

Boolean Modeling of Biological Network Applied to Protein–Protein Interaction Network of Autism Patients



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. The Clinical Applications and Description of the Biochemical Pathway
- All the patients that only have polymorphism mutations (mutations that are common in the population) are excluded.
- Any genes that are sex-related with regards to cellular metabolism, such as SMS and SEMG2, are excluded.
- Any genes that are not involved in Wnt or mTOR pathways are excluded
2.1.1. Mutation in SUMF1
2.1.2. Mutation in IDS
2.1.3. INTS6L/USP9X/RPS6KA6 and FLNA
2.2. Boolean Modeling of the PPI Network
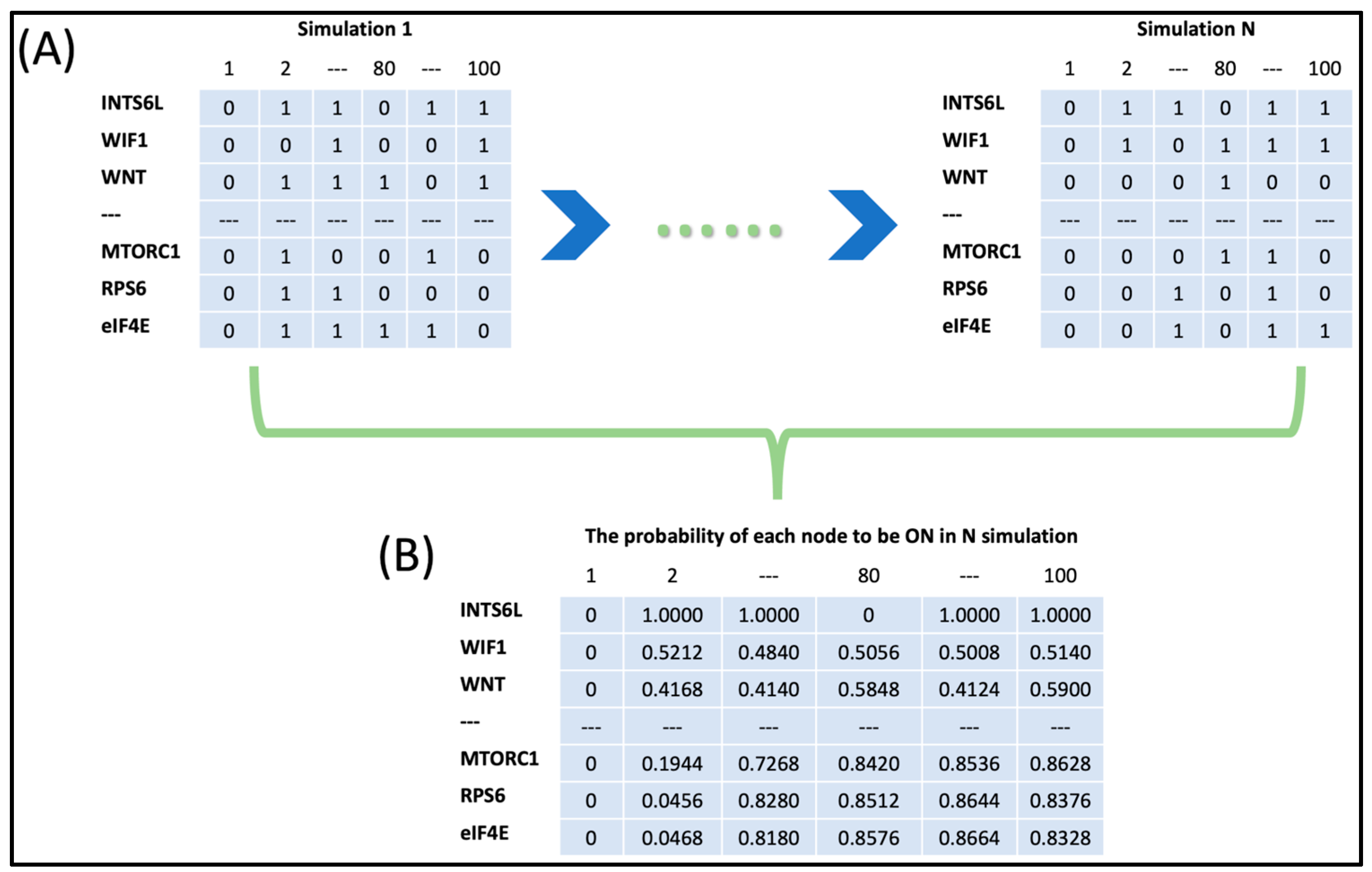
2.3. Boolean Network Simulation
2.4. Attractor Analysis
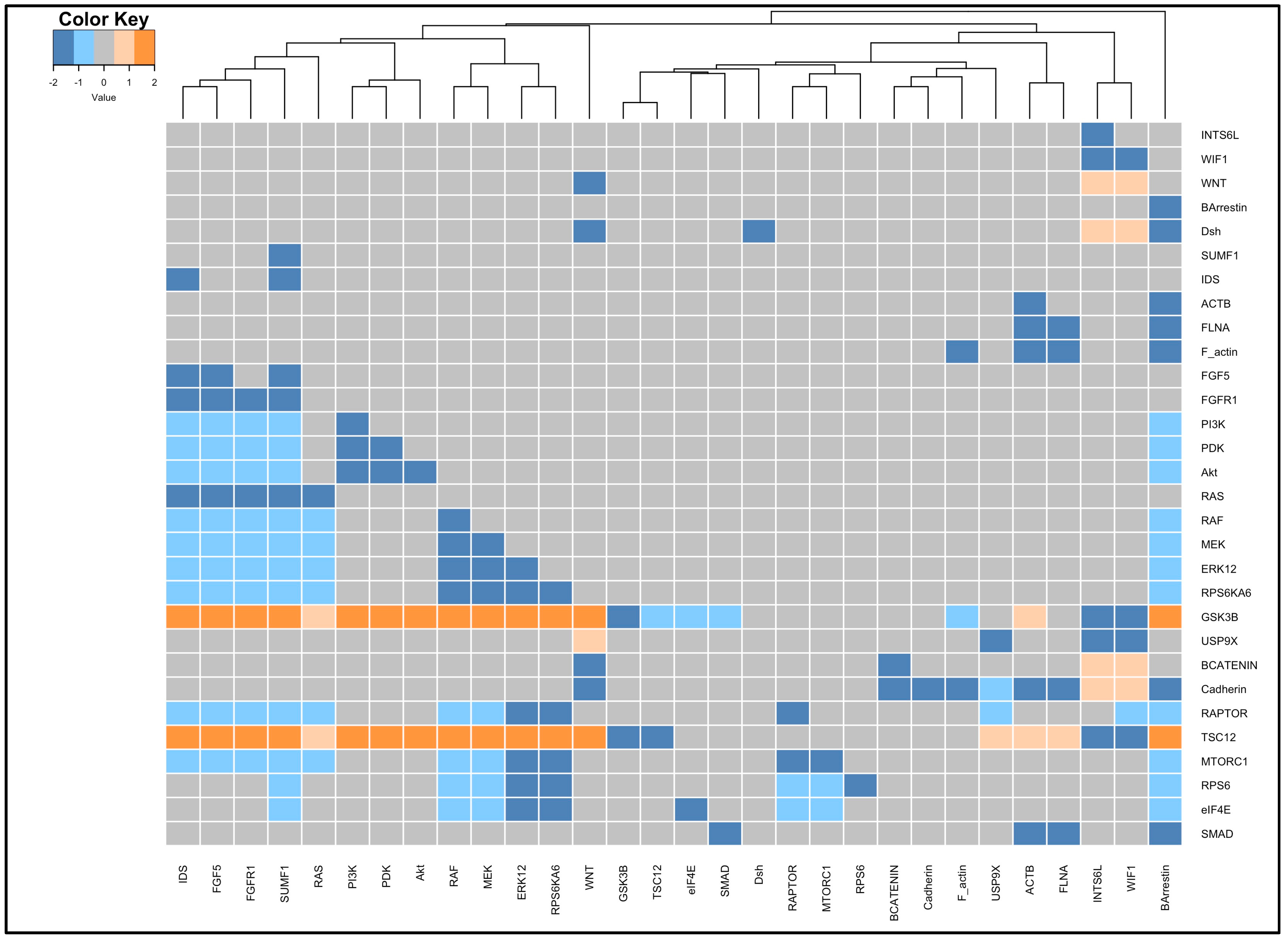
2.5. Sensitivity Analysis and Clustering
3. Results
3.1. Variants Annotation
3.2. The Dynamic of the Boolean Simulation
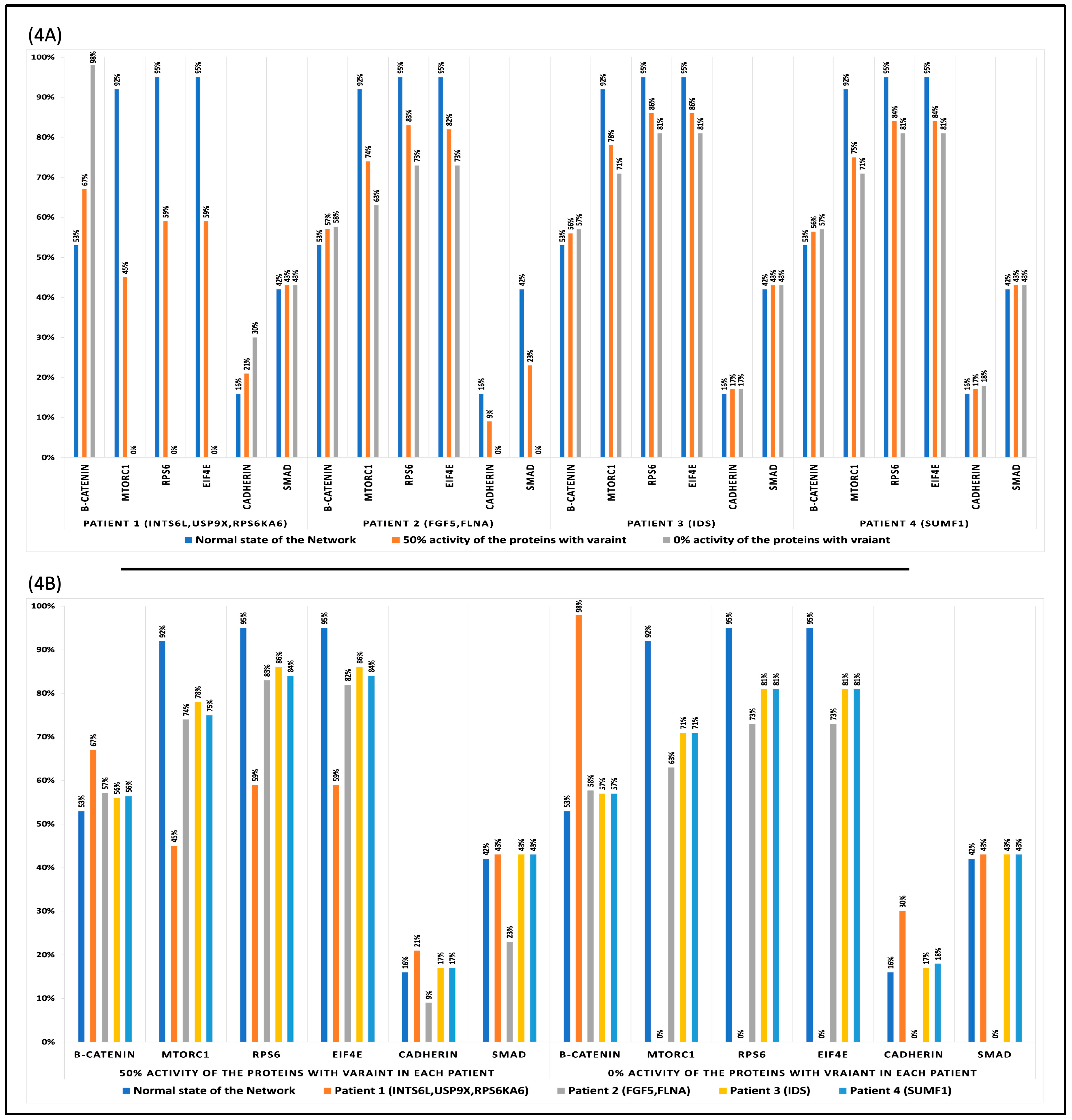
3.3. Attractor Analysis
3.4. Perturbation and Hierarchical Clustering Analysis
4. Discussion
4.1. Genetic Variants Annotations
4.2. Genetic Alterations’ Impact on Cellular Function
4.3. Possible Alternative Therapies
4.4. Model Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Node | Boolean Function | Explanation | Reference |
|---|---|---|---|
| INTS6L | INTS6L = ! (INTS6L & ANY_INTS6L[neg_modulator]) | To check if INTS6L is activated in any of the iterations of the simulation and not affected by any of its negative modulator | [21] |
| WIF1 | WIF1 = INTS6L &! (MOD_WIF1) | INTS6L is mediating the expression of WIF1 if WIF1 is activated | [34,35] |
| WNT | WNT = ! WIF1 | WNT is inhibited by WIF1 | [34,35] |
| BArrestin | BArrestin = ! (BArrestin & ANY_BArrestin[neg_modulator]) | To check if BArrestin is activated in any of the iterations of the simulation and not affected by any of its negative modulator | [53] |
| Dsh | Dsh = WNT & BArrestin | BArrestin binds to disheveled protein upon Wnt pathway activation | [120] |
| SUMF1 | SUMF1= ! (SUMF1& ANY_ SUMF1[neg_modulator]) | To check if SUMF1is activated in any of the iterations of the simulation and not affected by any of its negative modulator | [25] |
| IDS | IDS = SUMF1 | SUMF1 is a cofactor to enhance the activity of IDS | [57] |
| ACTB | ACTB = BArrestin &! (MOD_ACTB) | BArrestin mediates the activation of cofilin, severing existing filaments to release free actin monomers to assemble into a new filament | [121] |
| FLNA | FLNA = ACTB &! (MOD_FLAN) | FLNA binds ACTB to form a network of filaments that acts as the cell cytoskeleton. Also, it is involved in cell adhesion, migration, and determination of shape. | [62,122,123] |
| F_actin | F_actin = ACTB & FLNA | Filament protein that is assembled from actin binding to filamin A | [62,122,123] |
| FGF5 | FGF5 = IDS & !(MOD_FGF5) | Heparan sulfate, degraded by IDS, is a specific and central component in FGF/FGFR binding Mutation in IDS causes accumulation of heparan sulfate and leads to impaired FGF-FGFR binding | [55,73,74] |
| FGFR1 | FGFR1 = FGF5 | FGF5 binds specifically to FGFR1 | [124,125] |
| PI3K | PI3K = BArrestin|FGFR1 | PI3K activation induced either by BArrestin or FGFR1 activation | [66,126] |
| PDK | PDK = PI3K | PDK activated through PI3K | [66] |
| Akt | Akt = PDK | Akt activated by PDK | [66] |
| RAS | RAS = FGFR1 | RAS activated by FGFR1 activation | [66] |
| RAF | RAF = RAS|BArrestin | RAF activation induced either by BArrestin or RAS activation | [66,127] |
| MEK | MEK = RAF|(BArrestin & FLNA) | MEK activated either by RAF or BArrestin binding to FLNA | [53,66] |
| ERK1/2 | ERK1/2 = MEK|(BArrestin & FLNA) | ERK1/2 activated either by RAF or BArrestin binding to FLNA | [53,66] |
| RPS6KA6 | RPS6KA6 = ERK12 | RPS6KA6 is activated by ERK12 | [128] |
| GSK3B | GSK3B = ! (WNT|RPS6KA6 | Akt) | GSK3B is inhibited by the activation of WNT OR RPS6KA6 OR Akt | [44] |
| USP9X | USP9X = ! WNT|! (MOD_USP9X) | In the absence of WNT OR to check if USP9X is activated in any of the iterations of the simulation and not affected by any of its negative modulators | [21,37] |
