

Huperzine A Regulates the Physiological Homeostasis of Amyloid Precursor Protein Proteolysis and Tau Protein Conformation—A Computational and Experimental Investigation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and Treatment
2.3. Western Blotting Analysis
2.4. Molecular Docking
2.5. Enzymatic Activity Assay
2.6. Immunocytochemistry
2.7. Statistical Analysis
3. Results
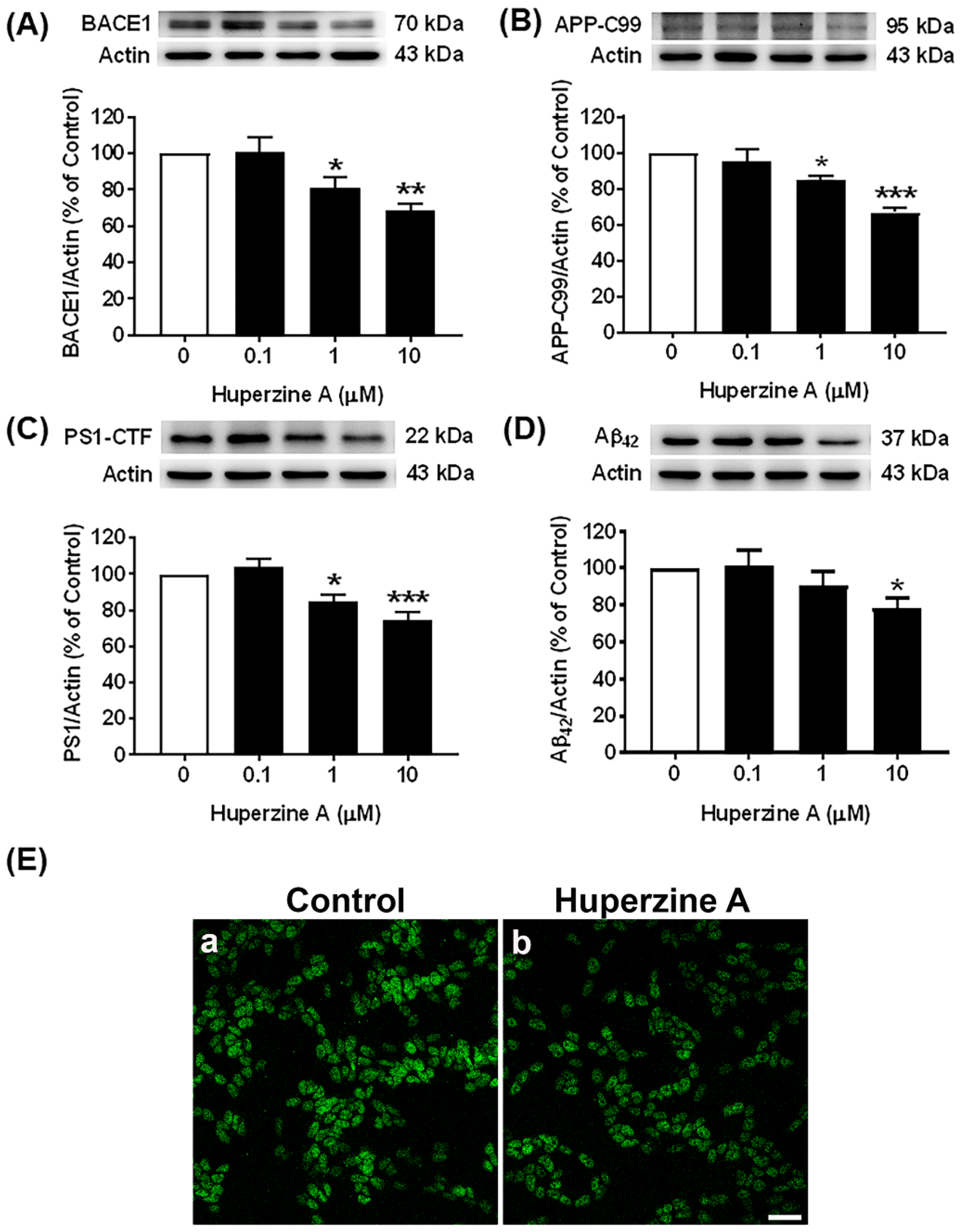
3.1. Effects of Hup A on the Amyloidogenic Pathway
3.2. Effects of Hup A on the Nonamyloidogenic Pathway
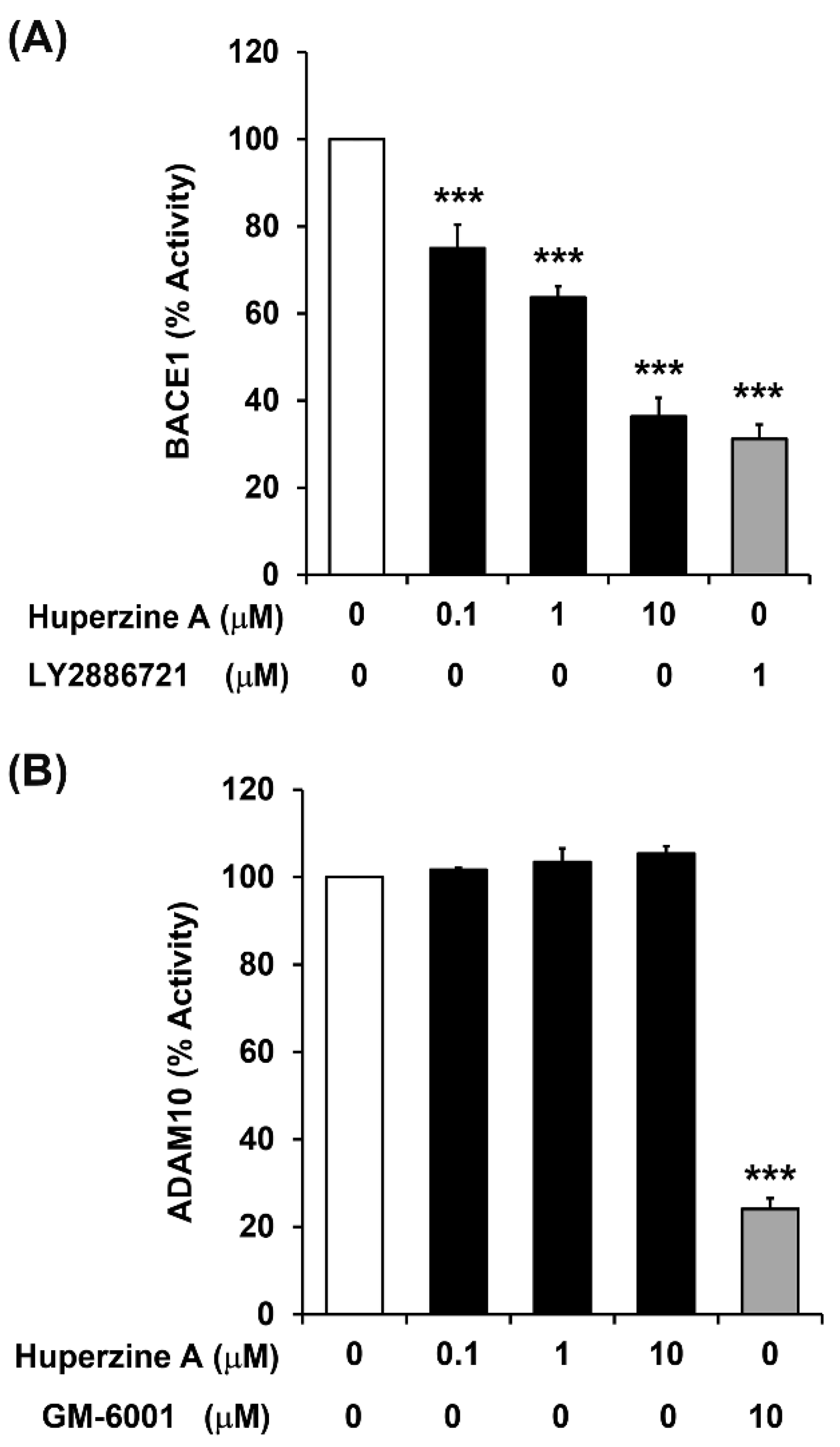
3.3. Regulation of BACE1 and ADAM10 Activity by Hup A
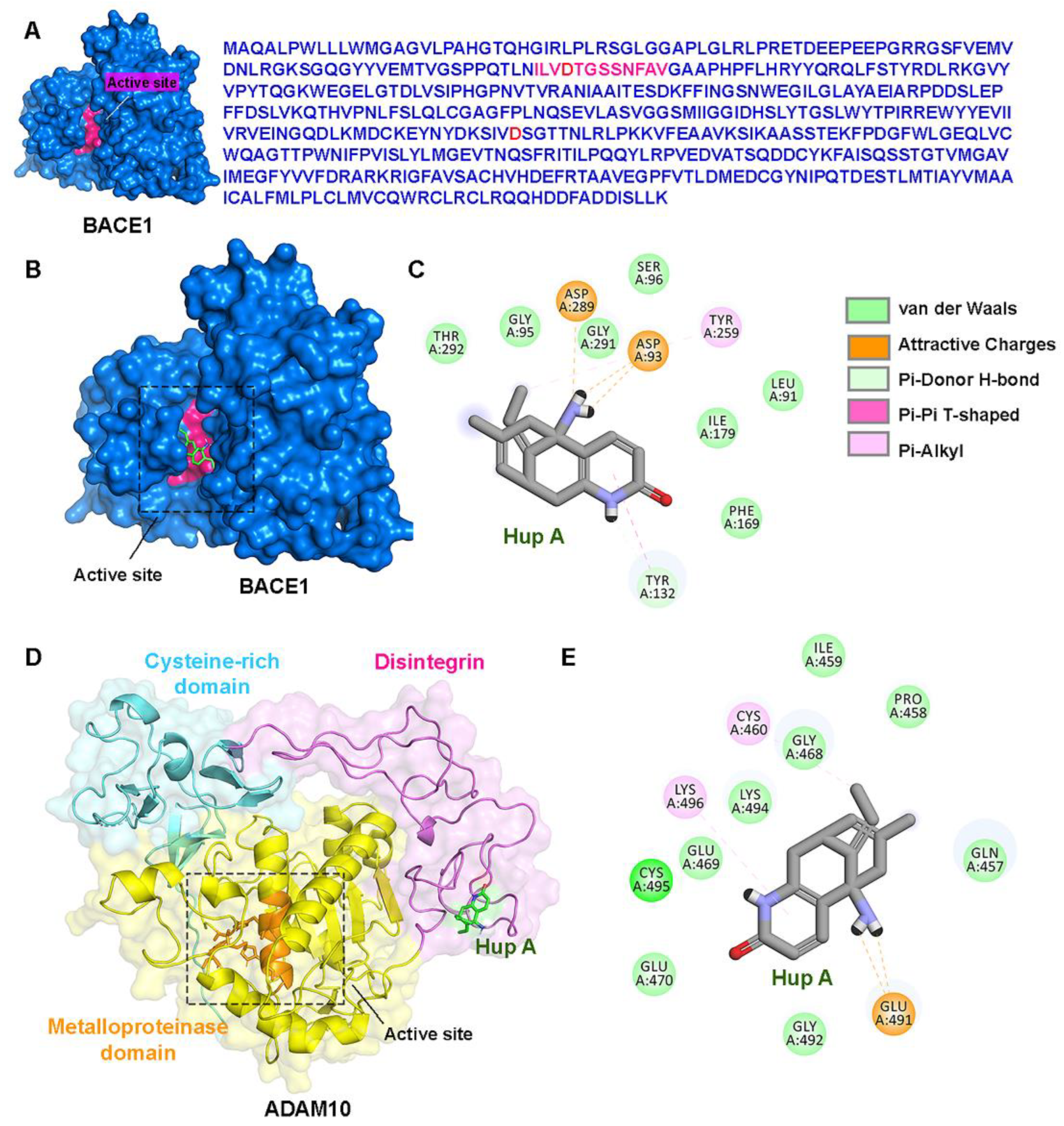
3.4. Direct Interactions of Hup A with the Active Site of BACE1/ADAM10
3.5. Effect of Hup A on the GSK3β and Tau Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Frozza, R.L.; Lourenco, M.V.; De Felice, F.G. Challenges for Alzheimer’s Disease Therapy: Insights from Novel Mechanisms Beyond Memory Defects. Front. Neurosci. 2018, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Wimo, A.; Jonsson, L.; Bond, J.; Prince, M.; Winblad, B.; Alzheimer Disease, I. The worldwide economic impact of dementia 2010. Alzheimers Dement 2013, 9, 1–11.e13. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.Z.; Sun, S.; Tan, C.C.; Yu, J.T.; Tan, L. The Role of ADAM10 in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 303–322. [Google Scholar] [CrossRef]
- Peron, R.; Vatanabe, I.P.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef]
- Dar, N.J.; Glazner, G.W. Deciphering the neuroprotective and neurogenic potential of soluble amyloid precursor protein alpha (sAPPalpha). Cell Mol. Life Sci. 2020, 77, 2315–2330. [Google Scholar] [CrossRef]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef]
