
Therapeutic Target Identification and Drug Discovery Driven by Chemical Proteomics

Abstract
:Simple Summary
Abstract
1. Introduction
2. The Methods in Chemical Proteomics
2.1. Probe-Based Chemical Proteomics
2.1.1. Classification of Chemical Probes
Immobilized Probes
Activity-Based Probes
Photoaffinity Probes
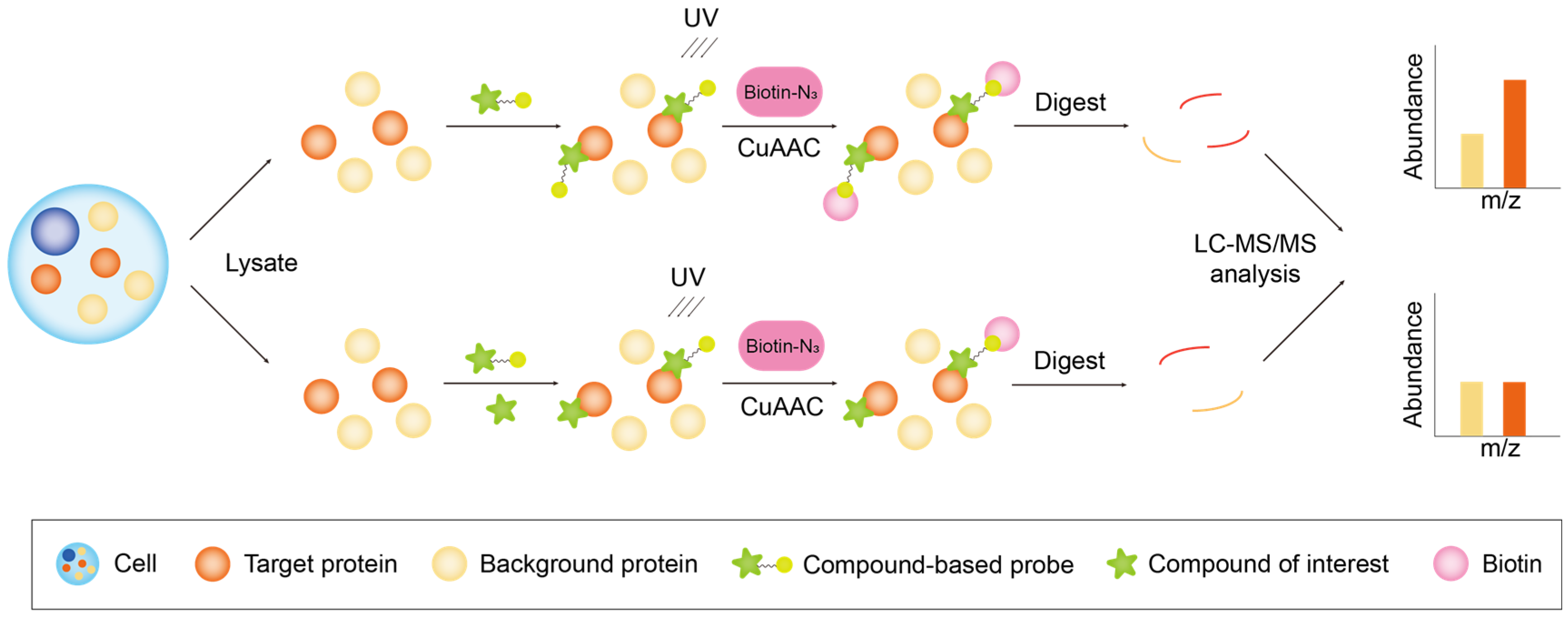
2.1.2. Workflow of Probe-Based Chemical Proteomics
Activity-Based Protein Profiling
Compound-Centric Chemical Proteomics
2.2. Probe-Free Chemical Proteomics
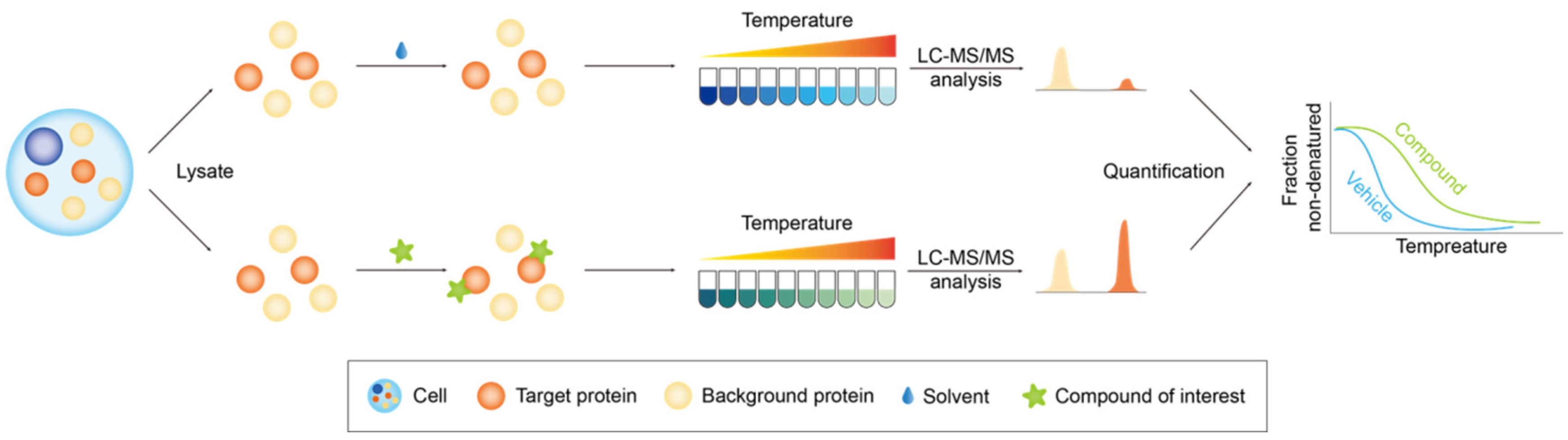
2.2.1. Thermal Proteome Profiling
2.2.2. Drug Affinity Responsive Target Stability
2.2.3. Stability of Proteins from Rates of Oxidation
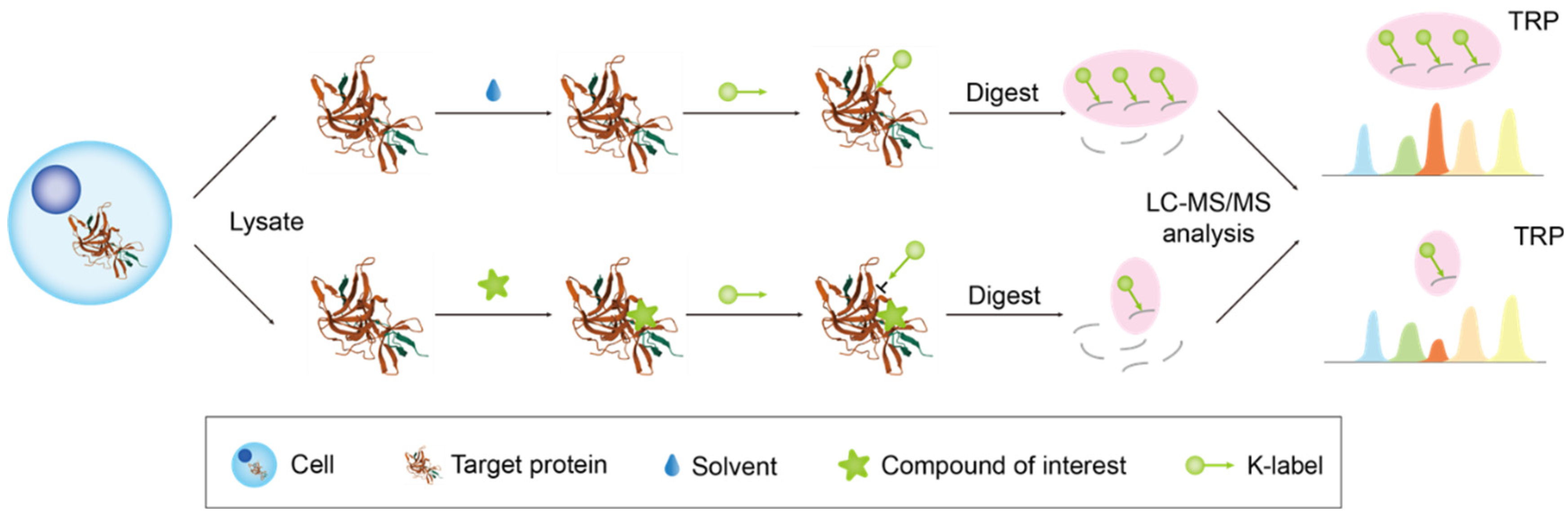
2.2.4. Target-Responsive Accessibility Profiling
2.3. Quantitative Proteomics
2.4. MS Acquisition Schemes
3. Target Identification of Small Molecules
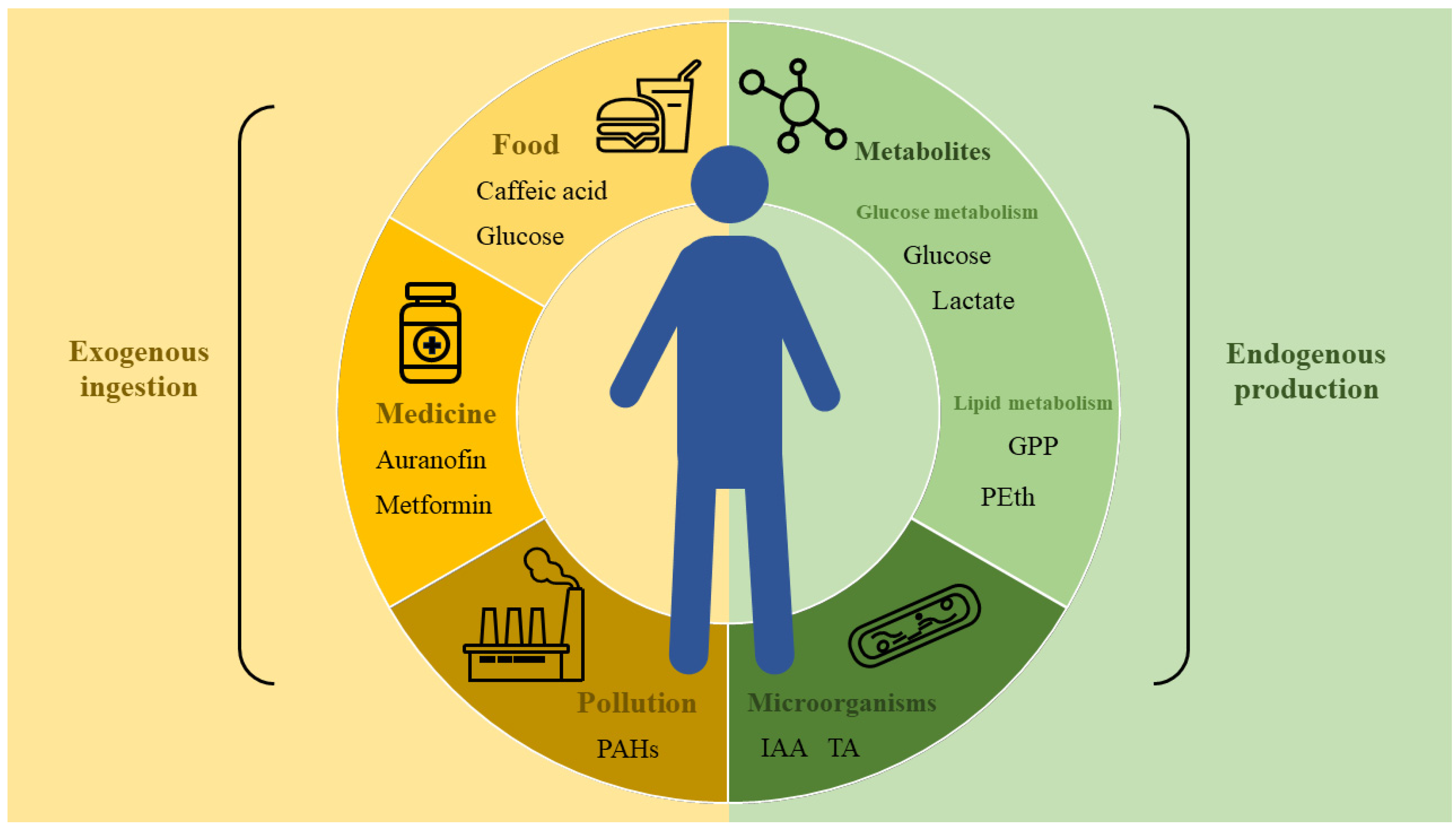
3.1. Target Proteins of Small Metabolite Molecules and Their Analogs
3.1.1. Small Molecules of Lipid Metabolism
3.1.2. Small Molecules of Carbohydrate Metabolism
3.1.3. Other Small Metabolite Molecules
3.1.4. Interactions between Microbe and Host
3.2. Annotation on the Target of Small-Molecule Drugs
3.2.1. Synthetic Small-Molecule Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small Molecule | Target Method | Target Protein | Probe Structure |
|---|---|---|---|
| Metformin | Photoactive metformin probe [133] | Presenilin enhancer 2 (PEN2) | 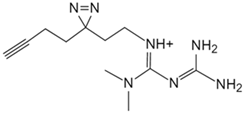 |
| Baicalin | Photoaffinity baicalin probe [134] | Carnitine palmitoyltransferase 1 (CPT1) | 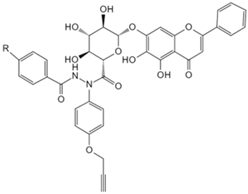 |
| Berberine | Biotinylated berberine probe [135] | Pyruvate kinase isozyme type M2 (PKM2) |  |
| Small Molecule | Target Method | Target Protein |
|---|---|---|
| Auranofin | Thermal proteomic profiling (TPP) [136] | Thioredoxin reductase 1 (TXNRD1) |
| Methotrexate | Cellular thermal shift assay (CETSA) [137] | Phosphoglycerate kinase 1 (PGK1) |
| Triptolide | TPP based on XGBoost (X-TPP) [138] | Heterogeneous nuclear ribonucleoprotein A2/B1 (HnRNP A2/B1) |
3.2.2. Natural Small-Molecule Drugs
4. Evaluation of the Off-Target Effects of Drugs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wan, F.; Zhong, R.; Wang, M.; Zhou, Y.; Chen, Y.; Yi, B.; Hou, F.; Liu, L.; Zhao, Y.; Chen, L.; et al. Caffeic Acid Supplement Alleviates Colonic Inflammation and Oxidative Stress Potentially through Improved Gut Microbiota Community in Mice. Front. Microbiol. 2021, 12, 784211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, J.; Xu, Y.; Tao, C.; Tong, J.; Luo, Y.; Chen, Y.; Liu, X.; Xu, T. Uncovering SOD3 and GPX4 as New Targets of Benzo[α]Pyrene-Induced Hepatotoxicity through Metabolomics and Chemical Proteomics. Redox Biol. 2023, 67, 102930. [Google Scholar] [CrossRef]
- Qiu, S.; Cai, Y.; Yao, H.; Lin, C.; Xie, Y.; Tang, S.; Zhang, A. Small Molecule Metabolites: Discovery of Biomarkers and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; Enerbäck, S. Lactate: The Ugly Duckling of Energy Metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef]
- Chen, L.; Chen, X.-W.; Huang, X.; Song, B.-L.; Wang, Y.; Wang, Y. Regulation of Glucose and Lipid Metabolism in Health and Disease. Sci. China Life Sci. 2019, 62, 1420–1458. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, E.; Mazzeo, A.; Lopatina, T.; Trento, M.; Porta, M. Thiamine Transporter 2 Is Involved in High Glucose-Induced Damage and Altered Thiamine Availability in Cell Models of Diabetic Retinopathy. Diabetes Vasc. Dis. Res. 2020, 17, 147916411987842. [Google Scholar] [CrossRef]
- Rafacho, A.; Gonçalves-Neto, L.M.; Santos-Silva, J.C.; Alonso-Magdalena, P.; Merino, B.; Taboga, S.R.; Carneiro, E.M.; Boschero, A.C.; Nadal, A.; Quesada, I. Pancreatic Alpha-Cell Dysfunction Contributes to the Disruption of Glucose Homeostasis and Compensatory Insulin Hypersecretion in Glucocorticoid-Treated Rats. PLoS ONE 2014, 9, e93531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, W.; Li, Y.; Wang, A.; Cao, H.; Fu, Y. Drug Development Advances in Human Genetics-based Targets. MedComm 2024, 5, e481. [Google Scholar] [CrossRef]
