
The Emerging Role of Ferroptosis in EBV-Associated Cancer: Implications for Cancer Therapy

Abstract
:Simple Summary
Abstract
1. Introduction
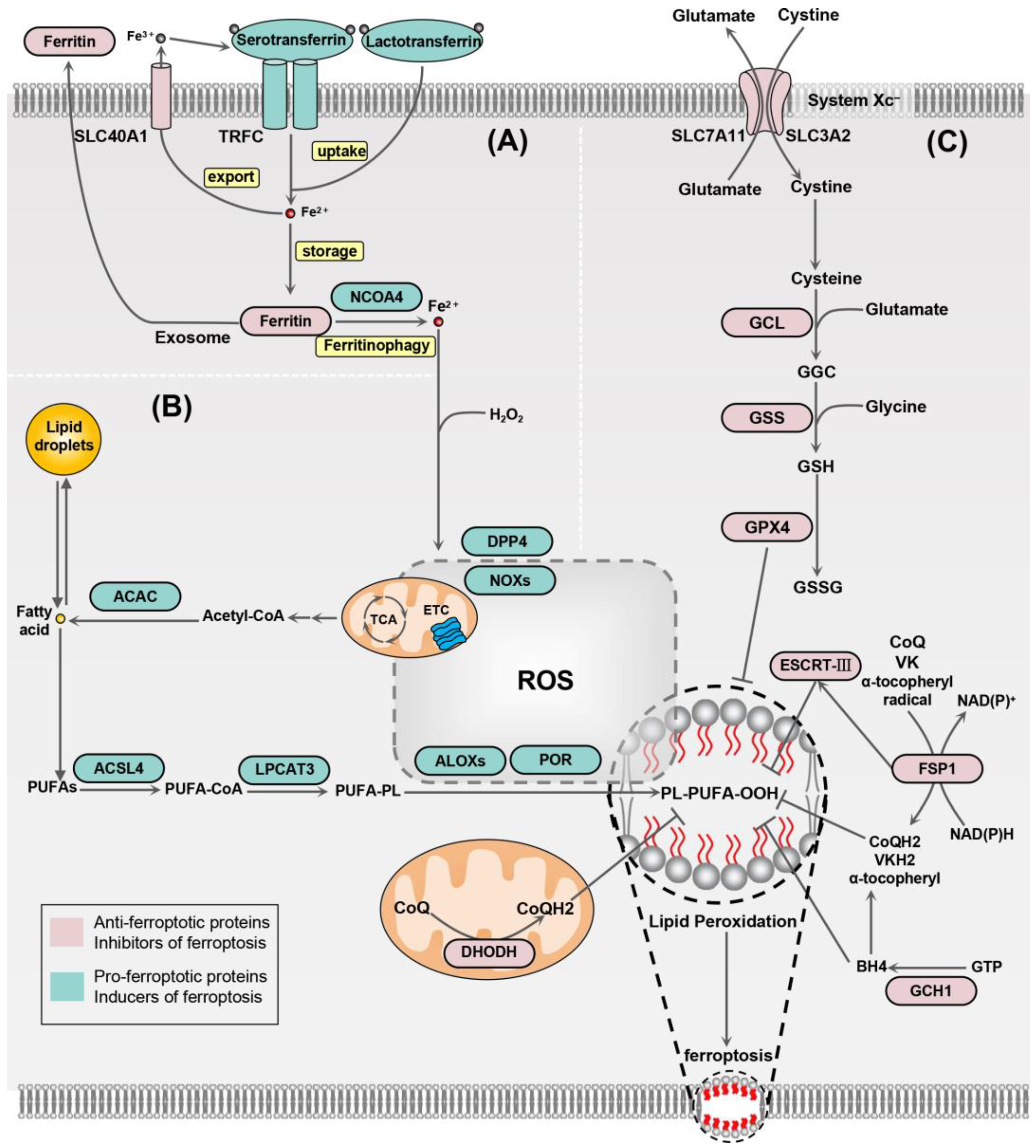
2. Mechanisms of Ferroptosis
2.1. Iron Homeostasis
2.2. Lipid Peroxidation
2.3. Antioxidant System
3. EBV Infection and EBV-Associated Tumors
4. The Crosstalk between EBV Infection and Ferroptosis Signaling Pathway
4.1. Nuclear Factor Erythroid 2-Related Factor-2 (NRF2)
4.2. TP53
4.3. Activation Transcription Factor 3 (ATF3)
4.4. Sterol Regulatory Element-Binding Protein 1 (SREBP1)
4.5. AMP-Activated Protein Kinase (AMPK)
4.6. Hypoxia-Inducible Factors (HIFs)
5. The Role of Ferroptosis in EBV-Associated Tumors
5.1. Nasopharyngeal Carcinoma (NPC)

{kind=link}
{kind=link}
| Tumor Type | Compounds | Effects | Refs. |
|---|---|---|---|
| NPC | Cucurbitacin B (CuB) | CuB induces widespread lipid peroxidation and downregulates the expression of GPX4, ultimately leading to the ferroptosis of NPC cells. | [16] |
| NPC | RSL3 | RSL3 plays a synergistic role with EGFR monoclonal antibody Cetuximab to inhibit the survival of NPC cells. However, the detailed operation of its mechanism is not fully clear. | [109] |
| NPC | Lupeol | Lupeol promotes the release of iron and lipid peroxidation in NPC cells, an effect that can be inhibited by the ferroptosis inhibitor Fer-1; at specific dosages, Lupeol suppresses the levels of GSH and GPX4, demonstrating the potential to induce ferroptosis. | [108] |
| NPC | Itraconazole | Itraconazole triggers ferroptosis and reduces cell viability while partially reversing radioresistance in NPC spherocytes. | [102] |
| NPC | Isoquercitrin | Isoquercitrin inhibits the proliferation of NPC cells and enhances oxidative stress and ferroptosis within these cells by suppressing the AMPK/NF-κB p65 pathway. | [115] |
| EBVaGC | Quercetin | Compared to EBV-negative gastric cancer cells, Quercetin has a greater effect on EBV-positive GC and can effectively induce the expression of anticancer factors, such as TP53, in cells. | [116,117] |
| DLBCL | Dimethyl fumarate (DMF) | DMF exhibits antitumor effects on both subtypes of DLBCL by inducing lipid peroxidation to trigger ferroptosis and is associated with high expression of 5-LOX in the germinal center B-like (GCB) DLBCL subtype. | [118] |
| DLBCL | APR-246 | APR-246 induces p53-dependent ferritinophagy in DLBCL cells and triggers ferroptosis in cells carrying wild-type TP53 or TP53 mutants. | [119] |
| DLBCL | Imidazole ketone erastin (IKE) | IKE induces ferroptosis in DLBCL cells both in vitro and in vivo by inhibiting System Xc−, leading to GSH depletion and lipid peroxidation. | [120] |
| DLBCL | BET inhibitors | BET inhibitors sensitize germinal center B-like (GCB) subtype DLBCL cells to ferroptosis induction, and their combined use with ferroptosis inducers, such as RSL3, enhances their cytotoxic effect on DLBCL cells both in vitro and in vivo. | [121] |
| DLBCL | Artesunate | Artesunate downregulates the levels of GPX4 and FTH1 in DLBCL cells via STAT3, which promotes ROS accumulation and ferroptosis. | [122] |
| DLBCL | Iron oxide nanoparticles (IONs) | IONs induce ferroptosis in DLBCL cells by accumulating intracellular iron ions and the onset of lipid peroxidation while inhibiting GPX4 and SLC40A1 expression. | [123] |
| NKTCL | Kayadiol | Kayadiol decreases GSH in NKTCL cells and induces ferroptosis in NKTCL cells by inhibiting SLC7A11 and GPX4 expression through the upregulation of TP53. | [124] |
| BL | Buthionine sulfoximine (BSO) | BSO significantly increases the level of lipid ROS in EBV-positive BL cells, and Fer-1 and GSH can inhibit BSO-induced BL cell death, indicating that BSO has the potential to induce ferroptosis in BL cells. | [125] |