| BCATENIN | BCATENIN= (GSK3B & USP9X)|(WNT &! GSK3B) | In the absence of WNT, where GSK3B is active and ubiquitinates B-CATENIN and USP9X deubiquitinates B-CATENIN and saves it from proteasomal degradation OR in the presence of WNT. | [44] |
| Cadherin | Cadherin = F_actin & BCATENIN | Alpha Catenin links BCATENIN to Actin and promotes BCATENIN binding to Cadherin for cell adhesion processes. | [62] |
| RAPTOR | RAPTOR = USP9X|RPS6KA6 | RAPTOR is activated by USP9X OR RPS6KA6 | [36,41] |
| TSC12 | TSC12 = GSK3B &! (RPS6KA6|Akt) | TSC12 is activated by GSK3B and inhibited by either RPS6KA6 OR Akt | [129,130] |
| MTORC1 | MTORC1 = RAPTOR &! TSC12 | MTORC1 is activated by RAPTOR and inhibited by TSC12 | [36,129,130] |
| RPS6 | RPS6 = MTORC1 | RPS6KA6 | RPS6 is activated by MTORC1 | [36] |
| eIF4E | eIF4E = MTORC1 | RPS6KA6 | eIF4E is activated by MTORC1 | [36] |
| SMAD | SMAD = FLNA | SMAD binds to FLNA to translocate to the nucleus and transcribe targeted genes | [51] |
References
- Chakravorty, S.; Hegde, M. Gene and Variant Annotation for Mendelian Disorders in the Era of Advanced Sequencing Technologies. Annu. Rev. Genom. Hum. Genet. 2017, 18, 229–256. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Hidalgo, R.; Veltman, J.A.; Hoischen, A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016, 17, 241. [Google Scholar] [CrossRef] [PubMed]
- Kuechler, A.; Willemsen, M.H.; Albrecht, B.; Bacino, C.A.; Bartholomew, D.W.; van Bokhoven, H.; van den Boogaard, M.J.; Bramswig, N.; Büttner, C.; Cremer, K.; et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: Expanding the mutational and clinical spectrum. Hum. Genet. 2015, 134, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Sleven, H.; Welsh, S.J.; Yu, J.; Churchill, M.E.A.; Wright, C.F.; Henderson, A.; Horvath, R.; Rankin, J.; Vogt, J.; Magee, A.; et al. De Novo Mutations in EBF3 Cause a Neurodevelopmental Syndrome. Am. J. Hum. Genet. 2017, 100, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Lal, D.; May, P.; Perez-Palma, E.; Samocha, K.E.; Kosmicki, J.A.; Robinson, E.B.; Møller, R.S.; Krause, R.; Nürnberg, P.; Weckhuysen, S.; et al. Gene family information facilitates variant interpretation and identification of disease-associated genes in neurodevelopmental disorders. Genome Med. 2020, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Palma, E.; May, P.; Iqbal, S.; Niestroj, L.M.; Du, J.; Heyne, H.O.; Castrillon, J.A.; O’Donnell-Luria, A.; Nürnberg, P.; Palotie, A.; et al. Identification of pathogenic variant enriched regions across genes and gene families. Genome Res. 2020, 30, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.D.; Kühlwein, S.D.; Ikonomi, N.; Kühl, M.; Kestler, H.A. Concepts in Boolean network modeling: What do they all mean? Comput. Struct. Biotechnol. J. 2020, 18, 571–582. [Google Scholar] [CrossRef]
- Bhardwaj, G.; Wells, C.P.; Albert, R.; van Rossum, D.B.; Patterson, R.L. Exploring phospholipase C-coupled Ca(2+) signalling networks using Boolean modelling. IET Syst. Biol. 2011, 5, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, S.A. Metabolic stability and epigenesis in randomly constructed genetic nets. J. Theor. Biol. 1969, 22, 437–467. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Imura, J.; Hiraishi, K. Polynomial-time algorithm for controllability test of a class of boolean biological networks. EURASIP J. Bioinform. Syst. Biol. 2010, 2010, 210685. [Google Scholar] [CrossRef] [PubMed]
- Mischnik, M.; Boyanova, D.; Hubertus, K.; Geiger, J.; Philippi, N.; Dittrich, M.; Wangorsch, G.; Timmer, J.; Dandekar, T. A Boolean view separates platelet activatory and inhibitory signalling as verified by phosphorylation monitoring including threshold behaviour and integrin modulation. Mol. Biosyst. 2013, 9, 1326–1339. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Bisht, S.S.; Kukreti, R.; Jain, S.; Brahmachari, S.K. Boolean network analysis of a neurotransmitter signaling pathway. J. Theor. Biol. 2007, 244, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Irurzun-Arana, I.; Pastor, J.M.; Trocóniz, I.F.; Gómez-Mantilla, J.D. Advanced Boolean modeling of biological networks applied to systems pharmacology. Bioinformatics 2017, 33, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Al-Mubarak, B.; Abouelhoda, M.; Omar, A.; AlDhalaan, H.; Aldosari, M.; Nester, M.; Alshamrani, H.A.; El-Kalioby, M.; Goljan, E.; Albar, R. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: A trio study from Saudi families. Sci. Rep. 2017, 7, 5679. [Google Scholar] [CrossRef] [PubMed]