- Vassar, R. BACE1: The beta-secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Hernandez-Zimbron, L.F.; Rivas-Arancibia, S. Deciphering an interplay of proteins associated with amyloid beta 1–42 peptide and molecular mechanisms of Alzheimer’s disease. Rev. Neurosci. 2014, 25, 773–783. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-beta Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qian, Y.P.; Jiang, K.D.; Yu, S.Y.; Wang, D.X.; Zhang, M.Y.; Jiang, S.D. [Expression levels of APP and PS1 genes in patients with Alzheimer’s disease]. Yi Chuan 2006, 28, 525–528. [Google Scholar] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A. Impairments in Brain Bioenergetics in Aging and Tau Pathology: A Chicken and Egg Situation? Cells 2021, 10, 2531. [Google Scholar] [CrossRef] [PubMed]
- Young, Z.T.; Mok, S.A.; Gestwicki, J.E. Therapeutic Strategies for Restoring Tau Homeostasis. Cold Spring Harb. Perspect. Med. 2018, 8, a024612. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Noguchi, K.; Michel, G.; Mercken, M.; Hoshi, M.; Ishiguro, K.; Imahori, K. Exposure of rat hippocampal neurons to amyloid beta peptide (25-35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci. Lett. 1996, 203, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Honda, T.; Yasutake, K.; Michel, G.; Murayama, O.; Murayama, M.; Ishiguro, K.; Yamaguchi, H. Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in hippocampal neurons. Neurosci. Res. 1998, 31, 317–323. [Google Scholar] [CrossRef]
- Bhat, R.V.; Budd, S.L. GSK3beta signalling: Casting a wide net in Alzheimer’s disease. Neurosignals 2002, 11, 251–261. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Kroemer, G. Hallmarks of health. Cell 2021, 184, 1929–1939. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Abeta: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Wang, S.; Qin, L. Homeostatic medicine: A strategy for exploring health and disease. Curr. Med. 2022, 1, 16. [Google Scholar] [CrossRef]
- Shukla, M.; Wongchitrat, P.; Govitrapong, P. A Synopsis of Multitarget Potential Therapeutic Effects of Huperzine A in Diverse Pathologies-Emphasis on Alzheimer’s Disease Pathogenesis. Neurochem. Res. 2022, 47, 1166–1182. [Google Scholar] [CrossRef]