| BL | Artesunate | Artesunate induces ferroptosis by depleting GSH through the upregulation of ATF4-related pathways. | [15] |
5.2. EBV-Associated Gastric Cancer (EBVaGC)
5.3. Diffuse Large B-Cell Lymphoma (DLBCL)
5.4. Natural Killer Cell and T-Cell Lymphomas (NKTCLs)
5.5. Burkitt’s Lymphoma (BL)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allocati, N.; Masulli, M.; Di Ilio, C.; De Laurenzi, V. Die for the Community: An Overview of Programmed Cell Death in Bacteria. Cell Death Dis. 2015, 6, e1609. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis Turns 10: Emerging Mechanisms, Physiological Functions, and Therapeutic Applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-Ras-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Xu, X.Q.; Xu, T.; Ji, W.; Wang, C.; Ren, Y.; Xiong, X.; Zhou, X.; Lin, S.H.; Xu, Y.; Qiu, Y. Herpes Simplex Virus 1-Induced Ferroptosis Contributes to Viral Encephalitis. mBio 2023, 14, e0237022. [Google Scholar] [CrossRef]
- Dai, L.; Cao, Y.; Chen, Y.; Parsons, C.; Qin, Z. Targeting Xct, a Cystine-Glutamate Transporter Induces Apoptosis and Tumor Regression for Kshv/Hiv-Associated Lymphoma. J. Hematol. Oncol. 2014, 7, 30. [Google Scholar] [CrossRef]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Cao, Y. Ebv Based Cancer Prevention and Therapy in Nasopharyngeal Carcinoma. Npj Precis. Oncol. 2017, 1, 10. [Google Scholar] [CrossRef]
- Yang, T.; You, C.; Meng, S.; Lai, Z.; Ai, W.; Zhang, J. Ebv Infection and Its Regulated Metabolic Reprogramming in Nasopharyngeal Tumorigenesis. Front. Cell Infect. Microbiol. 2022, 12, 935205. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, L.; Tsang, C.M.; Tsao, S.W. Ebv Infection and Glucose Metabolism in Nasopharyngeal Carcinoma. Adv. Exp. Med. Biol. 2017, 1018, 75–90. [Google Scholar]
- Cao, Y.; Xie, L.; Shi, F.; Tang, M.; Li, Y.; Hu, J.; Zhao, L.; Zhao, L.; Yu, X.; Luo, X.; et al. Targeting the Signaling in Epstein-Barr Virus-Associated Diseases: Mechanism, Regulation, and Clinical Study. Signal Transduct. Target. Ther. 2021, 6, 15. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by Gpx4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Wang, N.; Zeng, G.Z.; Yin, J.L.; Bian, Z.X. Artesunate Activates the Atf4-Chop-Chac1 Pathway and Affects Ferroptosis in Burkitt’s Lymphoma. Biochem. Biophys. Res. Commun. 2019, 519, 533–539. [Google Scholar] [CrossRef]
- Huang, S.; Cao, B.; Zhang, J.; Feng, Y.; Wang, L.; Chen, X.; Su, H.; Liao, S.; Liu, J.; Yan, J.; et al. Induction of Ferroptosis in Human Nasopharyngeal Cancer Cells by Cucurbitacin B: Molecular Mechanism and Therapeutic Potential. Cell Death Dis. 2021, 12, 237. [Google Scholar] [CrossRef]
- Yuan, L.; Li, S.; Chen, Q.; Xia, T.; Luo, D.; Li, L.; Liu, S.; Guo, S.; Liu, L.; Du, C.; et al. Ebv Infection-Induced Gpx4 Promotes Chemoresistance and Tumor Progression in Nasopharyngeal Carcinoma. Cell Death Differ. 2022, 29, 1513–1527. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. Bid Links Ferroptosis to Mitochondrial Cell Death Pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Lee, D.H.; Lee, Y.S.; Jo, M.J.; Jeong, Y.A.; Kwon, W.T.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Molecular Crosstalk between Ferroptosis and Apoptosis: Emerging Role of Er Stress-Induced P53-Independent Puma Expression. Oncotarget 2017, 8, 115164–115178. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening Horizons: The Role of Ferroptosis in Cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. Gtp Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. Dhodh-Mediated Ferroptosis Defence Is a Targetable Vulnerability in Cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Lee, N.K.; Cho, S.; Kim, I.S. Ferritin—A Multifaceted Protein Scaffold for Biotherapeutics. Exp. Mol. Med. 2022, 54, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Chung, S.W. Ros-Mediated Autophagy Increases Intracellular Iron Levels and Ferroptosis by Ferritin and Transferrin Receptor Regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. Fsp1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid Peroxidation in Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the Intersection of Lipid Metabolism and Cellular Signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef]