- Schlotawa, L.; Adang, L.; De Castro, M.; Ahrens-Nicklas, R. Multiple Sulfatase Deficiency. In GeneReviews® [Internet]; Adam, M.P., Fieldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Hirst, L.; Abou-Ameira, G.; Uudelepp, M.L. Multiple Sulfatase Deficiency (MSD): Review of the Literature and Case Reports of Two Siblings with Dental Caries and Trauma. Case Rep. Pediatr. 2021, 2021, 6611548. [Google Scholar] [CrossRef] [PubMed]
- Sabourdy, F.; Mourey, L.; Le Trionnaire, E.; Bednarek, N.; Caillaud, C.; Chaix, Y.; Delrue, M.A.; Dusser, A.; Froissart, R.; Garnotel, R.; et al. Natural disease history and characterisation of SUMF1 molecular defects in ten unrelated patients with multiple sulfatase deficiency. Orphanet J. Rare Dis. 2015, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, L.; Kashgari, A.; Alfadhel, M. Multiple Sulfatase Deficiency: A Case Series With a Novel Mutation. J. Child Neurol. 2018, 33, 820–824. [Google Scholar] [CrossRef] [PubMed]
- Sheth, J.; Shah, S.; Datar, C.; Bhatt, K.; Raval, P.; Nair, A.; Jain, D.; Shah, J.; Sheth, F.; Sheth, H. Late infantile form of multiple sulfatase deficiency with a novel missense variant in the SUMF1 gene: Case report and review. BMC Pediatr. 2023, 23, 133. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Beck, M.; Eng, C.; Giugliani, R.; Harmatz, P.; Muñoz, V.; Muenzer, J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008, 121, e377–e386. [Google Scholar] [CrossRef] [PubMed]
- De Risi, M.; Tufano, M.; Alvino, F.G.; Ferraro, M.G.; Torromino, G.; Gigante, Y.; Monfregola, J.; Marrocco, E.; Pulcrano, S.; Tunisi, L.; et al. Altered heparan sulfate metabolism during development triggers dopamine-dependent autistic-behaviours in models of lysosomal storage disorders. Nat. Commun. 2021, 12, 3495. [Google Scholar] [CrossRef] [PubMed]
- Kushima, I.; Aleksic, B.; Nakatochi, M.; Shimamura, T.; Okada, T.; Uno, Y.; Morikawa, M.; Ishizuka, K.; Shiino, T.; Kimura, H.; et al. Comparative Analyses of Copy-Number Variation in Autism Spectrum Disorder and Schizophrenia Reveal Etiological Overlap and Biological Insights. Cell Rep. 2018, 24, 2838–2856. [Google Scholar] [CrossRef] [PubMed]
- Kapp, L.D.; Abrams, E.W.; Marlow, F.L.; Mullins, M.C. The integrator complex subunit 6 (Ints6) confines the dorsal organizer in vertebrate embryogenesis. PLoS Genet. 2013, 9, e1003822. [Google Scholar] [CrossRef] [PubMed]
- Wald, A. Tests of statistical hypotheses concerning several parameters when the number of observations is large. Trans. Am. Math. Soc. 1943, 54, 426–482. [Google Scholar] [CrossRef]
- Nezamuldeen, L.; Jafri, M.S. Protein–Protein Interaction Network Extraction Using Text Mining Methods Adds Insight into Autism Spectrum Disorder. Biology 2023, 12, 1344. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shen, H.-X.; Lin, Y.-W.; Mao, Y.-Q.; Liu, B.; Xie, L.-P. Small RNA-induced INTS6 gene up-regulation suppresses castration-resistant prostate cancer cells by regulating β-catenin signaling. Cell Cycle 2018, 17, 1602–1613. [Google Scholar] [CrossRef] [PubMed]
- Lui, K.Y.; Zhao, H.; Qiu, C.; Li, C.; Zhang, Z.; Peng, H.; Fu, R.; Chen, H.A.; Lu, M.Q. Integrator complex subunit 6 (INTS6) inhibits hepatocellular carcinoma growth by Wnt pathway and serve as a prognostic marker. BMC Cancer 2017, 17, 644. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.R.; Tan, M.C.; Premarathne, S.; Nanayakkara, D.; Bellette, B.; Zencak, D.; Domingo, D.; Gecz, J.; Murtaza, M.; Jolly, L.A.; et al. USP9X deubiquitylating enzyme maintains RAPTOR protein levels, mTORC1 signalling and proliferation in neural progenitors. Sci. Rep. 2017, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- Taya, S.; Yamamoto, T.; Kanai-Azuma, M.; Wood, S.A.; Kaibuchi, K. The deubiquitinating enzyme Fam interacts with and stabilizes beta-catenin. Genes Cells 1999, 4, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhang, S.; Wang, Z.; Yang, C.; Ouyang, W.; Zhou, F.; Zhou, Y.; Xie, C. Deubiquitinase USP9X deubiquitinates β-catenin and promotes high grade glioma cell growth. Oncotarget 2016, 7, 79515–79525. