahfouz, A.; Dumas, S.J.; Giera, M.; et al. Spatial dynamic metabolomics identifies metabolic cell fate trajectories in human kidney differentiation. Cell Stem Cell 2022, 29, 1580–1593.e1587. [Google Scholar] [CrossRef]
- Zheng, P.; Zhang, N.; Ren, D.; Yu, C.; Zhao, B.; Zhang, Y. Integrated spatial transcriptome and metabolism study reveals metabolic heterogeneity in human injured brain. Cell Rep. Med. 2023, 4, 101057. [Google Scholar] [CrossRef]
- Cheng, S.J.; Li, Z.Y.; Gao, R.R.; Xing, B.C.; Gao, Y.N.; Yang, Y.; Qin, S.S.; Zhang, L.; Ouyang, H.Q.; Du, P.; et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 2021, 184, 792–809.e23. [Google Scholar] [CrossRef]
- Herring, C.A.; Simmons, R.K.; Freytag, S.; Poppe, D.; Moffet, J.J.D.; Pflueger, J.; Buckberry, S.; Vargas-Landin, D.B.; Clément, O.; Echeverría, E.G.; et al. Human prefrontal cortex gene regulatory dynamics from gestation to adulthood at single-cell resolution. Cell 2022, 185, 4428–4447.e4428. [Google Scholar] [CrossRef]
- Xia, K.; Sun, H.X.; Li, J.; Li, J.; Zhao, Y.; Chen, L.; Qin, C.; Chen, R.; Chen, Z.; Liu, G.; et al. The single-cell stereo-seq reveals region-specific cell subtypes and transcriptome profiling in Arabidopsis leaves. Dev Cell 2022, 57, 1299–1310.e1294. [Google Scholar] [CrossRef]
- Homberger, C.; Barquist, L.; Vogel, J. Ushering in a new era of single-cell transcriptomics in bacteria. Microlife 2022, 3, uqac020. [Google Scholar] [CrossRef]
- Lei, Y.L.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.J.; Shi, S. Applications of single-cell sequencing in cancer research: Progress and perspectives. J. Hematol. Oncol. 2021, 14, 91. [Google Scholar] [CrossRef]
- Li, H.G.; Qu, L.X.; Yang, Y.H.; Zhang, H.B.; Li, X.X.; Zhang, X.L. Single-cell Transcriptomic Architecture Unraveling the Complexity of Tumor Heterogeneity in Distal Cholangiocarcinoma. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1592–1609.e9. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.; Suh, Y.L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Kanton, S.; Boyle, M.J.; He, Z.; Santel, M.; Weigert, A.; Sanchís-Calleja, F.; Guijarro, P.; Sidow, L.; Fleck, J.S.; Han, D.; et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 2019, 574, 418–422. [Google Scholar] [CrossRef]
- Raj, B.; Farrell, J.A.; Liu, J.; El Kholtei, J.; Carte, A.N.; Navajas Acedo, J.; Du, L.Y.; McKenna, A.; Relić, Đ.; Leslie, J.M.; et al. Emergence of Neuronal Diversity during Vertebrate Brain Development. Neuron 2020, 108, 1058–1074.e1056. [Google Scholar] [CrossRef]
- Srivatsan, S.R.; Regier, M.C.; Barkan, E.; Franks, J.M.; Packer, J.S.; Grosjean, P.; Duran, M.; Saxton, S.; Ladd, J.J.; Spielmann, M.; et al. Embryo-scale, single-cell spatial transcriptomics. Science 2021, 373, 111–117. [Google Scholar] [CrossRef]
- Mittnenzweig, M.; Mayshar, Y.; Cheng, S.; Ben-Yair, R.; Hadas, R.; Rais, Y.; Chomsky, E.; Reines, N.; Uzonyi, A.; Lumerman, L.; et al. A single-embryo, single-cell time-resolved model for mouse gastrulation. Cell 2021, 184, 2825–2842.e2822. [Google Scholar] [CrossRef]
- Zhang, T.Q.; Chen, Y.; Wang, J.W. A single-cell analysis of the Arabidopsis vegetative shoot apex. Dev. Cell. 2021, 56, 1056–1074.e1058. [Google Scholar] [CrossRef]
- Satterlee, J.W.; Strable, J.; Scanlon, M.J. Plant stem-cell organization and differentiation at single-cell resolution. Proc. Natl. Acad. Sci. USA 2020, 117, 33689–33699. [Google Scholar] [CrossRef]