- Shui, S.; Zhao, Z.; Wang, H.; Conrad, M.; Liu, G. Non-Enzymatic Lipid Peroxidation Initiated by Photodynamic Therapy Drives a Distinct Ferroptosis-Like Cell Death Pathway. Redox Biol. 2021, 45, 102056. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor P53 Limits Ferroptosis by Blocking Dpp4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.X.; Li, C.; Yan, X.L.; Qu, Y.; Yang, Y.; Guo, Z.N. Crosstalk between Oxidative Stress and Ferroptosis/Oxytosis in Ischemic Stroke: Possible Targets and Molecular Mechanisms. Oxid. Med. Cell. Longev. 2021, 2021, 6643382. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Conrad, M.; Ursini, F. Gpx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal 2018, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liang, L.; Liu, S.; Yi, H.; Zhou, Y. Fsp1: A Key Regulator of Ferroptosis. Trends Mol. Med. 2023, 29, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Meng, L.; Kang, R.; Wang, X.; Tang, D. Escrt-Iii-Dependent Membrane Repair Blocks Ferroptosis. Biochem. Biophys. Res. Commun. 2020, 522, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Long, X.; Liu, P.S.; Xie, X. The Interplay of Oncogenic Signaling, Oxidative Stress and Ferroptosis in Cancer. Int. J. Cancer 2023, 153, 918–931. [Google Scholar] [CrossRef]
- McKenzie, J.; El-Guindy, A. Epstein-Barr Virus Lytic Cycle Reactivation. Curr. Top. Microbiol. Immunol. 2015, 391, 237–261. [Google Scholar]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr Virus Infection and Nasopharyngeal Carcinoma. Philos. Trans. R. Soc. B-Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Munz, C. Latency and Lytic Replication in Epstein-Barr Virus-Associated Oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D. The Cancer-Virus Cures. Nat. Med. 2014, 20, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr Virus: More Than 50 Years Old and Still Providing Surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Klein, G. Tumor Associations of Ebv–Historical Perspectives. Curr. Top. Microbiol. Immunol. 2015, 390 Pt 1, 17–22. [Google Scholar] [PubMed]
- Gilardini Montani, M.S.; Santarelli, R.; Granato, M.; Gonnella, R.; Torrisi, M.R.; Faggioni, A.; Cirone, M. Ebv Reduces Autophagy, Intracellular Ros and Mitochondria to Impair Monocyte Survival and Differentiation. Autophagy 2019, 15, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating Nrf2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and Proteomic Profiling of Keap1 Disrupted and Sulforaphane-Treated Human Breast Epithelial Cells Reveals Common Expression Profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile Response Element-Mediated Induction of the Cystine/Glutamate Exchange Transporter Gene Expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. Nrf2 Plays a Critical Role in Mitigating Lipid Peroxidation and Ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify Nadph as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef]