- Makley, L.N.; Gestwicki, J.E. Expanding the Number of ‘Druggable’ Targets: Non-Enzymes and Protein–Protein Interactions. Chem. Biol. Drug Des. 2013, 81, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-Positive Breast Cancer: Advances and Future Directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Yu, T.; Li, X.; Zhang, N.; Foster, L.J.; Peng, C.; Huang, W.; He, G. Recent Advances in Targeting the “Undruggable” Proteins: From Drug Discovery to Clinical Trials. Signal Transduct. Target. Ther. 2023, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Rodon, J. Taking Aim at the Undruggable. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e145–e152. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Geddes-McAlister, J.; Mann, M.; Bantscheff, M. The Emerging Role of Mass Spectrometry-Based Proteomics in Drug Discovery. Nat. Rev. Drug Discov. 2022, 21, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Drewes, G.; Knapp, S. Chemoproteomics and Chemical Probes for Target Discovery. Trends Biotechnol. 2018, 36, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Cuatrecasas, P.; Wilchek, M.; Anfinsen, C.B. Selective Enzyme Purification by Affinity Chromatography. Proc. Natl. Acad. Sci. USA 1968, 61, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Knockaert, M.; Lenormand, P.; Gray, N.; Schultz, P.; Pouysségur, J.; Meijer, L. P42/P44 MAPKs Are Intracellular Targets of the CDK Inhibitor Purvalanol. Oncogene 2002, 21, 6413–6424. [Google Scholar] [CrossRef] [PubMed]
- Siekierka, J.J.; Hung, S.H.Y.; Poe, M.; Lin, C.S.; Sigal, N.H. A Cytosolic Binding Protein for the Immunosuppressant FKS06 Has Peptidyl-Prolyl Isomerase Activity but Is Distinct from Cyclophilin. Nature 1989, 341, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Mao, F.; Liang, J.; Xue, M.; Wei, W.; Li, X.; Zhang, K.; Feng, D.; Liu, B.; Sun, Z. Transcriptomic Signature Associated with Carcinogenesis and Aggressiveness of Papillary Thyroid Carcinoma. Theranostics 2018, 8, 4345–4358. [Google Scholar] [CrossRef] [PubMed]
- Zon, L.I.; Peterson, R.T. In Vivo Drug Discovery in the Zebrafish. Nat. Rev. Drug Discov. 2005, 4, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Braun, P.; Yıldırım, M.A.; Lemmens, I.; Venkatesan, K.; Sahalie, J.; Hirozane-Kishikawa, T.; Gebreab, F.; Li, N.; Simonis, N.; et al. High-Quality Binary Protein Interaction Map of the Yeast Interactome Network. Science 2008, 322, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gao, L.; Lee, Y.M.; Kalesh, K.A.; Ong, Y.S.; Lim, J.; Jee, J.-E.; Sun, H.; Lee, S.S.; Hua, Z.-C.; et al. Target Identification of Natural and Traditional Medicines with Quantitative Chemical Proteomics Approaches. Pharmacol. Ther. 2016, 162, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Rix, U.; Superti-Furga, G. Target Profiling of Small Molecules by Chemical Proteomics. Nat. Chem. Biol. 2009, 5, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Drewes, G. Chemoproteomic Approaches to Drug Target Identification and Drug Profiling. Bioorg. Med. Chem. 2012, 20, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, B.; Zhou, S.; Zhao, X.; Bian, C.; Wei, Y. Chemical Proteomics: Terra Incognita for Novel Drug Target Profiling. Chin. J. Cancer 2012, 31, 507–518. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Ma, N.; Tian, J.; Shao, Y.; Zhu, B.; Wong, Y.K.; Liang, Z.; Zou, C.; Wang, J. Target Identification of Natural Medicine with Chemical Proteomics Approach: Probe Synthesis, Target Fishing and Protein Identification. Signal Transduct. Target. Ther. 2020, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ma, M.; Li, W.; Lei, X. Chemoproteomics, A Broad Avenue to Target Deconvolution. Adv. Sci. 2024, 11, e2305608. [Google Scholar] [CrossRef]
- Friese, A.; Kapoor, S.; Schneidewind, T.; Vidadala, S.R.; Sardana, J.; Brause, A.; Förster, T.; Bischoff, M.; Wagner, J.; Janning, P.; et al. Chemical Genetics Reveals a Role of dCTP Pyrophosphatase 1 in Wnt Signaling. Angew. Chem. Int. Ed. Engl. 2019, 58, 13009–13013. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Itoh, H.; Sakurai, K.; Dan, S.; Inoue, M. Target Identification of Yaku’amide B and Its Two Distinct Activities against Mitochondrial FoF1-ATP Synthase. J. Am. Chem. Soc. 2018, 140, 12189–12199. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Armstrong, Z.; Schröder, S.P.; de Boer, C.; Artola, M.; Aerts, J.M.; Overkleeft, H.S.; Davies, G.J. An Overview of Activity-Based Probes for Glycosidases. Curr. Opin. Chem. Biol. 2019, 53, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Cao, Z.; Sun, P.; Zhang, H.; Sun, Y.; Zhong, J.; Yin, W.; Fan, K.; Zheng, X.; Li, H.; et al. Target Discovery of Matrine against PRRSV in Marc-145 Cells via Activity-Based Protein Profiling. Int. J. Mol. Sci. 2023, 24, 11526. [Google Scholar] [CrossRef] [PubMed]
- Modrzycka, S.; Kołt, S.; Adams, T.E.; Potoczek, S.; Huntington, J.A.; Kasperkiewicz, P.; Drąg, M. Fluorescent Activity-Based Probe To Image and Inhibit Factor XIa Activity in Human Plasma. J. Med. Chem. 2023, 66, 3785–3797. [Google Scholar] [CrossRef] [PubMed]
