
Targeting Polyprotein to Design Potential Multiepitope Vaccine against Omsk Hemorrhagic Fever Virus (OHFV) by Evaluating Allergenicity, Antigenicity, and Toxicity Using Immunoinformatic Approaches

Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Retrieval of Sequence
2.2. Major Histocompatibility Complex-I (MHC-I) Epitopes Prediction
2.3. Epitope Prediction for Major Histocompatibility Complex-II (MHC-II)
2.4. B Cell Epitope Prediction
2.5. Epitopes Evaluation for Vaccine Development
2.6. Multiepitope Vaccine Construct
2.7. Physiochemical Properties Prediction
2.8. Prediction of Secondary and Tertiary Structure of the Vaccine Construct
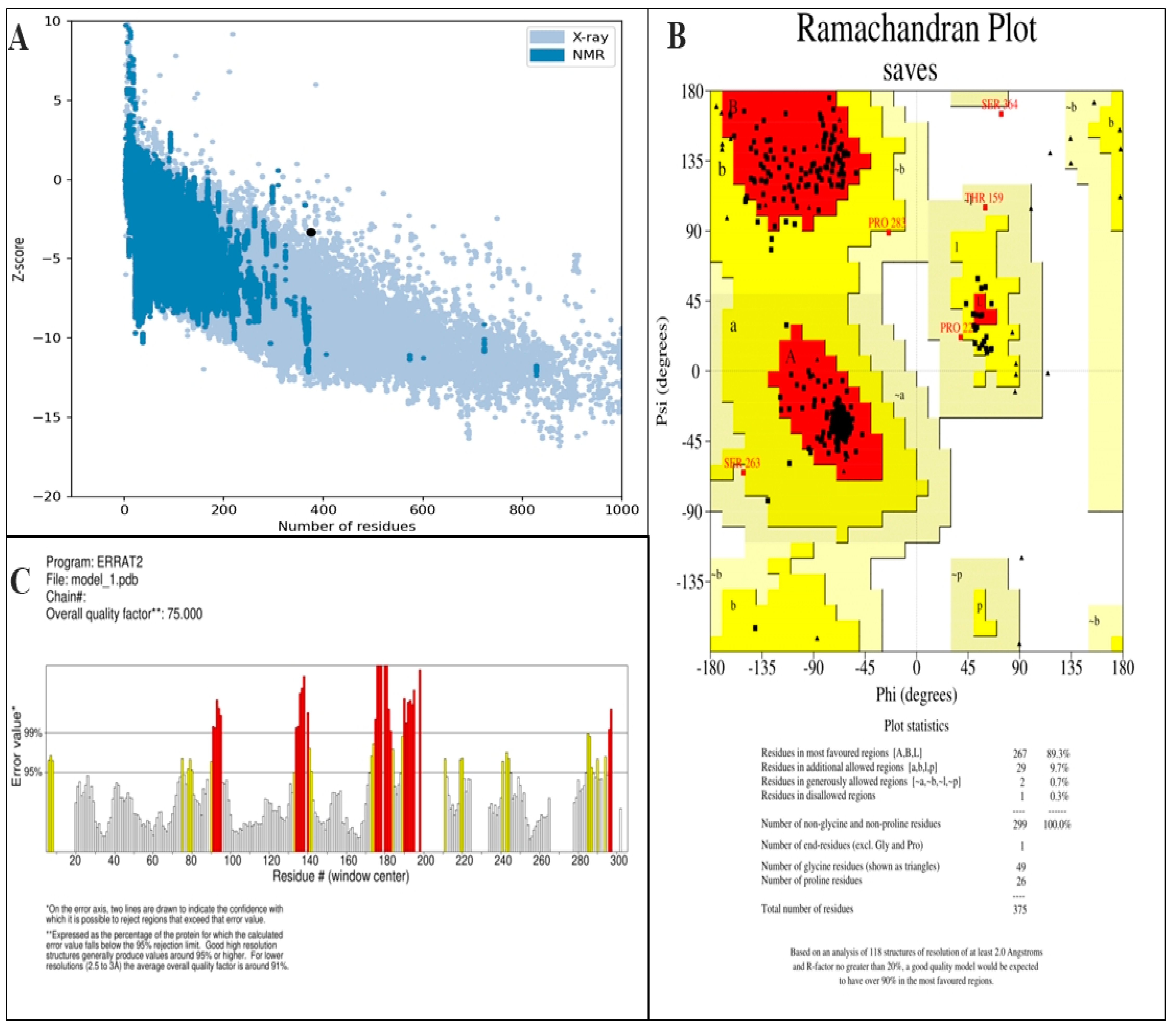
2.9. Validation of Tertiary Structure
2.10. Molecular Docking of TLR4 and Vaccine Construct
2.11. Codon Optimization of Vaccine Construct and In Silico Cloning
2.12. In-Silco Immune Responses Analysis
2.13. Molecular Dynamics Simulations
3. Results
3.1. Sequence Retrieval of Polyprotein
3.2. MHC-I Epitopes Prediction and Evaluation
3.3. MHC-II Epitope Prediction and Evaluation
3.4. B Cell Epitopes Prediction and Assessment
3.5. Construction of Multiepitope Vaccine and Assessment
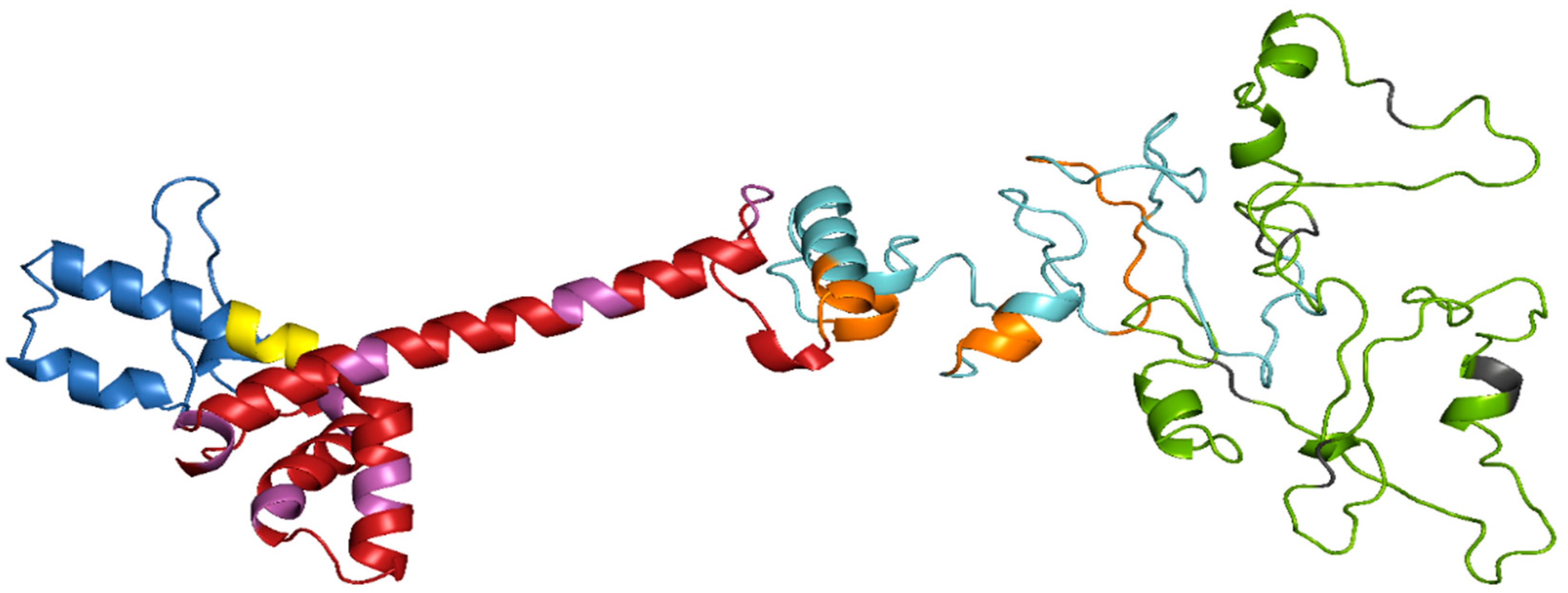
3.6. Prediction of Secondary and Tertiary Structure
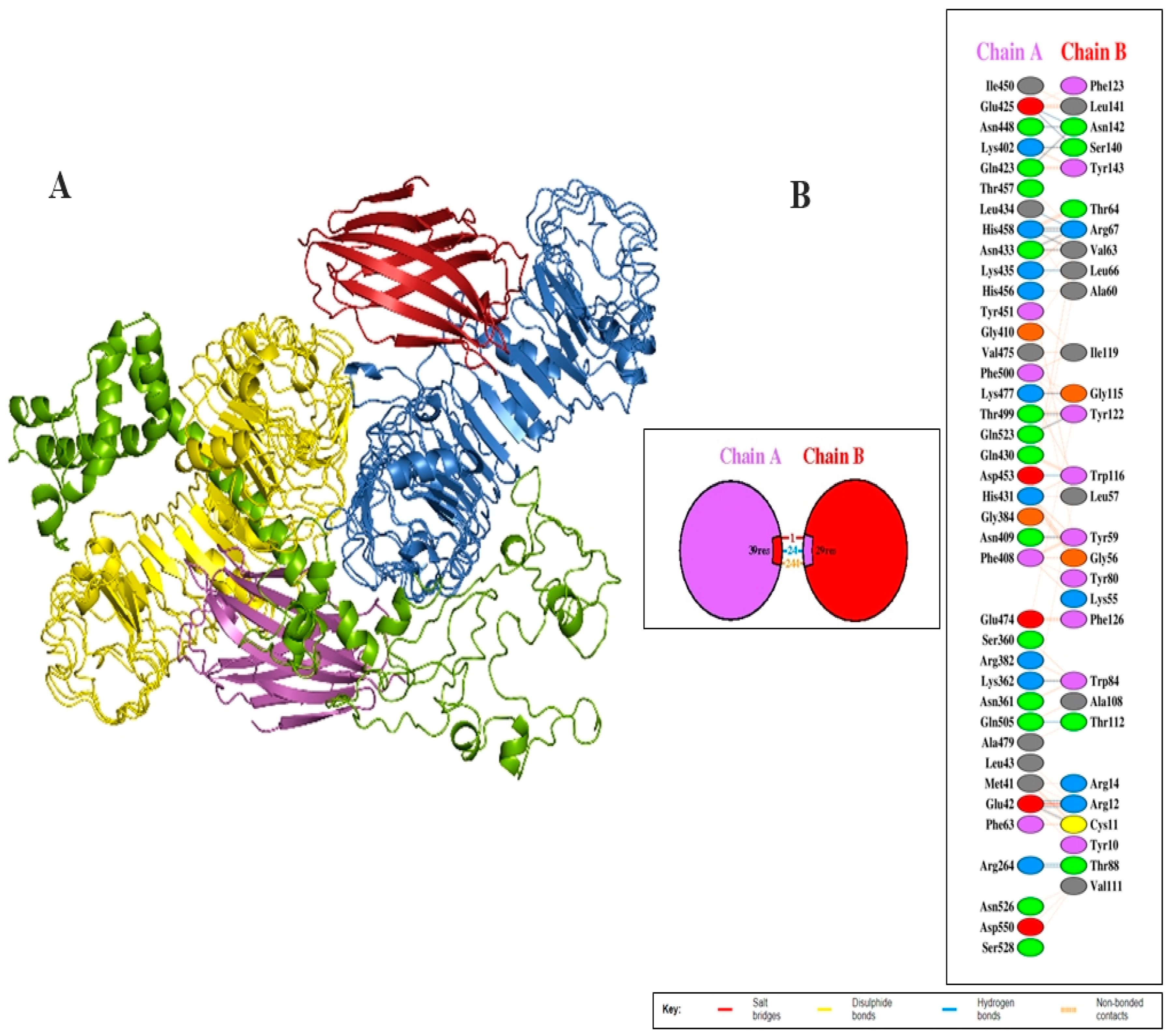