- Kang, Y.; Norris, M.H.; Zarzycki-Siek, J.; Nierman, W.C.; Donachie, S.P.; Hoang, T.T. Transcript amplification from single bacterium for transcriptome analysis. Genome Res. 2011, 21, 925–935. [Google Scholar] [CrossRef]
- Kang, Y.; McMillan, I.; Norris, M.H.; Hoang, T.T. Single prokaryotic cell isolation and total transcript amplification protocol for transcriptomic analysis. Nat. Protoc. 2015, 10, 974–984. [Google Scholar] [CrossRef]
- de Bekker, C.; Bruning, O.; Jonker, M.J.; Breit, T.M.; Wosten, H.A. Single cell transcriptomics of neighboring hyphae of Aspergillus niger. Genome Biol. 2011, 12, R71. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Chen, Z.; Zhang, W. RNA-seq based transcriptomic analysis of single bacterial cells. Integr. Biol. (Camb.) 2015, 7, 1466–1476. [Google Scholar] [CrossRef]
- Huang, X.; Li, Y.; Zhang, J.; Yan, L.; Zhao, H.; Ding, L.; Bhatara, S.; Yang, X.; Yoshimura, S.; Yang, W.; et al. Single-cell systems pharmacology identifies development-driven drug response and combination therapy in B cell acute lymphoblastic leukemia. Cancer Cell 2024, 42, 552–567.e556. [Google Scholar] [CrossRef]
- Wang, G.; Sun, J.; Zhang, J.; Zhu, Q.; Lu, J.; Gao, S.; Wang, F.; Yin, Q.; Wan, Y.; Li, Q. Single-cell transcriptional profiling uncovers the association between EOMES(+)CD8(+) T cells and acquired EGFR-TKI resistance. Drug Resist. Updates 2023, 66, 100910. [Google Scholar] [CrossRef]
- Du, R.; Zhang, X.; Lu, X.; Ma, X.; Guo, X.; Shi, C.; Ren, X.; Ma, X.; He, Y.; Gao, Y.; et al. PDPN positive CAFs contribute to HER2 positive breast cancer resistance to trastuzumab by inhibiting antibody-dependent NK cell-mediated cytotoxicity. Drug Resist. Updates 2023, 68, 100947. [Google Scholar] [CrossRef]
- Wu, H.; Guo, C.; Wang, C.; Xu, J.; Zheng, S.; Duan, J.; Li, Y.; Bai, H.; Xu, Q.; Ning, F.; et al. Single-cell RNA sequencing reveals tumor heterogeneity, microenvironment, and drug-resistance mechanisms of recurrent glioblastoma. Cancer Sci. 2023, 114, 2609–2621. [Google Scholar] [CrossRef]
- Xue, R.; Zhang, Q.; Cao, Q.; Kong, R.; Xiang, X.; Liu, H.; Feng, M.; Wang, F.; Cheng, J.; Li, Z.; et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 2022, 612, 141–147. [Google Scholar] [CrossRef]
- De Donno, C.; Hediyeh-Zadeh, S.; Moinfar, A.A.; Wagenstetter, M.; Zappia, L.; Lotfollahi, M.; Theis, F.J. Population-level integration of single-cell datasets enables multi-scale analysis across samples. Nat. Methods 2023, 20, 1683–1692. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Gogolewski, K.; Sykulski, M.; Chung, N.C.; Gambin, A. Truncated Robust Principal Component Analysis and Noise Reduction for Single Cell RNA Sequencing Data. J. Comput. Biol. 2019, 26, 782–793. [Google Scholar] [CrossRef]
- Haghverdi, L.; Lun, A.T.L.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Shangguan, Y.; Li, C.; Lin, H.; Ou, M.; Tang, D.; Dai, Y.; Yan, Q. Application of single-cell RNA sequencing in embryonic development. Genomics 2020, 112, 4547–4551. [Google Scholar] [CrossRef]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Tran, M.N.; Maynard, K.R.; Spangler, A.; Huuki, L.A.; Montgomery, K.D.; Sadashivaiah, V.; Tippani, M.; Barry, B.K.; Hancock, D.B.; Hicks, S.C.; et al. Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron 2021, 109, 3088–3103.e3085. [Google Scholar] [CrossRef]
- Du, H.Y.; Si, G.; Si, J.Q.; Song, X.J.; Si, F.C. Single-cell RNA sequencing analysis revealed malignant ductal cell heterogeneity and prognosis signatures in pancreatic cancer. Clin. Res. Hepatol. Gastroenterol. 2023, 47, 102200. [Google Scholar] [CrossRef]