- Dundas, C.M.; Demonte, D.; Park, S. Streptavidin-Biotin Technology: Improvements and Innovations in Chemical and Biological Applications. Appl. Microbiol. Biotechnol. 2013, 97, 9343–9353. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.G.; Pratt, M.R. Click Chemistry in Proteomic Investigations. Cell 2020, 180, 605–632. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejek, I.; Misas-Villamil, J.C.; Kaschani, F.; Clerc, J.; Gu, C.; Krahn, D.; Niessen, S.; Verdoes, M.; Willems, L.I.; Overkleeft, H.S.; et al. Proteasome Activity Imaging and Profiling Characterizes Bacterial Effector Syringolin A. Plant Physiol. 2011, 155, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Ciepla, P.; Konitsiotis, A.D.; Serwa, R.A.; Masumoto, N.; Leong, W.P.; Dallman, M.J.; Magee, A.I.; Tate, E.W. New Chemical Probes Targeting Cholesterylation of Sonic Hedgehog in Human Cells and Zebrafish. Chem. Sci. 2014, 5, 4249–4259. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Zhang, C.-J.; Wong, Y.K.; Lim, T.K.; Hua, Z.-C.; Liu, B.; Tannenbaum, S.R.; Shen, H.-M.; Lin, Q. In Situ Proteomic Profiling of Curcumin Targets in HCT116 Colon Cancer Cell Line. Sci. Rep. 2016, 6, 22146. [Google Scholar] [CrossRef] [PubMed]
- Husaini, A.M.; Morimoto, K.; Chandrasekar, B.; Kelly, S.; Kaschani, F.; Palmero, D.; Jiang, J.; Kaiser, M.; Ahrazem, O.; Overkleeft, H.S.; et al. Multiplex Fluorescent, Activity-Based Protein Profiling Identifies Active α-Glycosidases and Other Hydrolases in Plants. Plant Physiol. 2018, 177, 24–37. [Google Scholar] [CrossRef]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems [J. Am. Chem. Soc. 2004, 126, 15046−15047]. J. Am. Chem. Soc. 2005, 127, 11196. [Google Scholar] [CrossRef]
- Baskin, J.M.; Prescher, J.A.; Laughlin, S.T.; Agard, N.J.; Chang, P.V.; Miller, I.A.; Lo, A.; Codelli, J.A.; Bertozzi, C.R. Copper-Free Click Chemistry for Dynamic in Vivo Imaging. Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Donu, D.; Curry, A.M.; Barton, E.; Cen, Y. Multifunctional Activity-Based Chemical Probes for Sirtuins. RSC Adv. 2023, 13, 11771–11781. [Google Scholar] [CrossRef] [PubMed]
- Sumranjit, J.; Chung, S.J. Recent Advances in Target Characterization and Identification by Photoaffinity Probes. Molecules 2013, 18, 10425–10451. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Koh, M.; Koo, J.Y.; Lee, S.; Park, S.B. Investigation of Specific Binding Proteins to Photoaffinity Linkers for Efficient Deconvolution of Target Protein. ACS Chem. Biol. 2016, 11, 44–52. [Google Scholar] [CrossRef]
- Liu, Y.; Patricelli, M.P.; Cravatt, B.F. Activity-Based Protein Profiling: The Serine Hydrolases. Proc. Natl. Acad. Sci. USA 1999, 96, 14694–14699. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, C.-J.; Chia, W.N.; Loh, C.C.Y.; Li, Z.; Lee, Y.M.; He, Y.; Yuan, L.-X.; Lim, T.K.; Liu, M.; et al. Haem-Activated Promiscuous Targeting of Artemisinin in Plasmodium Falciparum. Nat. Commun. 2015, 6, 10111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Q.; Li, Y.; Ruan, C.; Wang, S.; Hu, L.; Ye, M. Solvent-Induced Protein Precipitation for Drug Target Discovery on the Proteomic Scale. Anal. Chem. 2020, 92, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; Zhang, C.-C.; Kast, J. Chemical Proteomics and Its Impact on the Drug Discovery Process. Expert Rev. Proteom. 2012, 9, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Charron, G.; Zhang, M.M.; Yount, J.S.; Wilson, J.; Raghavan, A.S.; Shamir, E.; Hang, H.C. Robust Fluorescent Detection of Protein Fatty-Acylation with Chemical Reporters. J. Am. Chem. Soc. 2009, 131, 4967–4975. [Google Scholar] [CrossRef] [PubMed]
- Agard, N.J.; Baskin, J.M.; Prescher, J.A.; Lo, A.; Bertozzi, C.R. A Comparative Study of Bioorthogonal Reactions with Azides. ACS Chem. Biol. 2006, 1, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Hang, H.C.; Wilson, J.P.; Charron, G. Bioorthogonal Chemical Reporters for Analyzing Protein Lipidation and Lipid Trafficking. Acc. Chem. Res. 2011, 44, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Tesseromatis, C.; Alevizou, A. The Role of the Protein-Binding on the Mode of Drug Action as Well the Interactions with Other Drugs. Eur. J. Drug Metab. Pharmacokinet. 2008, 33, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Martinez Molina, D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Martinez Molina, D.; Nordlund, P. The Cellular Thermal Shift Assay: A Novel Biophysical Assay for In Situ Drug Target Engagement and Mechanistic Biomarker Studies. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Tan, L.; Tao, H.; Li, Y.; Liu, H. CETSA and Thermal Proteome Profiling Strategies for Target Identification and Drug Discovery of Natural Products. Phytomedicine 2023, 116, 154862. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Jung, G.; Wohlschlegel, J.A.; Huang, J. Target Identification Using Drug Affinity Responsive Target Stability (DARTS). Curr. Protoc. Chem. Biol. 2011, 3, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.-S.; Li, H.-L.; Piao, X.-H.; Yang, Z.-Y.; Wang, S.-M.; Ge, Y.-W. Drug Affinity Responsive Target Stability (DARTS) Accelerated Small Molecules Target Discovery: Principles and Application. Biochem. Pharmacol. 2021, 194, 114798. [Google Scholar] [CrossRef] [PubMed]