3.7. Investigation of the Interaction between the Vaccine Design and TLR4
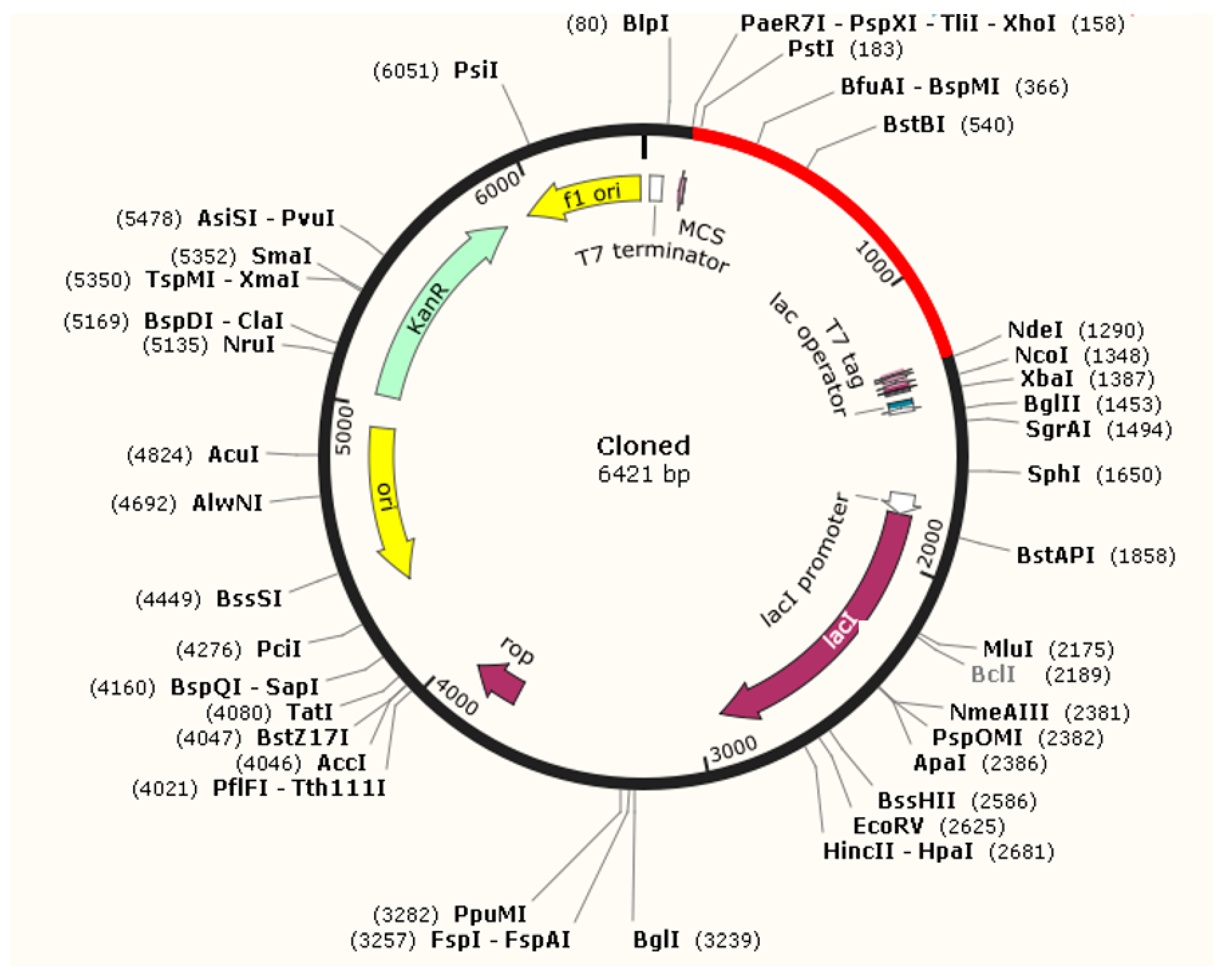
3.8. Optimization of Codon and Cloning
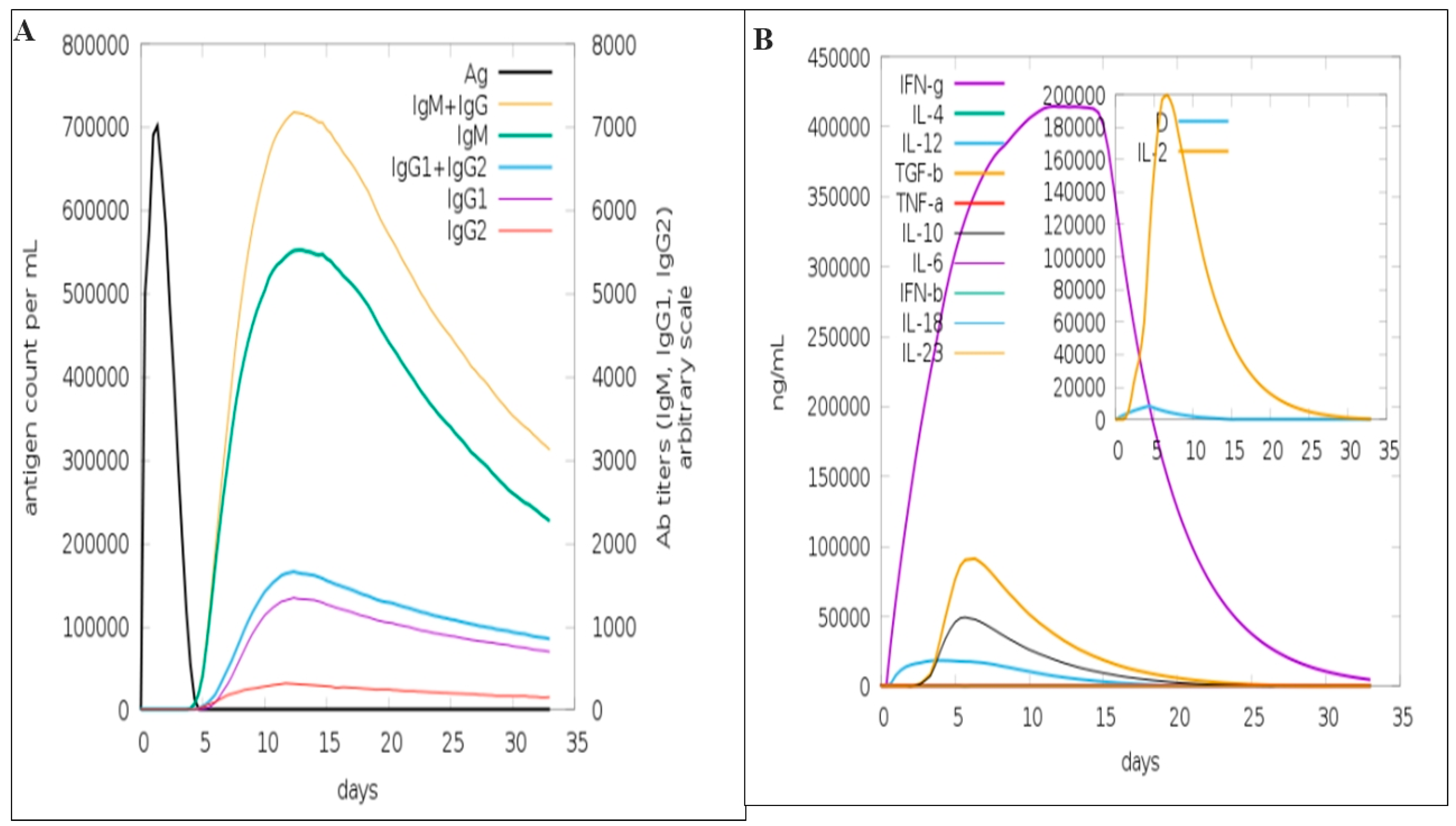
3.9. Immune Simulation
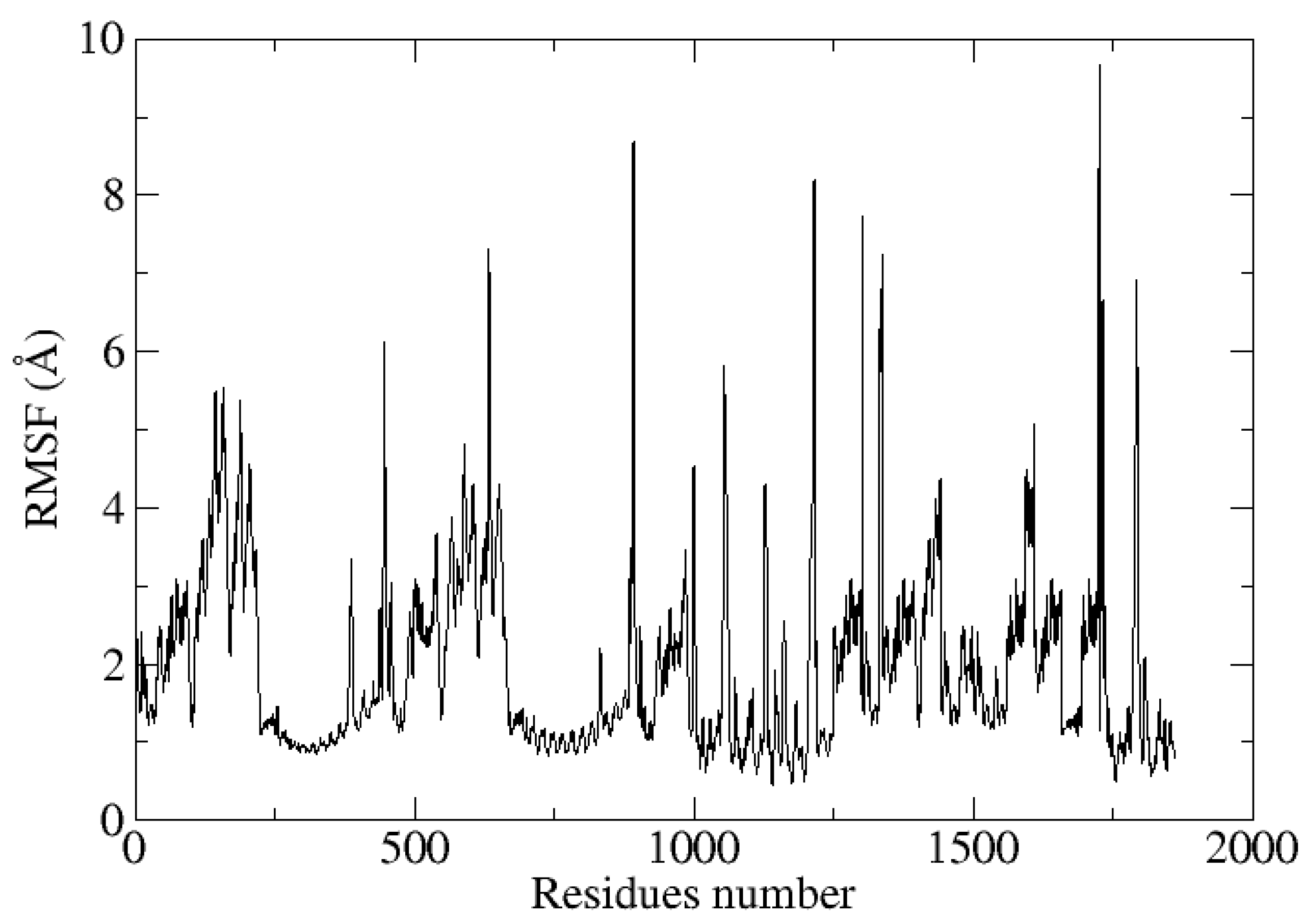
3.10. Molecular Dynamic Simulations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kovalev, S.Y.; Mazurina, E.A. Omsk Hemorrhagic Fever Virus Is a Tick-Borne Encephalitis Virus Adapted to Muskrat through Host-Jumping. J. Med. Virol. 2022, 94, 2510–2518. [Google Scholar] [CrossRef] [PubMed]
- Im, J.H.; Baek, J.-H.; Durey, A.; Kwon, H.Y.; Chung, M.-H.; Lee, J.-S. Geographic Distribution of Tick-Borne Encephalitis Virus Complex. J. Vector Borne Dis. 2020, 57, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Růžek, D.; Yakimenko, V.V.; Karan, L.S.; Tkachev, S.E. Omsk Haemorrhagic Fever. Lancet 2010, 376, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Shin, A.; Tukhanova, N.; Turebekov, N.; Nurmakhanov, T.; Sutyagin, V.; Berdibekov, A.; Maikanov, N.; Lezdinsh, I.; Shapiyeva, Z.; et al. First Indications of Omsk Haemorrhagic Fever Virus beyond Russia. Viruses 2022, 14, 754. [Google Scholar] [CrossRef]
- Liu, D. (Ed.) Molecular Detection of Human Viral Pathogens; CRC Press: Boca Raton, FL, USA, 2010; ISBN 978-0-429-13462-3. [Google Scholar]