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Frödin, M.; Jensen, C.J.; Merienne, K.; Gammeltoft, S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000, 19, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Carrière, A.; Cargnello, M.; Julien, L.A.; Gao, H.; Bonneil, E.; Thibault, P.; Roux, P.P. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296 Pt 1, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Esnafoglu, E.; Ayyıldız, S.N. Decreased levels of serum fibroblast growth factor-2 in children with autism spectrum disorder. Psychiatry Res. 2017, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Haub, O.; Drucker, B.; Goldfarb, M. Expression of the murine fibroblast growth factor 5 gene in the adult central nervous system. Proc. Natl. Acad. Sci. USA 1990, 87, 8022–8026. [Google Scholar] [CrossRef] [PubMed]
- Reuss, B.; von Bohlen und Halbach, O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003, 313, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Modarres, H.P.; Mofradt, M.R. Filamin: A structural and functional biomolecule with important roles in cell biology, signaling and mechanics. Mol. Cell. Biomech. 2014, 11, 39–65. [Google Scholar] [PubMed]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; de Leon, M.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Masuda, Y.; Ohta, Y.; Ikeda, K.; Watanabe, K. Filamin associates with Smads and regulates transforming growth factor-beta signaling. J. Biol. Chem. 2001, 276, 17871–17877. [Google Scholar] [CrossRef] [PubMed]
- Savoy, R.M.; Ghosh, P.M. The dual role of filamin A in cancer: Can’t live with (too much of) it, can’t live without it. Endocr. Relat. Cancer 2013, 20, R341–R356. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Pierotti, V.; Storez, H.; Lindberg, E.; Thuret, A.; Muntaner, O.; Labbé-Jullié, C.; Pitcher, J.A.; Marullo, S. Cooperative regulation of extracellular signal-regulated kinase activation and cell shape change by filamin A and beta-arrestins. Mol. Cell. Biol. 2006, 26, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A. The mucopolysaccharidoses: A success of molecular medicine. Expert Rev. Mol. Med. 2008, 10, e1. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. Bioessays 2000, 22, 108–112. [Google Scholar] [CrossRef]
- Fraldi, A.; Biffi, A.; Lombardi, A.; Visigalli, I.; Pepe, S.; Settembre, C.; Nusco, E.; Auricchio, A.; Naldini, L.; Ballabio, A.; et al. SUMF1 enhances sulfatase activities in vivo in five sulfatase deficiencies. Biochem. J. 2007, 403, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Annunziata, I.; Roma, G.; Ballabio, A. Sulfatases and sulfatase modifying factors: An exclusive and promiscuous relationship. Hum. Mol. Genet. 2005, 14, 3203–3217. [Google Scholar] [CrossRef] [PubMed]
- Robeva, R.; Yildirim, N. Bistability in the lactose operon of Escherichia coli: A comparison of differential equation and boolean network models. In Mathematical Concepts and Methods in Modern Biology: Using Modern Discrete Models; Academic Press: Cambridge, MA, USA, 2013; pp. 37–74. [Google Scholar]
- Veliz-Cuba, A.; Murrugarra, D. Steady State Analysis of Boolean Models: A Dimension Reduction Approach. In Algebraic and Discrete Mathematical Methods for Modern Biology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 121–139. [Google Scholar]
- Harvey, I.; Bossomaier, T. Time out of joint: Attractors in asynchronous random boolean networks. In Proceedings of the Fourth European Conference on Artificial Life, Brighton, UK, 26–31 July 1997; pp. 67–75. [Google Scholar]