- West, G.M.; Tang, L.; Fitzgerald, M.C. Thermodynamic Analysis of Protein Stability and Ligand Binding Using a Chemical Modification- and Mass Spectrometry-Based Strategy. Anal. Chem. 2008, 80, 4175–4185. [Google Scholar] [CrossRef]
- Meng, H.; Ma, R.; Fitzgerald, M.C. Chemical Denaturation and Protein Precipitation Approach for Discovery and Quantitation of Protein-Drug Interactions. Anal. Chem. 2018, 90, 9249–9255. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wan, N.; Zhang, H.; Shao, C.; Ding, M.; Bao, Q.; Hu, H.; Sun, H.; Liu, C.; Zhou, K.; et al. Chemoproteomic Mapping of the Glycolytic Targetome in Cancer Cells. Nat. Chem. Biol. 2023, 19, 1480–1491. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Martinović, S.; Veenstra, T.D.; Anderson, G.A.; Pasa-Tolić, L.; Smith, R.D. Selective Incorporation of Isotopically Labeled Amino Acids for Identification of Intact Proteins on a Proteome-Wide Level. J. Mass Spectrom. 2002, 37, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative Mass Spectrometry in Proteomics: Critical Review Update from 2007 to the Present. Anal. Bioanal. Chem. 2012, 404, 939–965. [Google Scholar] [CrossRef] [PubMed]
- Choe, L.; D’Ascenzo, M.; Relkin, N.R.; Pappin, D.; Ross, P.; Williamson, B.; Guertin, S.; Pribil, P.; Lee, K.H. 8-Plex Quantitation of Changes in Cerebrospinal Fluid Protein Expression in Subjects Undergoing Intravenous Immunoglobulin Treatment for Alzheimer’s Disease. Proteomics 2007, 7, 3651–3660. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces Cerevisiae Using Amine-Reactive Isobaric Tagging Reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Nahnsen, S.; Bielow, C.; Reinert, K.; Kohlbacher, O. Tools for Label-Free Peptide Quantification. Mol. Cell Proteom. 2013, 12, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Megger, D.A.; Bracht, T.; Meyer, H.E.; Sitek, B. Label-Free Quantification in Clinical Proteomics. Biochim. Biophys. Acta 2013, 1834, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Li, K.W.; Gonzalez-Lozano, M.A.; Koopmans, F.; Smit, A.B. Recent Developments in Data Independent Acquisition (DIA) Mass Spectrometry: Application of Quantitative Analysis of the Brain Proteome. Front. Mol. Neurosci. 2020, 13, 564446. [Google Scholar] [CrossRef] [PubMed]
- Abbiss, H.; Maker, G.L.; Trengove, R.D. Metabolomics Approaches for the Diagnosis and Understanding of Kidney Diseases. Metabolites 2019, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Steiner, C.; Othman, A.; Saely, C.; Rein, P.; Drexel, H.; von Eckardstein, A.; Rentsch, K. Bile Acid Metabolites in Serum: Intraindividual Variation and Associations with Coronary Heart Disease, Metabolic Syndrome and Diabetes Mellitus. PLoS ONE 2011, 6, e25006. [Google Scholar] [CrossRef] [PubMed]
- Piazza, I.; Kochanowski, K.; Cappelletti, V.; Fuhrer, T.; Noor, E.; Sauer, U.; Picotti, P. A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication. Cell 2018, 172, 358–372.e23. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Gray, N.S. Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Losada de la Lastra, A.; Hassan, S.; Tate, E.W. Deconvoluting the Biology and Druggability of Protein Lipidation Using Chemical Proteomics. Curr. Opin. Chem. Biol. 2021, 60, 97–112. [Google Scholar] [CrossRef]
- Yang, J.; Gupta, V.; Carroll, K.S.; Liebler, D.C. Site-Specific Mapping and Quantification of Protein S-Sulphenylation in Cells. Nat. Commun. 2014, 5, 4776. [Google Scholar] [CrossRef] [PubMed]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A Metabolite-Derived Protein Modification Integrates Glycolysis with KEAP1–NRF2 Signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.-Y. Chemoproteomics: Towards Global Drug Target Profiling. Chembiochem 2020, 21, 3189–3191. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Yang, X.; Lee, H.; Garst, E.H.; Valencia, E.; Chandran, K.; Im, W.; Hang, H.C. S-Palmitoylation and Sterol Interactions Mediate Antiviral Specificity of IFITMs. ACS Chem. Biol. 2022, 17, 2109–2120. [Google Scholar] [CrossRef]
- Cai, R.; Dong, X.; Yu, K.; He, X.; Liu, X.; Wang, Y. Chemical Proteomic Profiling of the Interacting Proteins of Isoprenoid Pyrophosphates. Anal. Chem. 2020, 92, 8031–8036. [Google Scholar] [CrossRef]
- Yu, W.; Lin, Z.; Woo, C.M.; Baskin, J.M. A Chemoproteomics Approach to Profile Phospholipase D-Derived Phosphatidyl Alcohol Interactions. ACS Chem. Biol. 2022, 17, 3276–3283. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tan, J.; Li, M.; Gao, C.; Li, W.; Xu, J.; Guo, C.; Chen, Z.; Cai, R. Clickable Photoreactive ATP-Affinity Probe for Global Profiling of ATP-Binding Proteins. Anal. Chem. 2023, 95, 17533–17540. [Google Scholar] [CrossRef] [PubMed]