- Ruan, Q.; Hu, X.; Ao, H.; Ma, H.; Gao, Z.; Liu, F.; Kong, D.; Bao, Z.; Yu, Z. The neurovascular protective effects of huperzine A on D-galactose-induced inflammatory damage in the rat hippocampus. Gerontology 2014, 60, 424–439. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Stanzione, F.; Giangreco, I.; Cole, J.C. Use of molecular docking computational tools in drug discovery. Prog. Med. Chem. 2021, 60, 273–343. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Sengoku, R. Aging and Alzheimer’s disease pathology. Neuropathology 2020, 40, 22–29. [Google Scholar] [CrossRef]
- Panmanee, J.; Nopparat, C.; Chavanich, N.; Shukla, M.; Mukda, S.; Song, W.; Vincent, B.; Govitrapong, P. Melatonin regulates the transcription of betaAPP-cleaving secretases mediated through melatonin receptors in human neuroblastoma SH-SY5Y cells. J. Pineal Res. 2015, 59, 308–320. [Google Scholar] [CrossRef]
- Bell, M.; Zempel, H. SH-SY5Y-derived neurons: A human neuronal model system for investigating TAU sorting and neuronal subtype-specific TAU vulnerability. Rev. Neurosci. 2022, 33, 1–15. [Google Scholar] [CrossRef]
- Peng, Y.; Jiang, L.; Lee, D.Y.; Schachter, S.C.; Ma, Z.; Lemere, C.A. Effects of huperzine A on amyloid precursor protein processing and beta-amyloid generation in human embryonic kidney 293 APP Swedish mutant cells. J. Neurosci. Res. 2006, 84, 903–911. [Google Scholar] [CrossRef]
- Peng, Y.; Lee, D.Y.; Jiang, L.; Ma, Z.; Schachter, S.C.; Lemere, C.A. Huperzine A regulates amyloid precursor protein processing via protein kinase C and mitogen-activated protein kinase pathways in neuroblastoma SK-N-SH cells over-expressing wild type human amyloid precursor protein 695. Neuroscience 2007, 150, 386–395. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, X.C.; Zhang, H.Y. Huperzine A alleviates synaptic deficits and modulates amyloidogenic and nonamyloidogenic pathways in APPswe/PS1dE9 transgenic mice. J. Neurosci. Res. 2012, 90, 508–517. [Google Scholar] [CrossRef]