- Jelínková-Skalová, E.; Tesarová, J.; Buresová, D.; Kouba, K.; Hronovský, V. [Laboratory infection with virus of Omsk haemorrhagic fever with neurological and psychiatric symptomatology (author’s transl)]. Cesk. Epidemiol. Mikrobiol. Imunol. 1974, 23, 290–293. [Google Scholar]
- Madani, T.A.; Abuelzein, E.-T.M.E. Alkhumra Hemorrhagic Fever Virus Infection. Arch. Virol. 2021, 166, 2357–2367. [Google Scholar] [CrossRef]
- Gagarina, A.V.; Netsky, G.I. Data on distribution and vectors of hemorrhagic fever in western Siberia. In Prirod Ochag Bolezn Chelovek Krayev Epidemiol; State Publishing House of Medical Literature: Moscow, Russia, 1955. [Google Scholar]
- Rubel, F.; Brugger, K.; Belova, O.A.; Kholodilov, I.S.; Didyk, Y.M.; Kurzrock, L.; García-Pérez, A.L.; Kahl, O. Vectors of Disease at the Northern Distribution Limit of the Genus Dermacentor in Eurasia: D. reticulatus and D. silvarum. Exp. Appl. Acarol. 2020, 82, 95–123. [Google Scholar] [CrossRef]
- Shah, T.; Li, Q.; Wang, B.; Baloch, Z.; Xia, X. Geographical Distribution and Pathogenesis of Ticks and Tick-Borne Viral Diseases. Front. Microbiol. 2023, 14, 1185829. [Google Scholar] [CrossRef]
- Kondrashova, Z.N. [A study of the survival of the virus of Omsk hemorrhagic fever in Ixodes persulcatus in conditions of their massive infection]. Med. Parazitol. (Mosk) 1970, 39, 274–278. [Google Scholar]