- Šileikytė, J.; Sundalam, S.; David, L.L.; Cohen, M.S. Chemical Proteomics Approach for Profiling the NAD Interactome. J. Am. Chem. Soc. 2021, 143, 6787–6791. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Jose, G.P.; Shanbhag, C.; Tagad, N.; Kalia, J. Metabolic Labeling-Based Chemoproteomics Establishes Choline Metabolites as Protein Function Modulators. ACS Chem. Biol. 2022, 17, 2272–2283. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhuang, S.; Tian, R.; Liu, Y.; Wang, Y.; Lei, X.; Wang, C. Chemoproteomic Profiling Reveals the Mechanism of Bile Acid Tolerance in Bacteria. ACS Chem. Biol. 2022, 17, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Pedicord, V.A.; Peng, T.; Hang, H.C. Site-Specific Acylation of a Bacterial Virulence Regulator Attenuates Infection. Nat. Chem. Biol. 2020, 16, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Stein, K.R.; Chen, V.; Griffin, M.E.; Lairson, L.L.; Hang, H.C. Chemoproteomics Reveals Microbiota-Derived Aromatic Monoamine Agonists for GPRC5A. Nat. Chem. Biol. 2023, 19, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.J.; Montgomery, D.C.; Sardiu, M.E.; Montano, J.L.; Bergholtz, S.E.; Nance, K.D.; Thorpe, A.L.; Fox, S.D.; Lin, Q.; Andresson, T.; et al. A Systems Chemoproteomic Analysis of Acyl-CoA/Protein Interaction Networks. Cell Chem. Biol. 2020, 27, 322–333.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, Y.; Bozi, L.H.M.; Fischer, P.D.; Jedrychowski, M.P.; Xiao, H.; Wu, T.; Darabedian, N.; He, X.; Mills, E.L.; et al. Lactate Regulates Cell Cycle by Remodelling the Anaphase Promoting Complex. Nature 2023, 616, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.G. Lipid–Protein Interactions. Biochem. Soc. Trans. 2011, 39, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Tate, E.W.; Kalesh, K.A.; Lanyon-Hogg, T.; Storck, E.M.; Thinon, E. Global Profiling of Protein Lipidation Using Chemical Proteomic Technologies. Curr. Opin. Chem. Biol. 2015, 24, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Hannoush, R.N.; Sun, J. The Chemical Toolbox for Monitoring Protein Fatty Acylation and Prenylation. Nat. Chem. Biol. 2010, 6, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Bolla, J.R.; Agasid, M.T.; Mehmood, S.; Robinson, C.V. Membrane Protein–Lipid Interactions Probed Using Mass Spectrometry. Annu. Rev. Biochem. 2019, 88, 85–111. [Google Scholar] [CrossRef] [PubMed]
- Storck, E.M.; Serwa, R.A.; Tate, E.W. Chemical Proteomics: A Powerful Tool for Exploring Protein Lipidation. Biochem. Soc. Trans. 2013, 41, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Thinon, E.; Hang, H.C. Proteomic Analysis of Fatty-Acylated Proteins. Curr. Opin. Chem. Biol. 2016, 30, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. IFITMs Restrict the Replication of Multiple Pathogenic Viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef] [PubMed]
- Savidis, G.; Perreira, J.M.; Portmann, J.M.; Meraner, P.; Guo, Z.; Green, S.; Brass, A.L. The IFITMs Inhibit Zika Virus Replication. Cell Rep. 2016, 15, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Viel, G.; Boscolo-Berto, R.; Cecchetto, G.; Fais, P.; Nalesso, A.; Ferrara, S.D. Phosphatidylethanol in Blood as a Marker of Chronic Alcohol Use: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2012, 13, 14788–14812. [Google Scholar] [CrossRef] [PubMed]
- Dashty, M. A Quick Look at Biochemistry: Carbohydrate Metabolism. Clin. Biochem. 2013, 46, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pajares, V.; Bhaduri, A.; Zhao, Y.; Gowrishankar, G.; Donohue, L.; Guo, M.G.; Siprashvili, Z.; Miao, W.; Nguyen, D.T.; Li, A.M.; et al. Glucose Modulates Transcription Factor Dimerization to Enable Tissue Differentiation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Miao, W.; Porter, D.F.; Lopez-Pajares, V.; Siprashvili, Z.; Meyers, R.M.; Bai, Y.; Nguyen, D.T.; Ko, L.A.; Zarnegar, B.J.; Ferguson, I.D.; et al. Glucose Dissociates DDX21 Dimers to Regulate mRNA Splicing and Tissue Differentiation. Cell 2023, 186, 80–97.e26. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Shestov, A.A.; Lai, L.; Locasale, J.W. A Flux Balance of Glucose Metabolism Clarifies the Requirements of the Warburg Effect. Biophys. J. 2016, 111, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S. Interplay between Compartmentalized NAD+ Synthesis and Consumption: A Focus on the PARP Family. Genes Dev. 2020, 34, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The Hallmarks of Cancer Metabolism: Still Emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Chelakkot, V.S.; Shin, Y.; Song, K. Modulating Glycolysis to Improve Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2606. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Beyazal Polat, H.; Beyazal, M.; Beyazal Çeliker, F. A Case Report of Acute Acalculous Cholecystitis and Acute Hemorrhagic Cystitis Due to Salmonella Typhi. Case Rep. Med. 2014, 2014, e758583. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, Q.; Luo, F.; Shang, D.; Wu, D.; Zhang, H.; Shang, X.; Kang, X.; Abdo, M.; Liu, B.; et al. Acute Cholecystitis Associated with Infection of Enterobacteriaceae from Gut Microbiota. Clin. Microbiol. Infect. 2015, 21, 851.e1–851.e9. [Google Scholar] [CrossRef] [PubMed]