- Barao, S.; Zhou, L.; Adamczuk, K.; Vanhoutvin, T.; van Leuven, F.; Demedts, D.; Vijverman, A.C.; Bossuyt, X.; Vandenberghe, R.; De Strooper, B. BACE1 levels correlate with phospho-tau levels in human cerebrospinal fluid. Curr. Alzheimer Res. 2013, 10, 671–678. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The beta-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef]
- Mosser, S.; Gerber, H.; Fraering, P.C. Identification of truncated C-terminal fragments of the Alzheimer’s disease amyloid protein precursor derived from sequential proteolytic pathways. J. Neurochem. 2021, 156, 943–956. [Google Scholar] [CrossRef]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Becot, A.; Checler, F. Does Intraneuronal Accumulation of Carboxyl-terminal Fragments of the Amyloid Precursor Protein Trigger Early Neurotoxicity in Alzheimer’s Disease? Curr. Alzheimer Res. 2019, 16, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, J.; Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Agrawal, R.R.; Velasco, K.R.; Yun, T.D.; Stavrovskaya, I.G.; Xu, Y.; Koo, S.Y.; et al. The Alzheimer’s disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. EMBO J. 2020, 39, e103791. [Google Scholar] [CrossRef]
- Shukla, M.; Htoo, H.H.; Wintachai, P.; Hernandez, J.F.; Dubois, C.; Postina, R.; Xu, H.; Checler, F.; Smith, D.R.; Govitrapong, P.; et al. Melatonin stimulates the nonamyloidogenic processing of betaAPP through the positive transcriptional regulation of ADAM10 and ADAM17. J. Pineal Res. 2015, 58, 151–165. [Google Scholar] [CrossRef]
- Sogorb-Esteve, A.; Garcia-Ayllon, M.S.; Gobom, J.; Alom, J.; Zetterberg, H.; Blennow, K.; Saez-Valero, J. Levels of ADAM10 are reduced in Alzheimer’s disease CSF. J. Neuroinflammation 2018, 15, 213. [Google Scholar] [CrossRef] [PubMed]
- Manzine, P.R.; Barham, E.J.; Vale Fde, A.; Selistre-de-Araujo, H.S.; Iost Pavarini, S.C.; Cominetti, M.R. Correlation between mini-mental state examination and platelet ADAM10 expression in Alzheimer’s disease. J. Alzheimers Dis. 2013, 36, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Manzine, P.R.; Barham, E.J.; Vale, F.A.; Selistre-de-Araujo, H.S.; Pavarini, S.C.; Cominetti, M.R. Platelet