- Thomas, R. Regulatory networks seen as asynchronous automata: A logical description. J. Theor. Biol. 1991, 153, 1–23. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.; Zheng, G. E-cadherin/β-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kimelman, D. Mechanistic insights from structural studies of beta-catenin and its binding partners. J. Cell Sci. 2007, 120, 3337–3344. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.M.; Hong, J.Y. The Wnt Signaling Pathway and Related Therapeutic Drugs in Autism Spectrum Disorder. Clin. Psychopharmacol. Neurosci. 2018, 16, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, H.; Balasubramanian, V.; Iyer, M.; Venugopal, A.; Subramaniam, M.D.; Cho, S.G.; Vellingiri, B. mTOR signalling pathway—A root cause for idiopathic autism? BMB Rep. 2019, 52, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Dailey, L.; Ambrosetti, D.; Mansukhani, A.; Basilico, C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005, 16, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000, 101, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.N.; Eliseenkova, A.V.; Ibrahimi, O.A.; Shriver, Z.; Sasisekharan, R.; Lemmon, M.A.; Mohammadi, M. Crystal structure of fibroblast growth factor 9 reveals regions implicated in dimerization and autoinhibition. J. Biol. Chem. 2001, 276, 4322–4329. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins: Organizers of cell structure and function. Cell Adhes. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Walsh, C.A. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 2004, 6, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Tirupula, K.C.; Ithychanda, S.S.; Mohan, M.L.; Naga Prasad, S.V.; Qin, J.; Karnik, S.S. G protein-coupled receptors directly bind filamin A with high affinity and promote filamin phosphorylation. Biochemistry 2015, 54, 6673–6683. [Google Scholar] [CrossRef]
- Dwyer, C.; Lin, Y.; Dozier, L.; Schnaar, R.; Allen, N.; Patrick, G.; Esko, J. Lysosomal degradation of heparan sulfate is required for normal development of the neural circuitry. Mol. Genet. Metab. 2016, 2, S42. [Google Scholar] [CrossRef]
- De Pasquale, V.; Sarogni, P.; Pistorio, V.; Cerulo, G.; Paladino, S.; Pavone, L.M. Targeting Heparan Sulfate Proteoglycans as a Novel Therapeutic Strategy for Mucopolysaccharidoses. Mol. Ther. Methods Clin. Dev. 2018, 10, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Nelson, M.S.; Reyes, M.; Koodie, L.; Brazil, J.J.; Stephenson, E.J.; Zhao, R.C.; Peters, C.; Selleck, S.B.; Stringer, S.E.; et al. Functional abnormalities of heparan sulfate in mucopolysaccharidosis-I are associated with defective biologic activity of FGF-2 on human multipotent progenitor cells. Blood 2005, 106, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Appel, M.J.; Bertozzi, C.R. Formylglycine, a post-translationally generated residue with unique catalytic capabilities and biotechnology applications. ACS Chem. Biol. 2015, 10, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Demydchuk, M.; Hill, C.H.; Zhou, A.; Bunkóczi, G.; Stein, P.E.; Marchesan, D.; Deane, J.E.; Read, R.J. Insights into Hunter syndrome from the structure of iduronate-2-sulfatase. Nat. Commun. 2017, 8, 15786. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kim, D.; Kim, H.-L.; Choi, M.; Korbel, J.O.; Yoon, S.-S.; Koh, Y. Oncogenic effects of germline mutations in lysosomal storage disease genes. bioRxiv 2018, 380121. [Google Scholar] [CrossRef]
- Dierks, T.; Dickmanns, A.; Preusser-Kunze, A.; Schmidt, B.; Mariappan, M.; von Figura, K.; Ficner, R.; Rudolph, M.G. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell 2005, 121, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Tchevkina, E.; Komelkov, A. Protein phosphorylation as a key mechanism of mTORC1/2 signaling pathways. In Protein Phosphorylation in Human Health; IntechOpen: London, UK, 2012; pp. 3–50. [Google Scholar]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Reynolds, K.; Ji, Y.; Gu, R.; Rai, S.; Zhou, C.J. Impaired neurodevelopmental pathways in autism spectrum disorder: A review of signaling mechanisms and crosstalk. J. Neurodev. Disord. 2019, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Kwan, V.; Unda, B.K.; Singh, K.K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.A.; Andrade, V.M.; Caracci, M.O.; Avila, M.E.; Verdugo, D.A.; Vargas, M.F.; Ugarte, G.D.; Reyes, A.E.; Opazo, C.; De Ferrari, G.V. Wnt/β-catenin signaling stimulates the expression and synaptic clustering of the autism-associated Neuroligin 3 gene. Transl. Psychiatry 2018, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Wisniewska, M.B.; Nagalski, A.; Dabrowski, M.; Misztal, K.; Kuznicki, J. Novel β-catenin target genes identified in thalamic neurons encode modulators of neuronal excitability. BMC Genom. 2012, 13, 635. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef] [PubMed]