- Ogunrinola, G.A.; Oyewale, J.O.; Oshamika, O.O.; Olasehinde, G.I. The Human Microbiome and Its Impacts on Health. Int. J. Microbiol. 2020, 2020, 8045646. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut Microbial Metabolites as Multi-Kingdom Intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Gatsios, A.; Kim, C.S.; Crawford, J.M. Escherichia Coli Small Molecule Metabolism at the Host–Microorganism Interface. Nat. Chem. Biol. 2021, 17, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Baud, D.; Zuber, A.; Peric, A.; Pluchino, N.; Vulliemoz, N.; Stojanov, M. Impact of Semen Microbiota on the Composition of Seminal Plasma. Microbiol. Spectr. 2024, 12, e02911-23. [Google Scholar] [CrossRef] [PubMed]
- Łaniewski, P.; Ilhan, Z.E.; Herbst-Kralovetz, M.M. The Microbiome and Gynaecological Cancer Development, Prevention and Therapy. Nat. Rev. Urol. 2020, 17, 232–250. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Hatzios, S.K. Activity-Based Tools for Interrogating Host Biology During Infection. Isr. J. Chem. 2023, 63, e202200095. [Google Scholar] [CrossRef] [PubMed]
- Malarney, K.P.; Chang, P.V. Chemoproteomic Approaches for Unraveling Prokaryotic Biology. Isr. J. Chem. 2023, 63, e202200076. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.H. Chemical Biology Tools for Protein Labelling: Insights into Cell–Cell Communication. Biochem. J. 2023, 480, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, Y.; Williams, B.B.; Battaglioli, E.J.; Whitaker, W.R.; Till, L.; Grover, M.; Linden, D.R.; Akiba, Y.; Kandimalla, K.K.; Zachos, N.C.; et al. Gut Microbiota-Produced Tryptamine Activates an Epithelial G-Protein-Coupled Receptor to Increase Colonic Secretion. Cell Host Microbe 2018, 23, 775–785.e5. [Google Scholar] [CrossRef] [PubMed]
- Hezaveh, K.; Shinde, R.S.; Klötgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-Derived Microbial Metabolites Activate the Aryl Hydrocarbon Receptor in Tumor-Associated Macrophages to Suppress Anti-Tumor Immunity. Immunity 2022, 55, 324–340.e8. [Google Scholar] [CrossRef] [PubMed]
- Tintelnot, J.; Xu, Y.; Lesker, T.R.; Schönlein, M.; Konczalla, L.; Giannou, A.D.; Pelczar, P.; Kylies, D.; Puelles, V.G.; Bielecka, A.A.; et al. Microbiota-Derived 3-IAA Influences Chemotherapy Efficacy in Pancreatic Cancer. Nature 2023, 615, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Halip, L.; Curpan, R.; Oprea, T.I. Novel Drug Targets in 2019. Nat. Rev. Drug Discov. 2020, 19, 300. [Google Scholar] [CrossRef]
- Avram, S.; Halip, L.; Curpan, R.; Oprea, T.I. Novel Drug Targets in 2020. Nat. Rev. Drug Discov. 2021, 20, 333. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Halip, L.; Curpan, R.; Oprea, T.I. Novel Drug Targets in 2021. Nat. Rev. Drug Discov. 2022, 21, 328. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Halip, L.; Curpan, R.; Oprea, T.I. Novel Drug Targets in 2022. Nat. Rev. Drug Discov. 2023, 22, 437. [Google Scholar] [CrossRef]
- Avram, S.; Halip, L.; Curpan, R.; Oprea, T.I. Novel Drug Targets in 2023. Nat. Rev. Drug Discov. 2024, 23, 330. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Xiang, Y.; Kang, X. Small-Molecule PROTACs for Cancer Immunotherapy. Molecules 2022, 27, 5439. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Qu, B.; Yang, H.; Hu, S.; Dong, X. If Small Molecules Immunotherapy Comes, Can the Prime Be Far Behind? Eur. J. Med. Chem. 2021, 218, 113356. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Song, Z.; Zhang, A. Small-Molecule Immuno-Oncology Therapy: Advances, Challenges and New Directions. Curr. Top. Med. Chem. 2019, 19, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Yuan, W.-E.; Su, J.; Liu, Y.; Chen, J. Recent Advances in Small Molecule Based Cancer Immunotherapy. Eur. J. Med. Chem. 2018, 157, 582–598. [Google Scholar] [CrossRef]
- Beck, H.; Härter, M.; Haß, B.; Schmeck, C.; Baerfacker, L. Small Molecules and Their Impact in Drug Discovery: A Perspective on the Occasion of the 125th Anniversary of the Bayer Chemical Research Laboratory. Drug Discov. Today 2022, 27, 1560–1574. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-Dose Metformin Targets the Lysosomal AMPK Pathway through PEN2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Liang, K.; Zhao, S.; Jia, W.; Liu, Y.; Wu, H.; Lv, J.; Cao, C.; Chen, T.; Zhuang, S.; et al. Chemoproteomics Reveals Baicalin Activates Hepatic CPT1 to Ameliorate Diet-Induced Obesity and Hepatic Steatosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5896–E5905. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.-H.; Hu, L.-M.; Hao, X.-H.; Liu, J.; Tan, X.-Y.; Geng, Z.-R.; Ma, J.; Wang, Z.-L. Chemoproteomics Reveals Berberine Directly Binds to PKM2 to Inhibit the Progression of Colorectal Cancer. iScience 2022, 25, 104773. [Google Scholar] [CrossRef] [PubMed]