- Shah, S.Z.; Jabbar, B.; Ahmed, N.; Rehman, A.; Nasir, H.; Nadeem, S.; Jabbar, I.; ur Rahman, Z.; Azam, S. Epidemiology, Pathogenesis, and Control of a Tick-Borne Disease- Kyasanur Forest Disease: Current Status and Future Directions. Front. Cell Infect. Microbiol. 2018, 8, 149. [Google Scholar] [CrossRef]
- Li, L.; Rollin, P.E.; Nichol, S.T.; Shope, R.E.; Barrett, A.D.T.; Holbrook, M.R. Molecular Determinants of Antigenicity of Two Subtypes of the Tick-Borne Flavivirus Omsk Haemorrhagic Fever Virus. J. Gen. Virol. 2004, 85, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.H. Further Studies on Antigenic Relationships among the Viruses of the Group B Tick-Borne Complex. Bull. World Health Organ. 1964, 31, 45–56. [Google Scholar] [PubMed]
- Lin, D.; Li, L.; Dick, D.; Shope, R.E.; Feldmann, H.; Barrett, A.D.T.; Holbrook, M.R. Analysis of the Complete Genome of the Tick-Borne Flavivirus Omsk Hemorrhagic Fever Virus. Virology 2003, 313, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, N.; Deng, C.; Zhang, Z.; Li, X.; Yoshii, K.; Ye, H.; Zhang, B. Trans Complementation of Replication-Defective Omsk Hemorrhagic Fever Virus for Antiviral Study. Virol. Sin. 2019, 34, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, M.R.; Aronson, J.F.; Campbell, G.A.; Jones, S.; Feldmann, H.; Barrett, A.D.T. An Animal Model for the Tickborne Flavivirus—Omsk Hemorrhagic Fever Virus. J. Infect. Dis. 2005, 191, 100–108. [Google Scholar] [CrossRef]