- He, D.; Wang, D.; Lu, P.; Yang, N.; Xue, Z.G.; Zhu, X.M.; Zhang, P.; Fan, G.P. Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations. Oncogene 2021, 40, 355–368. [Google Scholar] [CrossRef]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.P.; Wiederspahn, J.; Uhlitz, F.; Liang, X.Z.; Lehmann, A.; Jurmeister, P.; Elsner, A.; et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef]
- Jiang, H.P.; Yu, D.Y.; Yang, P.H.; Guo, R.F.; Kong, M.; Gao, Y.; Yu, X.F.; Lu, X.Y.; Fan, X.H. Revealing the transcriptional heterogeneity of organ-specific metastasis in human gastric cancer using single-cell RNA Sequencing. Clin. Transl. Med. 2022, 12, e730. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Wen, X.; Luo, Z.; Zhao, W.; Calandrelli, R.; Nguyen, T.C.; Wan, X.; Charles Richard, J.L.; Zhong, S. Single-cell multiplex chromatin and RNA interactions in ageing human brain. Nature 2024, 628, 648–656. [Google Scholar] [CrossRef]
- Chen, A.F.; Parks, B.; Kathiria, A.S.; Ober-Reynolds, B.; Goronzy, J.J.; Greenleaf, W.J. NEAT-seq: Simultaneous profiling of intra-nuclear proteins, chromatin accessibility and gene expression in single cells. Nat. Methods 2022, 19, 547–553. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Name | Method | Single-Cell Resolution | Analysis Area | Time Required | Advantage | Disadvantage |
|---|---|---|---|---|---|---|
| 10 × Genomics Visium | Sequencing-based | ~55 µm/spot | (6.5 mm × 6.5 mm) × 4 | <1 h | Easy and convenient operation, short test time, high cell flux and cell capture efficiency, and good cell compatibility | Only analyzes RNA with poly A, high sample size requirements and non-full-length information |
| Stereo-seq | Sequencing-based | Close to single cell | 10 mm × 10 mm (13 cm × 13 cm) | <1 h | Nanoscale resolution, high cell capture rate, panoramic field of view | Requires specific equipment and optimized sample handling |
| MERFISH | Imaging-based | ≤100 nm/spot | Full slide (theoretically) | <24 h | High spatial resolution, large area imaging, high sensitivity, diverse applicability, multi-omics research | Specialized equipment required, high-cost, low detection efficacy |
| seqFISH | Imaging-based | Subcellular level | 0.5 mm × 0.5 mm | 2–3 days | Highly specific, limiting off-target effects | Expensive and time-consuming |
| DBiT-seq | Microfluidic chip Sequencing-based | ~10 μm/pixel | Full slide (theoretically) | 5–7 h | Simultaneous detection of mRNA and protein | Large inter batch differences, and low capture efficiency |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, K.; Xu, Y.; Feng, T.; Lan, H.; Ling, F.; Xiang, H.; Liu, Q. The Advancement and Application of the Single-Cell Transcriptome in Biological and Medical Research. Biology 2024, 13, 451. https://doi.org/10.3390/biology13060451
Huang K, Xu Y, Feng T, Lan H, Ling F, Xiang H, Liu Q. The Advancement and Application of the Single-Cell Transcriptome in Biological and Medical Research. Biology. 2024; 13(6):451. https://doi.org/10.3390/biology13060451
Chicago/Turabian StyleHuang, Kongwei, Yixue Xu, Tong Feng, Hong Lan, Fei Ling, Hai Xiang, and Qingyou Liu. 2024. "The Advancement and Application of the Single-Cell Transcriptome in Biological and Medical Research" Biology 13, no. 6: 451. https://doi.org/10.3390/biology13060451
APA StyleHuang, K., Xu, Y., Feng, T., Lan, H., Ling, F., Xiang, H., & Liu, Q. (2024). The Advancement and Application of the Single-Cell Transcriptome in Biological and Medical Research. Biology, 13(6), 451. https://doi.org/10.3390/biology13060451