- Saei, A.A.; Gullberg, H.; Sabatier, P.; Beusch, C.M.; Johansson, K.; Lundgren, B.; Arvidsson, P.I.; Arnér, E.S.J.; Zubarev, R.A. Comprehensive Chemical Proteomics for Target Deconvolution of the Redox Active Drug Auranofin. Redox Biol. 2020, 32, 101491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, X.; Zhang, X.; Dong, Y.; Hu, L. Probing the Methotrexate-Protein Interactions by Proteomics and Thermostability Assay for Drug Resistance Study. Anal. Methods 2021, 13, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhao, P.; Hu, M.; Wang, L.; Lei, T.; Liu, B.; Li, L.; Shi, J.; Lu, C. HnRNP A2/B1 as a Potential Anti-Tumor Target for Triptolide Based on a Simplified Thermal Proteome Profiling Method Using XGBoost. Phytomedicine 2023, 117, 154929. [Google Scholar] [CrossRef] [PubMed]
- Pessetto, Z.Y.; Weir, S.J.; Sethi, G.; Broward, M.A.; Godwin, A.K. Drug Repurposing for Gastrointestinal Stromal Tumor. Mol. Cancer Ther. 2013, 12, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an Old Drug for a Golden New Age. Drugs R D 2015, 15, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Rios Perez, M.V.; Roife, D.; Dai, B.B.; Kang, Y.; Li, X.; Pratt, M.; Fleming, J.B. Auranofin to Prevent Progression of Pancreatic Ductal Adenocarcinoma. JCO 2016, 34, 236. [Google Scholar] [CrossRef]
- Chen, S.; Cai, J.; Zhang, W.; Zheng, X.; Hu, S.; Lu, J.; Xing, J.; Dong, Y. Proteomic Identification of Differentially Expressed Proteins Associated with the Multiple Drug Resistance in Methotrexate-Resistant Human Breast Cancer Cells. Int. J. Oncol. 2014, 45, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.-P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single Phosphorylation Sites in Acc1 and Acc2 Regulate Lipid Homeostasis and the Insulin-Sensitizing Effects of Metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Duca, F.A.; Côté, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K.T. Metformin Activates a Duodenal Ampk–Dependent Pathway to Lower Hepatic Glucose Production in Rats. Nat. Med. 2015, 21, 506–511. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on Natural Products for Drug Design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, D.; Ma, Y.; Liu, J.; Wang, Y.; Du, Z.; Wang, X.; Shen, J.; Peng, H. Long-Term Baicalin Administration Ameliorates Metabolic Disorders and Hepatic Steatosis in Rats given a High-Fat Diet. Acta Pharmacol. Sin. 2009, 30, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Wu, M.; Li, H.; Dong, S.; Luo, E.; Gu, M.; Shen, X.; Jiang, Y.; Liu, Y.; Liu, H. Baicalin Attenuates High Fat Diet-Induced Obesity and Liver Dysfunction: Dose-Response and Potential Role of CaMKKβ/AMPK/ACC Pathway. Cell. Physiol. Biochem. 2015, 35, 2349–2359. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Gao, Q.-Y.; Zou, T.-H.; Wang, B.-M.; Liu, S.-D.; Sheng, J.-Q.; Ren, J.-L.; Zou, X.-P.; Liu, Z.-J.; Song, Y.-Y.; et al. Berberine versus Placebo for the Prevention of Recurrence of Colorectal Adenoma: A Multicentre, Double-Blinded, Randomised Controlled Study. Lancet Gastroenterol. Hepatol. 2020, 5, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Chan, A.T. Extracting the Benefits of Berberine for Colorectal Cancer. Lancet Gastroenterol. Hepatol. 2020, 5, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Xu, M.; Wang, J.; Zhou, S.; Liu, Y.; Liu, S.; Huang, Y.; Chen, Y.; Chen, L.; Song, Q.; et al. Sequential Delivery of Nanoformulated α-Mangostin and Triptolide Overcomes Permeation Obstacles and Improves Therapeutic Effects in Pancreatic Cancer. Biomaterials 2020, 241, 119907. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Xu, X.; Li, L.; Chen, C.; Gong, K.; Guo, Q.; Liu, F.; Wang, Y.; Duan, Y.; Li, H. Functional Exosome-Mediated Delivery of Triptolide Endowed with Targeting Properties as Chemotherapy Carriers for Ovarian Carcinoma. J. Biomed. Nanotechnol. 2021, 17, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Kong, F.; Liu, S.; Liu, X.; Pei, D.; Miao, X. Membrane Protein-Chimeric Liposome-Mediated Delivery of Triptolide for Targeted Hepatocellular Carcinoma Therapy. Drug Deliv. 2021, 28, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Palve, V.; Liao, Y.; Remsing Rix, L.L.; Rix, U. Turning Liabilities into Opportunities: Off-Target Based Drug Repurposing in Cancer. Semin. Cancer Biol. 2021, 68, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.-Y.; Liu, K.; Ngai, M.H.; Lear, M.J.; Wenk, M.R.; Yao, S.Q. Activity-Based Proteome Profiling of Potential Cellular Targets of Orlistat—An FDA-Approved Drug with Anti-Tumor Activities. J. Am. Chem. Soc. 2010, 132, 656–666. [Google Scholar] [CrossRef]