- Forés-Martos, J.; Catalá-López, F.; Sánchez-Valle, J.; Ibáñez, K.; Tejero, H.; Palma-Gudiel, H.; Climent, J.; Pancaldi, V.; Fañanás, L.; Arango, C.; et al. Transcriptomic metaanalyses of autistic brains reveals shared gene expression and biological pathway abnormalities with cancer. Mol. Autism 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Frei, J.A.; Niescier, R.F.; Bridi, M.S.; Durens, M.; Nestor, J.E.; Kilander, M.B.C.; Yuan, X.; Dykxhoorn, D.M.; Nestor, M.W.; Huang, S.; et al. Regulation of Neural Circuit Development by Cadherin-11 Provides Implications for Autism. eNeuro 2021, 8, ENEURO.0066-21.2021. [Google Scholar] [CrossRef] [PubMed]
- Redies, C.; Hertel, N.; Hübner, C.A. Cadherins and neuropsychiatric disorders. Brain Res. 2012, 1470, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Parrini, E.; Mei, D.; Pisanti, M.A.; Catarzi, S.; Pucatti, D.; Bianchini, C.; Mascalchi, M.; Bertini, E.; Morrone, A.; Cavaliere, M.L.; et al. Familial periventricular nodular heterotopia, epilepsy and Melnick-Needles Syndrome caused by a single FLNA mutation with combined gain-of-function and loss-of-function effects. J. Med. Genet. 2015, 52, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, P.J.; Zhang, M.; Silverman-Gavrila, R.V.; Bendeck, M.P. Cadherins at cell-autonomous membrane contacts control macropinocytosis. J. Cell Sci. 2011, 124, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L.; Attisano, L. The Smad pathway. Cytokine Growth Factor Rev. 2000, 11, 5–13. [Google Scholar] [CrossRef]
- Ashwood, P.; Enstrom, A.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.L.; Croen, L.A.; Ozonoff, S.; Pessah, I.N.; Van de Water, J. Decreased transforming growth factor beta1 in autism: A potential link between immune dysregulation and impairment in clinical behavioral outcomes. J. Neuroimmunol. 2008, 204, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Vojinovic, D.; Brison, N.; Ahmad, S.; Noens, I.; Pappa, I.; Karssen, L.C.; Tiemeier, H.; van Duijn, C.M.; Peeters, H.; Amin, N. Variants in TTC25 affect autistic trait in patients with autism spectrum disorder and general population. Eur. J. Hum. Genet. 2017, 25, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Winden, K.D.; Ebrahimi-Fakhari, D.; Sahin, M. Abnormal mTOR Activation in Autism. Annu. Rev. Neurosci. 2018, 41, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Jean-Charles, P.Y.; Kaur, S.; Shenoy, S.K. G Protein-Coupled Receptor Signaling Through β-Arrestin-Dependent Mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Weïwer, M.; Xu, Q.; Gale, J.P.; Lewis, M.; Campbell, A.J.; Schroeder, F.A.; Van de Bittner, G.C.; Walk, M.; Amaya, A.; Su, P.; et al. Functionally Biased D2R Antagonists: Targeting the β-Arrestin Pathway to Improve Antipsychotic Treatment. ACS Chem. Biol. 2018, 13, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Fukuchi, M.; Habata, Y. Potential Utility of Biased GPCR Signaling for Treatment of Psychiatric Disorders. Int. J. Mol. Sci. 2019, 20, 3207. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Polter, A.M.; Li, X. Glycogen Synthase Kinase-3 is an Intermediate Modulator of Serotonin Neurotransmission. Front. Mol. Neurosci. 2011, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Del’guidice, T.; Sotnikova, T.D.; Lemasson, M.; Gainetdinov, R.R. Beyond cAMP: The Regulation of Akt and GSK3 by Dopamine Receptors. Front. Mol. Neurosci. 2011, 4, 38. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Sotnikova, T.D.; Marion, S.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 2005, 122, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Krishnankutty, A.; Kimura, T.; Saito, T.; Aoyagi, K.; Asada, A.; Takahashi, S.I.; Ando, K.; Ohara-Imaizumi, M.; Ishiguro, K.; Hisanaga, S.I. In vivo regulation of glycogen synthase kinase 3β activity in neurons and brains. Sci. Rep. 2017, 7, 8602. [Google Scholar] [CrossRef] [PubMed]