a disintegrin and metallopeptidase 10 expression correlates with clock drawing test scores in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2014, 29, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Zheng, W.; Wang, T.; Xie, J.W.; Wang, S.L.; Zhao, B.L.; Teng, W.P.; Wang, Z.Y. Huperzine A activates Wnt/beta-catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology 2011, 36, 1073–1089. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.P.; Kozikowski, A.P. Prediction of the binding sites of huperzine A in acetylcholinesterase by docking studies. J. Comput. Aided Mol. Des. 1994, 8, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Sigalapalli, D.K.; Rangaswamy, R.; Tangellamudi, N.D. Novel huperzine A based NMDA antagonists: Insights from molecular docking, ADME/T and molecular dynamics simulation studies. RSC Adv. 2020, 10, 25446–25455. [Google Scholar] [CrossRef]
- Shamsi, A.; Shahwan, M.; Khan, M.S.; Alhumaydhi, F.A.; Alsagaby, S.A.; Al Abdulmonem, W.; Abdullaev, B.; Yadav, D.K. Mechanistic Insight into Binding of Huperzine A with Human Serum Albumin: Computational and Spectroscopic Approaches. Molecules 2022, 27, 797. [Google Scholar] [CrossRef]
- Atiya, A.; Alhumaydhi, F.A.; Shamsi, A.; Olatunde, A.; Alsagaby, S.A.; Al Abdulmonem, W.; Sharaf, S.E.; Shahwan, M. Mechanistic Insight into the Binding of Huperzine a with Human Transferrin: Computational, Spectroscopic and Calorimetric Approaches. ACS Omega 2022, 7, 38361–38370. [Google Scholar] [CrossRef]
- Stoothoff, W.H.; Johnson, G.V. Tau phosphorylation: Physiological and pathological consequences. Biochim. Biophys. Acta 2005, 1739, 280–297. [Google Scholar] [CrossRef]
- Abner, E.L.; Neltner, J.H.; Jicha, G.A.; Patel, E.; Anderson, S.L.; Wilcock, D.M.; Van Eldik, L.J.; Nelson, P.T. Diffuse Amyloid-beta Plaques, Neurofibrillary Tangles, and the Impact of APOE in Elderly Persons’ Brains Lacking Neuritic Amyloid Plaques. J. Alzheimers Dis. 2018, 64, 1307–1324. [Google Scholar] [CrossRef]