- Eddleston, M.; Cohen, A.F.; Webb, D.J. Implications of the BIA-102474-101 Study for Review of First-into-Human Clinical Trials. Br. J. Clin. Pharmacol. 2016, 81, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.E.; Beliaev, A.; Ferreira, H.S.; Rosa, C.P.; Bonifácio, M.J.; Loureiro, A.I.; Pires, N.M.; Palma, P.N.; Soares-da-Silva, P. Discovery of a Potent, Long-Acting, and CNS-Active Inhibitor (BIA 10-2474) of Fatty Acid Amide Hydrolase. ChemMedChem 2018, 13, 2177–2188. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.-F.; Santos, A.; Gama, H.; Moser, P.; Falcão, A.; Pressman, P.; Wallace Hayes, A.; Soares-da-Silva, P. Safety, Tolerability, and Pharmacokinetics of FAAH Inhibitor BIA 10-2474: A Double-Blind, Randomized, Placebo-Controlled Study in Healthy Volunteers. Clin. Pharmacol. Ther. 2022, 111, 391–403. [Google Scholar] [CrossRef]
- Huang, Z.; Ogasawara, D.; Seneviratne, U.I.; Cognetta, A.B.I.; am Ende, C.W.; Nason, D.M.; Lapham, K.; Litchfield, J.; Johnson, D.S.; Cravatt, B.F. Global Portrait of Protein Targets of Metabolites of the Neurotoxic Compound BIA 10–2474. ACS Chem. Biol. 2019, 14, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.; Benjamin, N.; Jackson, N.; Allen, M.J. Effects of Sildenafil Citrate on Human Hemodynamics. Am. J. Cardiol. 1999, 83, 13–20. [Google Scholar] [CrossRef]
- Schmidt, A.F.; Hingorani, A.D.; Finan, C. Human Genomics and Drug Development. Cold Spring Harb. Perspect. Med. 2022, 12, a039230. [Google Scholar] [CrossRef] [PubMed]

| Method | Advantages | Disadvantages |
|---|---|---|
| ABPP | 1. Binds to the active site 2. Reveals enzyme activity | 1. Probe synthesizing is cumbersome 2. Support for chemical synthesis required |
| CCCP | 1. Targets proteins based on strong affinity 2. Extensive screening available | |
| TPP | 1. No synthetic probes required 2. Simple process | 1. Low resolution; 2. Unable to locate binding site |
| TRAP | 1. No synthetic probes required 2. Simple process 3. High resolution 4. Locate the binding site in peptide |
| Small Molecule | Method | Target | Probe Structure |
|---|---|---|---|
| Cholesterol | Cholesterol photoaffinity probe [78] | S-palmitoylated Interferon-induced transmembrane proteins |  |
| Geranyl pyrophosphate | Desthiobiotin-Geranyl pyrophosphate acyl phosphate probe [79] | Histone deacetylase 1 | 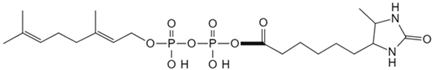 |
| Phosphatidylethanol | Clickable photoaffinity phosphatidyl alcohol probe [80] | Basigin/CD147 | 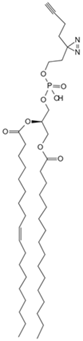 |
| Adenosine triphosphate | Clickable Adenosine triphosphate photoaffinity probe [81] | Adenosine triphosphate-binding proteins |  |
| Nicotinamide adenine dinucleotide | Photoaffinity 2-ad-BAD probe [82] | Nicotinamide adenine dinucleotide-binding proteins |  |
| Nicotinamide adenine dinucleotide | Photoaffinity 6-ad-BAD probe [82] | Nicotinamide adenine dinucleotide-binding proteins |  |
| Choline | Photocrosslinkable choline probe [83] | p32 | 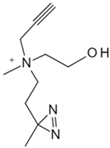 |
| Bile acids | Clickable photoaffinity Bile acids probe [84] | EnvZ |  |
| Short-chain fatty acids | Alkyne butyrate analog probe [85] | HilA |  |
| Indole-3-acetic acid | Indole-3-acetic acid analog probe [86] | G Protein-coupled receptor class C group 5 member A (GPRC5A) |  |
| Tryptamine | Tryptamine analog probe [86] | G Protein-coupled receptor class C group 5 member A (GPRC5A) | 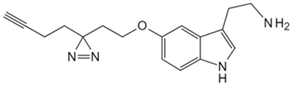 |
| Small Molecule | Method | Target |
|---|---|---|
| Acetyl-CoA | CoA/AcetylTraNsferase Interaction Profiling (CATNIP) [87] | Acetyl-CoA-binding proteins |
| Lactate | Thermal proteomic profiling (TPP) [88] | Ubiquitin conjugating enzyme E2 C (UBE2C) |
| Glycolysis metabolite group | Target-responsive accessibility profiling (TRAP) [61] | Glycolysis metabolite-binding protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, M.; Zhou, H.; Gu, L.; Zhang, J.; Fang, L. Therapeutic Target Identification and Drug Discovery Driven by Chemical Proteomics. Biology 2024, 13, 555. https://doi.org/10.3390/biology13080555
Zou M, Zhou H, Gu L, Zhang J, Fang L. Therapeutic Target Identification and Drug Discovery Driven by Chemical Proteomics. Biology. 2024; 13(8):555. https://doi.org/10.3390/biology13080555
Chicago/Turabian StyleZou, Mingjie, Haiyuan Zhou, Letian Gu, Jingzi Zhang, and Lei Fang. 2024. "Therapeutic Target Identification and Drug Discovery Driven by Chemical Proteomics" Biology 13, no. 8: 555. https://doi.org/10.3390/biology13080555