- Bowling, H.; Zhang, G.; Bhattacharya, A.; Pérez-Cuesta, L.M.; Deinhardt, K.; Hoeffer, C.A.; Neubert, T.A.; Gan, W.B.; Klann, E.; Chao, M.V. Antipsychotics activate mTORC1-dependent translation to enhance neuronal morphological complexity. Sci. Signal. 2014, 7, ra4. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Konkalmatt, P.; Mokashi, C.; Kumar, M.; Zhang, Y.; Ko, A.; Farino, Z.J.; Asico, L.D.; Xu, G.; Gildea, J.; et al. Dopamine D. Sci. Rep. 2019, 9, 16861. [Google Scholar] [CrossRef]
- Han, J.M.; Sahin, M. TSC1/TSC2 signaling in the CNS. FEBS Lett. 2011, 585, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Mines, M.A.; Jope, R.S. Brain region differences in regulation of Akt and GSK3 by chronic stimulant administration in mice. Cell. Signal. 2012, 24, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Giaime, E.; Narayan, S.; Hahm, S.; Howell, J.; O’Neill, D.; Vlasuk, G.P.; Saiah, E. Discovery of NV-5138, the first selective Brain mTORC1 activator. Sci. Rep. 2019, 9, 4107. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Seto, T.; Kadono, C.; Morimoto, H.; Kida, S.; Suga, M.; Nakamura, M.; Kataoka, Y.; Hamazaki, T.; Shintaku, H. Autophagy in the Central Nervous System and Effects of Chloroquine in Mucopolysaccharidosis Type II Mice. Int. J. Mol. Sci. 2019, 20, 5829. [Google Scholar] [CrossRef] [PubMed]
- Ahrens-Nicklas, R.; Schlotawa, L.; Ballabio, A.; Brunetti-Pierri, N.; De Castro, M.; Dierks, T.; Eichler, F.; Ficicioglu, C.; Finglas, A.; Gaertner, J.; et al. Complex care of individuals with multiple sulfatase deficiency: Clinical cases and consensus statement. Mol. Genet. Metab. 2018, 123, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Schlotawa, L.; Dierks, T.; Christoph, S.; Cloppenburg, E.; Ohlenbusch, A.; Korenke, G.C.; Gärtner, J. Severe neonatal multiple sulfatase deficiency presenting with hydrops fetalis in a preterm birth patient. JIMD Rep. 2019, 49, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Vorstman, J.A.S.; Freitag, C.M.; Persico, A.M. From Genes to Therapy in Autism Spectrum Disorder. Genes 2022, 13, 1377. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, Z. A novel boolean network for analyzing the p53 gene regulatory network. Curr. Bioinform. 2016, 11, 13–21. [Google Scholar] [CrossRef]
- Sgariglia, D.; Conforte, A.J.; Pedreira, C.E.; Vidal de Carvalho, L.A.; Carneiro, F.R.G.; Carels, N.; Silva, F.A.B.D. Data-Driven Modeling of Breast Cancer Tumors Using Boolean Networks. Front. Big Data 2021, 4, 656395. [Google Scholar] [CrossRef] [PubMed]
- Anda-Jáuregui, G.d.; Espinal-Enríquez, J.; Sandoval-Motta, S.; Hernández-Lemus, E. A Boolean Network Approach to Estrogen Transcriptional Regulation. Complexity 2019, 2019, 8740279. [Google Scholar] [CrossRef]
- Müssel, C.; Hopfensitz, M.; Kestler, H.A. BoolNet—An R package for generation, reconstruction and analysis of Boolean networks. Bioinformatics 2010, 26, 1378–1380. [Google Scholar] [CrossRef] [PubMed]
- Albert, I.; Thakar, J.; Li, S.; Zhang, R.; Albert, R. Boolean network simulations for life scientists. Source Code Biol. Med. 2008, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Oles, V.; Kukushkin, A. BoolSi: A tool for distributed simulations and analysis of Boolean networks. arXiv 2019, arXiv:1910.03736. [Google Scholar] [CrossRef]
- Schwab, J.; Burkovski, A.; Siegle, L.; Müssel, C.; Kestler, H.A. ViSiBooL—Visualization and simulation of Boolean networks with temporal constraints. Bioinformatics 2017, 33, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Furukawa, K.; Hamamura, K.; Furukawa, K. Positive Feedback Loop Between PI3K-Akt-mTORC1 Signaling and the Lipogenic Pathway Boosts Akt Signaling: Induction of the Lipogenic Pathway by a Melanoma Antigen. Cancer Res. 2011, 71, 4989–4997. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Gradl, D.; Schambony, A.; Arenas, E.; Schulte, G. Beta-arrestin is a necessary component of Wnt/beta-catenin signaling in vitro and in vivo. Proc Natl Acad Sci U S A 2007, 104, 6690–6695. [Google Scholar] [CrossRef] [PubMed]
- DeFea, K.A. Stop that cell! Beta-arrestin-dependent chemotaxis: A tale of localized actin assembly and receptor desensitization. Annual review of physiology 2007, 69, 535–560. [Google Scholar] [CrossRef] [PubMed]