- Hu, J.; Li, Y.; Li, H.; Shi, F.; Xie, L.; Zhao, L.; Tang, M.; Luo, X.; Jia, W.; Fan, J.; et al. Targeting Epstein-Barr Virus Oncoprotein Lmp1-Mediated High Oxidative Stress Suppresses Ebv Lytic Reactivation and Sensitizes Tumors to Radiation Therapy. Theranostics 2020, 10, 11921–11937. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; He, Q.; Zeng, M.; Chen, Y.; Liu, Y. Activation of Mek1/2/Nrf-2 Signaling Pathway by Epstein-Barr Virus-Latent Membrane Protein 1 Enhances Autophagy and Cisplatin Resistance in T-Cell Lymphoma. Anal. Cell. Pathol. 2021, 2021, 6668947. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.M.; Kim, Y.S.; Hur, D.Y. Lmp1 and 2a Induce the Expression of Nrf2 through Akt Signaling Pathway in Epstein-Barr Virus-Transformed B Cells. Transl. Oncol. 2019, 12, 775–783. [Google Scholar] [CrossRef]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins Activate Nrf2 by Promoting P62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef]
- Cirone, M. Ebv and Kshv Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis. Viruses 2018, 10, 599. [Google Scholar] [CrossRef]
- Kang, R.; Kroemer, G.; Tang, D. The Tumor Suppressor Protein P53 and the Ferroptosis Network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity During Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Kon, N.; Yi, J.; Zhao, H.; Zhang, W.; Tang, Q.; Li, H.; Kobayashi, H.; Li, Z.; Duan, S.; et al. Specific Regulation of Bach1 by the Hotspot Mutant P53(R175h) Reveals a Distinct Gain-of-Function Mechanism. Nat. Cancer 2023, 4, 564–581. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of Sat1 Engages Polyamine Metabolism with P53-Mediated Ferroptotic Responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. P53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef]
- Rodencal, J.; Kim, N.; He, A.; Li, V.L.; Lange, M.; He, J.; Tarangelo, A.; Schafer, Z.T.; Olzmann, J.A.; Long, J.Z.; et al. Sensitization of Cancer Cells to Ferroptosis Coincident with Cell Cycle Arrest. Cell Chem. Biol. 2024, 31, 234–248.e13. [Google Scholar] [CrossRef]
- Yi, F.; Saha, A.; Murakami, M.; Kumar, P.; Knight, J.S.; Cai, Q.; Choudhuri, T.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3c Targets P53 and Modulates Its Transcriptional and Apoptotic Activities. Virology 2009, 388, 236–247. [Google Scholar] [CrossRef]
- Chevallier-Greco, A.; Manet, E.; Chavrier, P.; Mosnier, C.; Daillie, J.; Sergeant, A. Both Epstein-Barr Virus (Ebv)-Encoded Trans-Acting Factors, Eb1 and Eb2, Are Required to Activate Transcription from an Ebv Early Promoter. EMBO J. 1986, 5, 3243–3249. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, M.L.; Annunziata, C.; Tornesello, A.L.; Buonaguro, L.; Buonaguro, F.M. Human Oncoviruses and P53 Tumor Suppressor Pathway Deregulation at the Origin of Human Cancers. Cancers 2018, 10, 213. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.M.; Kong, Y.L.; Wang, L.; Zhu, H.Y.; Wu, J.Z.; Xia, Y.; Li, Y.; Qin, S.C.; Fan, L.; Li, J.Y.; et al. Ebv-Mir-Bhrf1-1 Targets P53 Gene: Potential Role in Epstein-Barr Virus Associated Chronic Lymphocytic Leukemia. Cancer Res. Treat. 2020, 52, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y.; Sun, L.; Liu, S.; Zhao, M.; Luo, B. Lmp1 Induces P53 Protein Expression Via the H19/Mir-675-5p Axis. Microbiol. Spectr. 2022, 10, e0000622. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, W.; Xiao, L.; Xu, J.; Chen, X.; Tang, M.; Dong, Z.; Tao, Q.; Cao, Y. Viral Oncoprotein Lmp1 Disrupts P53-Induced Cell Cycle Arrest and Apoptosis through Modulating K63-Linked Ubiquitination of P53. Cell Cycle 2012, 11, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Hai, T.; Wolfgang, C.D.; Marsee, D.K.; Allen, A.E.; Sivaprasad, U. Atf3 and Stress Responses. Gene Expr. 1999, 7, 321–335. [Google Scholar]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.F.; Zhang, J.; Wang, H.; Chen, X.; et al. Atf3 Promotes Erastin-Induced Ferroptosis by Suppressing System Xc−. Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef]
- Fu, D.; Wang, C.; Yu, L.; Yu, R. Induction of Ferroptosis by Atf3 Elevation Alleviates Cisplatin Resistance in Gastric Cancer by Restraining Nrf2/Keap1/Xct Signaling. Cell Mol. Biol. Lett. 2021, 26, 26. [Google Scholar] [CrossRef]