- Guo, Q.; Wu, G.; Huang, F.; Wei, Z.; Wang, J.Z.; Zhang, B.; Lui, R.; Yang, Y.; Wang, X.; Li, H.L. Novel small molecular compound 2JY-OBZ4 alleviates AD pathology in cell models via regulating multiple targets. Aging 2022, 14, 8077–8094. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas Ruiz, S.M.; Eldar-Finkelman, H. Glycogen Synthase Kinase-3 Inhibitors: Preclinical and Clinical Focus on CNS-A Decade Onward. Front. Mol. Neurosci. 2021, 14, 792364. [Google Scholar] [CrossRef] [PubMed]


 = increase,
= increase,  = decrease).
= increase, = decrease).
= decrease).
= increase, = decrease).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wongjaikam, S.; Nopparat, C.; Boontem, P.; Panmanee, J.; Thasana, N.; Shukla, M.; Govitrapong, P. Huperzine A Regulates the Physiological Homeostasis of Amyloid Precursor Protein Proteolysis and Tau Protein Conformation—A Computational and Experimental Investigation. Biology 2024, 13, 518. https://doi.org/10.3390/biology13070518
Wongjaikam S, Nopparat C, Boontem P, Panmanee J, Thasana N, Shukla M, Govitrapong P. Huperzine A Regulates the Physiological Homeostasis of Amyloid Precursor Protein Proteolysis and Tau Protein Conformation—A Computational and Experimental Investigation. Biology. 2024; 13(7):518. https://doi.org/10.3390/biology13070518
Chicago/Turabian StyleWongjaikam, Suwakon, Chutikorn Nopparat, Parichart Boontem, Jiraporn Panmanee, Nopporn Thasana, Mayuri Shukla, and Piyarat Govitrapong. 2024. "Huperzine A Regulates the Physiological Homeostasis of Amyloid Precursor Protein Proteolysis and Tau Protein Conformation—A Computational and Experimental Investigation" Biology 13, no. 7: 518. https://doi.org/10.3390/biology13070518
APA StyleWongjaikam, S., Nopparat, C., Boontem, P., Panmanee, J., Thasana, N., Shukla, M., & Govitrapong, P. (2024). Huperzine A Regulates the Physiological Homeostasis of Amyloid Precursor Protein Proteolysis and Tau Protein Conformation—A Computational and Experimental Investigation. Biology, 13(7), 518. https://doi.org/10.3390/biology13070518