- Jahed, Z.; Shams, H.; Mehrbod, M.; Mofrad, M.R. Mechanotransduction pathways linking the extracellular matrix to the nucleus. International review of cell and molecular biology 2014, 310, 171–220. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, S. Actin filament association at adherens junctions. The journal of medical investigation: JMI 2017, 64, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wu, H.; Lian, Z. Bioinformatics analysis of evolutionary characteristics and biochemical structure of FGF5 Gene in sheep. Gene 2019, 702, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Allerstorfer, S.; Sonvilla, G.; Fischer, H.; Spiegl-Kreinecker, S.; Gauglhofer, C.; Setinek, U.; Czech, T.; Marosi, C.; Buchroithner, J.; Pichler, J.; et al. FGF5 as an oncogenic factor in human glioblastoma multiforme: Autocrine and paracrine activities. Oncogene 2008, 27, 4180–4190. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Ji, Q.; Li, Q. The role and mechanism of β-arrestins in cancer invasion and metastasis (Review). International journal of molecular medicine 2018, 41, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Bologna, Z.; Teoh, J.P.; Bayoumi, A.S.; Tang, Y.; Kim, I.M. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomolecules & therapeutics 2017, 25, 12–25. [Google Scholar] [CrossRef]
- Carriere, A.; Ray, H.; Blenis, J.; Roux, P.P. The RSK factors of activating the Ras/MAPK signaling cascade. Frontiers in bioscience: A journal and virtual library 2008, 13, 4258–4275. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends in biochemical sciences 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]



| Gender | Clinical Demographic Information | Protein Name | Variant Position | Effect of the Variant | ||
|---|---|---|---|---|---|---|
| Mutation Taster | PolyPhen | |||||
| Patient 1 | F | Language delay and regression | DDX26B /INTS6L | p:E435V | DC | PD/0.843 |
| USP9X | p:Y1268C | DC | B/0.007 | |||
| RPS6KA6 /RSK4 | p:Q512R | DC | B/0.195 | |||
| Patient 2 | M | NR | FGF5 | p:S84L | DC | D/1.0 |
| FLNA | p:Y2360A | DC | D/0.971 | |||
| Patient 3 | M | Language delay | IDS | p:D175E | DC | PD/0.94 |
| Patient 4 | M | Language delay | SUMF1 | p:Q237R | DC | D/1.0 |
| Low | High | Value | Color |
|---|---|---|---|
| ≤0.5 | −2 | Dark Blue | |
| 0.5 | 0.8 | −1 | Light Blue |
| 0.8 | 1.25 | 0 | Gray |
| 1.25 | 2.0 | 1 | Light Orange |
| ≥2.0 | 2 | Dark Orange | |
| Running the dynamic evolution function in the physiological state of the protein | |||||||||||||
| Steps | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | --- | 99 | 100 |
| INTS6L | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | --- | 0 | 1 |
| Running the dynamic evolution function when the mutation-like effect was delaying the protein activity with a 50% rate | |||||||||||||
| INTS6L | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | --- | 0 | 1 |
| Patient1 (INTS6L, USP9X, RPS6KA6) | Pateint2 (FLNA, FGF5) | Pateint3 (IDS) | Pteint4 (SUMF1) | |
|---|---|---|---|---|
| Normal conditions of the network | 321 | 321 | 321 | 321 |
| 50% activity of proteins with variant | 103 | 158 | 185 | 102 |
| 0% activity of proteins with variant | 1229 | 224 | 243 | 236 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nezamuldeen, L.; Jafri, M.S. Boolean Modeling of Biological Network Applied to Protein–Protein Interaction Network of Autism Patients. Biology 2024, 13, 606. https://doi.org/10.3390/biology13080606
Nezamuldeen L, Jafri MS. Boolean Modeling of Biological Network Applied to Protein–Protein Interaction Network of Autism Patients. Biology. 2024; 13(8):606. https://doi.org/10.3390/biology13080606
Chicago/Turabian StyleNezamuldeen, Leena, and Mohsin Saleet Jafri. 2024. "Boolean Modeling of Biological Network Applied to Protein–Protein Interaction Network of Autism Patients" Biology 13, no. 8: 606. https://doi.org/10.3390/biology13080606
APA StyleNezamuldeen, L., & Jafri, M. S. (2024). Boolean Modeling of Biological Network Applied to Protein–Protein Interaction Network of Autism Patients. Biology, 13(8), 606. https://doi.org/10.3390/biology13080606