- Asakawa, Y.; Okabe, A.; Fukuyo, M.; Li, W.; Ikeda, E.; Mano, Y.; Funata, S.; Namba, H.; Fujii, T.; Kita, K.; et al. Epstein-Barr Virus-Positive Gastric Cancer Involves Enhancer Activation through Activating Transcription Factor 3. Cancer Sci. 2020, 111, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; Dawson, C.W.; Lo, K.W.; Yu, Y.; Young, L.S. Upregulation of Id1 by Epstein-Barr Virus-Encoded Lmp1 Confers Resistance to Tgfbeta-Mediated Growth Inhibition. Mol. Cancer 2010, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; Lung, R.W.; Dawson, C.W.; Young, L.S.; Ko, C.W.; Yeung, W.W.; Kang, W.; To, K.F.; Lo, K.W. Activation of Sterol Regulatory Element-Binding Protein 1 (Srebp1)-Mediated Lipogenesis by the Epstein-Barr Virus-Encoded Latent Membrane Protein 1 (Lmp1) Promotes Cell Proliferation and Progression of Nasopharyngeal Carcinoma. J. Pathol. 2018, 246, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic Activation of Pi3k-Akt-Mtor Signaling Suppresses Ferroptosis Via Srebp-Mediated Lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qi, Q.; Wu, N.; Wang, Y.; Feng, Q.; Jin, R.; Jiang, L. Aspirin Promotes Rsl3-Induced Ferroptosis by Suppressing Mtor/Srebp-1/Scd1-Mediated Lipogenesis in Pik3ca-Mutatnt Colorectal Cancer. Redox Biol. 2022, 55, 102426. [Google Scholar] [CrossRef] [PubMed]
- Parlani, M.; Jorgez, C.; Friedl, P. Plasticity of Cancer Invasion and Energy Metabolism. Trends Cell Biol. 2023, 33, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. Ampk: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. Ampk: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Li, W.; Fang, D.; Xiao, C.; Wu, X.; Li, M.; Luo, Z. Emerging Roles of Energy Metabolism in Ferroptosis Regulation of Tumor Cells. Adv. Sci. 2021, 8, e2100997. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-Stress-Mediated Ampk Activation Inhibits Ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Zhou, W.; Men, H.; Bao, T.; Sun, Y.; Wang, Q.; Tan, Y.; Keller, B.B.; Tong, Q.; et al. Ferroptosis Is Essential for Diabetic Cardiomyopathy and Is Prevented by Sulforaphane Via Ampk/Nrf2 Pathways. Acta Pharm. Sin. B 2022, 12, 708–722. [Google Scholar] [CrossRef]
- Wang, Z.; Yao, M.; Jiang, L.; Wang, L.; Yang, Y.; Wang, Q.; Qian, X.; Zhao, Y.; Qian, J. Dexmedetomidine Attenuates Myocardial Ischemia/Reperfusion-Induced Ferroptosis Via Ampk/Gsk-3β/Nrf2 Axis. Biomed. Pharmacother. 2022, 154, 113572. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. Ampk-Mediated Becn1 Phosphorylation Promotes Ferroptosis by Directly Blocking System XC− Activity. Curr. Biol. 2018, 28, 2388–2399.e5. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tang, M.; Li, H.; Xu, Z.; Weng, X.; Li, J.; Yu, X.; Zhao, L.; Liu, H.; Hu, Y.; et al. Ebv-Lmp1 Suppresses the DNA Damage Response through DNA-Pk/Ampk Signaling to Promote Radioresistance in Nasopharyngeal Carcinoma. Cancer Lett. 2016, 380, 191–200. [Google Scholar] [CrossRef]
- Lo, A.K.; Lo, K.W.; Ko, C.W.; Young, L.S.; Dawson, C.W. Inhibition of the Lkb1-Ampk Pathway by the Epstein-Barr Virus-Encoded Lmp1 Promotes Proliferation and Transformation of Human Nasopharyngeal Epithelial Cells. J. Pathol. 2013, 230, 336–346. [Google Scholar] [CrossRef]
- Lyu, X.; Wang, J.; Guo, X.; Wu, G.; Jiao, Y.; Faleti, O.D.; Liu, P.; Liu, T.; Long, Y.; Chong, T.; et al. Ebv-Mir-Bart1-5p Activates Ampk/Mtor/Hif1 Pathway Via a Pten Independent Manner to Promote Glycolysis and Angiogenesis in Nasopharyngeal Carcinoma. PLoS Pathog. 2018, 14, e1007484. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, L.; Li, X.F. Hypoxia and the Tumor Microenvironment. Technol. Cancer Res. Treat. 2021, 20, 15330338211036304. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. Hif1α and Hif2α: Sibling Rivalry in Hypoxic Tumour Growth and Progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Lin, Z.; Song, J.; Gao, Y.; Huang, S.; Dou, R.; Zhong, P.; Huang, G.; Han, L.; Zheng, J.; Zhang, X.; et al. Hypoxia-Induced Hif-1α/Lncrna-Pman Inhibits Ferroptosis by Promoting the Cytoplasmic Translocation of Elavl1 in Peritoneal Dissemination from Gastric Cancer. Redox Biol. 2022, 52, 102312. [Google Scholar] [CrossRef]