- Shestopalova, N.M.; Reingold, V.N.; Gagarina, A.V.; Kornilova, E.A.; Popov, G.V.; Chumakov, M.P. Electron Microscopic Study of the Central Nervous System in Mice Infected by Omsk Hemorrhagic Fever (OHF) Virus. Virus Reproduction in Cerebellum Neurons. J. Ultrastruct. Res. 1972, 40, 458–469. [Google Scholar] [CrossRef]
- Orlinger, K.K.; Hofmeister, Y.; Fritz, R.; Holzer, G.W.; Falkner, F.G.; Unger, B.; Loew-Baselli, A.; Poellabauer, E.-M.; Ehrlich, H.J.; Barrett, P.N.; et al. A Tick-Borne Encephalitis Virus Vaccine Based on the European Prototype Strain Induces Broadly Reactive Cross-Neutralizing Antibodies in Humans. J. Infect. Dis. 2011, 203, 1556–1564. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing Multi-Epitope Vaccine against Staphylococcus Aureus by Employing Subtractive Proteomics, Reverse Vaccinology and Immuno-Informatics Approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Ong, E.; Wong, M.U.; Huffman, A.; He, Y. COVID-19 Coronavirus Vaccine Design Using Reverse Vaccinology and Machine Learning. bioRxiv 2020, 2020.03.20.000141. [Google Scholar] [CrossRef]
- Folaranmi, T.; Rubin, L.; Martin, S.W.; Patel, M.; MacNeil, J.R. Use of Serogroup B Meningococcal Vaccines in Persons Aged ≥10 Years at Increased Risk for Serogroup B Meningococcal Disease: Recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 608–612. [Google Scholar]
- Park, S.E. Prevention of Neonatal Group B Streptococcal Disease. Infect. Chemother. 2013, 45, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Rappuoli, R. Reverse Vaccinology: Developing Vaccines in the Era of Genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Zeb, A.; Ali, S.S.; Azad, A.K.; Safdar, M.; Anwar, Z.; Suleman, M.; Nizam-Uddin, N.; Khan, A.; Wei, D.-Q. Genome-Wide Screening of Vaccine Targets Prioritization and Reverse Vaccinology Aided Design of Peptides Vaccine to Enforce Humoral Immune Response against Campylobacter Jejuni. Comput. Biol. Med. 2021, 133, 104412. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.; Mahita, J.; Blazeska, N.; Greenbaum, J.; Ha, B.; Wheeler, K.; Wang, J.; Shackelford, D.; Sette, A.; Peters, B. IEDB-3D 2.0: Structural Data Analysis within the Immune Epitope Database. Protein Sci. 2023, 32, e4605. [Google Scholar] [CrossRef] [PubMed]
- Ghafoor, D.; Zeb, A.; Ali, S.S.; Ali, M.; Akbar, F.; Ud Din, Z.; Ur Rehman, S.; Suleman, M.; Khan, W. Immunoinformatic Based Designing of Potential Immunogenic Novel mRNA and Peptide-Based Prophylactic Vaccines against H5N1 and H7N9 Avian Influenza Viruses. J. Biomol. Struct. Dyn. 2024, 42, 3641–3658. [Google Scholar] [CrossRef]