- Yuan, S.; Wei, C.; Liu, G.; Zhang, L.; Li, J.; Li, L.; Cai, S.; Fang, L. Sorafenib Attenuates Liver Fibrosis by Triggering Hepatic Stellate Cell Ferroptosis Via Hif-1α/Slc7a11 Pathway. Cell Prolif. 2022, 55, e13158. [Google Scholar] [CrossRef]
- Singhal, R.; Mitta, S.R.; Das, N.K.; Kerk, S.A.; Sajjakulnukit, P.; Solanki, S.; Andren, A.; Kumar, R.; Olive, K.P.; Banerjee, R.; et al. Hif-2α Activation Potentiates Oxidative Cell Death in Colorectal Cancers by Increasing Cellular Iron. J. Clin. Investig. 2021, 131, e143691. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A Gpx4-Dependent Cancer Cell State Underlies the Clear-Cell Morphology and Confers Sensitivity to Ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Lin, Y.X.; Ma, W.; Zhang, H.J.; Chen, K.M.; He, G.P.; Zhang, X.; Xu, M.; Feng, Q.S.; Chen, M.Y.; et al. Vasculogenic Mimicry Formation in Ebv-Associated Epithelial Malignancies. Nat. Commun. 2018, 9, 5009. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.W.; Chu, Y.C.; Chen, P.R.; Liao, M.H.; Lee, J.W. Positive Regulation of Hif-1a Expression by Ebv Oncoprotein Lmp1 in Nasopharyngeal Carcinoma Cells. Cancer Lett. 2016, 382, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Yu, X.; Cordes, B.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-Inducible Factor-1α Plays Roles in Epstein-Barr Virus’s Natural Life Cycle and Tumorigenesis by Inducing Lytic Infection through Direct Binding to the Immediate-Early Bzlf1 Gene Promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Chan, A.T.C.; Le, Q.T.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal Carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef]
- Tsao, S.W.; Yip, Y.L.; Tsang, C.M.; Pang, P.S.; Lau, V.M.; Zhang, G.; Lo, K.W. Etiological Factors of Nasopharyngeal Carcinoma. Oral. Oncol. 2014, 50, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Du, J.; Shi, J.; Huang, Y.; Zhao, Y.; Ma, L. Ferroptosis-Related Gene Atg5 Is a Novel Prognostic Biomarker in Nasopharyngeal Carcinoma and Head and Neck Squamous Cell Carcinoma. Front. Bioeng. Biotechnol. 2022, 10, 1006535. [Google Scholar] [CrossRef]
- Zhou, J.; Guo, T.; Zhou, L.; Bao, M.; Wang, L.; Zhou, W.; Tan, S.; Li, G.; He, B.; Guo, Z. The Ferroptosis Signature Predicts the Prognosis and Immune Microenvironment of Nasopharyngeal Carcinoma. Sci. Rep. 2023, 13, 1861. [Google Scholar] [CrossRef]
- Li, B.; Liao, Z.; Mo, Y.; Zhao, W.; Zhou, X.; Xiao, X.; Cui, W.; Feng, G.; Zhong, S.; Liang, Y.; et al. Inactivation of 3-Hydroxybutyrate Dehydrogenase Type 2 Promotes Proliferation and Metastasis of Nasopharyngeal Carcinoma by Iron Retention. Br. J. Cancer 2020, 122, 102–110. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Q.; Li, X.; Chen, Y.; Xu, G. Itraconazole Attenuates the Stemness of Nasopharyngeal Carcinoma Cells Via Triggering Ferroptosis. Environ. Toxicol. 2021, 36, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Wu, X.R.; Ren, Z.; Li, Y.L.; Zou, W.L.; Chen, J.C.; Wang, H.Q. Overcoming Cancer Chemotherapy Resistance by the Induction of Ferroptosis. Drug Resist. Updates 2023, 66, 100916. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Metabolic Reprogramming: The Emerging Concept and Associated Therapeutic Strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the Crossroads of Cancer-Acquired Drug Resistance and Immune Evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yao, Q.; Fan, D.; Duan, L.; You, Y.; Liang, W.; Zhou, Z.; Teng, S.; Liang, Z.; Hall, D.D.; et al. Cephalosporin Antibiotics Specifically and Selectively Target Nasopharyngeal Carcinoma through Hmox1-Induced Ferroptosis. Life Sci. 2021, 277, 119457. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Berghe, T.V. Targeting Ferroptosis to Iron out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.C.; Wu, B.; Zhang, J.J.; Zhang, W. Lupeol Triggers Oxidative Stress, Ferroptosis, Apoptosis and Restrains Inflammation in Nasopharyngeal Carcinoma Via Ampk/Nf-Κb Pathway. Immunopharmacol. Immunotoxicol. 2022, 44, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yan, S.; Zhu, J.; Lu, R.; Kang, C.; Tang, K.; Zeng, J.; Ding, M.; Guo, Z.; Lai, X.; et al. Combination Rsl3 Treatment Sensitizes Ferroptosis- and Egfr-Inhibition-Resistant Hnsccs to Cetuximab. Int. J. Mol. Sci. 2022, 23, 9014. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Deng, N.H.; Xiao, J.X.; He, X.S. Cross-Link between Ferroptosis and Nasopharyngeal Carcinoma: New Approach to Radiotherapy Sensitization. Oncol. Lett. 2021, 22, 770. [Google Scholar] [CrossRef]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis Via Synergistic Repression of Slc7a11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef]
- Zhou, J.; Ding, J.; Ma, X.; Zhang, M.; Huo, Z.; Yao, Y.; Li, D.; Wang, Z. The Nrf2/Keap1 Pathway Modulates Nasopharyngeal Carcinoma Cell Radiosensitivity Via Ros Elimination. Onco Targets Ther. 2020, 13, 9113–9122. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xie, Y.; Kang, R.; Hou, W.; Sun, X.; Epperly, M.W.; Greenberger, J.S.; Tang, D. Fancd2 Protects against Bone Marrow Injury from Ferroptosis. Biochem. Biophys. Res. Commun. 2016, 480, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Feng, H.; Zhao, F.; Zhang, L.; Xu, S.; Zhang, C.; Zhao, C.; Qin, G. Fancd2 Knockdown with Shrna Interference Enhances the Ionizing Radiation Sensitivity of Nasopharyngeal Carcinoma Cne-2 Cells. Neoplasma 2021, 68, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Gong, Y.; Jiang, Q.; Wang, Q.; Li, S.; Liu, L. Isoquercitrin Promotes Ferroptosis and Oxidative Stress in Nasopharyngeal Carcinoma Via the Ampk/Nf-Κb Pathway. J. Biochem. Mol. Toxicol. 2024, 38, e23542. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K. Quercetin and Ferroptosis. Life 2023, 13, 1730. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Lee, S.; Shin, Y.S.; Cho, M.; Kang, H.; Cho, H. Anti-Cancer Effect of Quercetin in Xenograft Models with Ebv-Associated Human Gastric Carcinoma. Molecules 2016, 21, 1286. [Google Scholar] [CrossRef]
- Schmitt, A.; Xu, W.; Bucher, P.; Grimm, M.; Konantz, M.; Horn, H.; Zapukhlyak, M.; Berning, P.; Brändle, M.; Jarboui, M.A.; et al. Dimethyl Fumarate Induces Ferroptosis and Impairs Nf-Κb/Stat3 Signaling in Dlbcl. Blood 2021, 138, 871–884. [Google Scholar] [CrossRef]
- Hong, Y.; Ren, T.; Wang, X.; Liu, X.; Fei, Y.; Meng, S.; Han, X.; Sun, C.; Shen, H.; Li, L.; et al. Apr-246 Triggers Ferritinophagy and Ferroptosis of Diffuse Large B-Cell Lymphoma Cells with Distinct Tp53 Mutations. Leukemia 2022, 36, 2269–2280. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e9. [Google Scholar] [CrossRef]
- Schmitt, A.; Grimm, M.; Kreienkamp, N.; Junge, H.; Labisch, J.; Schuhknecht, L.; Schönfeld, C.; Görsch, E.S.; Tibello, A.; Menck, K.; et al. Brd4 Inhibition Sensitizes Diffuse Large B-Cell Lymphoma Cells to Ferroptosis. Blood 2023, 142, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, F.; Wu, P.; Gong, S.; Gao, J.; Tao, H.; Shen, Q.; Wang, S.; Zhou, Z.; Jia, Y. Artesunate Induces Apoptosis, Autophagy and Ferroptosis in Diffuse Large B Cell Lymphoma Cells by Impairing Stat3 Signaling. Cell. Signal. 2021, 88, 110167. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.T.; Hu, Q.Q.; Wen, Z.F.; Li, Y.L. Iron Oxide Nanoparticles Inhibit Tumor Growth by Ferroptosis in Diffuse Large B-Cell Lymphoma. Am. J. Cancer Res. 2023, 13, 498–508. [Google Scholar]
- He, C.; Wang, C.; Liu, H.; Shan, B. Kayadiol Exerted Anticancer Effects through P53-Mediated Ferroptosis in Nktcl Cells. BMC Cancer 2022, 22, 724. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.M.; Voyer, J.; Gewurz, B.E. Epstein-Barr Virus Latency Programs Dynamically Sensitize B Cells to Ferroptosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2118300119. [Google Scholar] [CrossRef] [PubMed]
- Matsusaka, K.; Kaneda, A.; Nagae, G.; Ushiku, T.; Kikuchi, Y.; Hino, R.; Uozaki, H.; Seto, Y.; Takada, K.; Aburatani, H.; et al. Classification of Epstein-Barr Virus-Positive Gastric Cancers by Definition of DNA Methylation Epigenotypes. Cancer Res. 2011, 71, 7187–7197. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Z.; Zeng, B.; Hu, G.; Gan, R. Epstein-Barr Virus-Associated Gastric Cancer: A Distinct Subtype. Cancer Lett. 2020, 495, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.S.; Pal, A.D.; Banerjee, S. Epstein-Barr Virus-Encoded Small Non-Coding Rnas Induce Cancer Cell Chemoresistance and Migration. Virology 2013, 443, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Li, H.; Lou, L.; Huang, Q.; Zhang, Z.; Mo, J.; Li, M.; Lu, J.; Zhu, K.; Chu, Y.; et al. Inhibition of Stat3-Ferroptosis Negative Regulatory Axis Suppresses Tumor Growth and Alleviates Chemoresistance in Gastric Cancer. Redox Biol. 2022, 52, 102317. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, J.; Wei, L.; Peng, Q.; Gao, Y.; Fu, Y.; Lu, Y.; Qin, Z.; Zhang, X.; Lu, J.; et al. Epstein-Barr Virus Microrna Mir-Bart5-3p Inhibits P53 Expression. J. Virol. 2018, 92, e01022-18. [Google Scholar] [CrossRef]
- Kraus, R.J.; Cordes, B.A.; Sathiamoorthi, S.; Patel, P.; Yuan, X.; Iempridee, T.; Yu, X.; Lee, D.L.; Lambert, P.F.; Mertz, J.E. Reactivation of Epstein-Barr Virus by Hif-1α Requires P53. J. Virol. 2020, 94, e00722-20. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Burkitt’s Lymphoma. N. Engl. J. Med. 2022, 387, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Banjac, A.; Perisic, T.; Sato, H.; Seiler, A.; Bannai, S.; Weiss, N.; Kölle, P.; Tschoep, K.; Issels, R.D.; Daniel, P.T.; et al. The Cystine/Cysteine Cycle: A Redox Cycle Regulating Susceptibility Versus Resistance to Cell Death. Oncogene 2008, 27, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Shibata, D.; Weiss, L.M. Epstein-Barr Virus-Associated Gastric Adenocarcinoma. Am. J. Pathol. 1992, 140, 769–774. [Google Scholar]
- Mungrue, I.N.; Pagnon, J.; Kohannim, O.; Gargalovic, P.S.; Lusis, A.J. Chac1/Mgc4504 Is a Novel Proapoptotic Component of the Unfolded Protein Response, Downstream of the Atf4-Atf3-Chop Cascade. J. Immunol. 2009, 182, 466–476. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, S.; Luo, C.; Shi, F.; Zhou, J.; Shang, L. The Emerging Role of Ferroptosis in EBV-Associated Cancer: Implications for Cancer Therapy. Biology 2024, 13, 543. https://doi.org/10.3390/biology13070543
He S, Luo C, Shi F, Zhou J, Shang L. The Emerging Role of Ferroptosis in EBV-Associated Cancer: Implications for Cancer Therapy. Biology. 2024; 13(7):543. https://doi.org/10.3390/biology13070543
Chicago/Turabian StyleHe, Shan, Cheng Luo, Feng Shi, Jianhua Zhou, and Li Shang. 2024. "The Emerging Role of Ferroptosis in EBV-Associated Cancer: Implications for Cancer Therapy" Biology 13, no. 7: 543. https://doi.org/10.3390/biology13070543
APA StyleHe, S., Luo, C., Shi, F., Zhou, J., & Shang, L. (2024). The Emerging Role of Ferroptosis in EBV-Associated Cancer: Implications for Cancer Therapy. Biology, 13(7), 543. https://doi.org/10.3390/biology13070543