- Faisal, A.-R.M.; Imtiaz, S.H.; Zerin, T.; Rahman, T.; Shekhar, H.U. Computer Aided Epitope Design as a Peptide Vaccine Component against Lassa Virus. Bioinformation 2017, 13, 417–429. [Google Scholar] [CrossRef]
- Dörner, T.; Radbruch, A. Antibodies and B Cell Memory in Viral Immunity. Immunity 2007, 27, 384–392. [Google Scholar] [CrossRef]
- Ahmad, T.A.; Eweida, A.E.; Sheweita, S.A. B-Cell Epitope Mapping for the Design of Vaccines and Effective Diagnostics. Trials Vaccinol. 2016, 5, 71–83. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Yang, J.; Chou, K.-C. Prediction of Linear B-Cell Epitopes Using Amino Acid Pair Antigenicity Scale. Amino Acids 2007, 33, 423–428. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. AlgPred: Prediction of Allergenic Proteins and Mapping of IgE Epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Naorem, L.D.; Jain, S.; Raghava, G.P.S. ToxinPred2: An Improved Method for Predicting Toxicity of Proteins. Brief. Bioinform. 2022, 23, bbac174. [Google Scholar] [CrossRef] [PubMed]
- ProtParam, E. ExPASy-ProtParam Tool; SIB: Lausanne, Switzerland, 2017. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Humana Press Inc.: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED Protein Structure Prediction Server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein Structure Prediction and Analysis Using the Robetta Server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. ERRAT: An Empirical Atom-Based Method for Validating Protein Structures. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Wallner, B.; Elofsson, A. Can Correct Protein Models Be Identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef]
- Khan, S.; Rizwan, M.; Zeb, A.; Eldeen, M.A.; Hassan, S.; Ur Rehman, A.; Eid, R.A.; Samir, A.; Zaki, M.; Albadrani, G.M.; et al. Identification of a Potential Vaccine against Treponema pallidum Using Subtractive Proteomics and Reverse-Vaccinology Approaches. Vaccines 2023, 11, 72. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein–Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania Secretory Proteins to Design B and T Cell Multi-Epitope Subunit Vaccine Using Immunoinformatics Approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- Suleman, M.; Rashid, F.; Ali, S.; Sher, H.; Luo, S.; Xie, L.; Xie, Z. Immunoinformatic-Based Design of Immune-Boosting Multiepitope Subunit Vaccines against Monkeypox Virus and Validation through Molecular Dynamics and Immune Simulation. Front. Immunol. 2022, 13, 1042997. [Google Scholar] [CrossRef] [PubMed]
- Chander, S.; Pandey, R.K.; Penta, A.; Choudhary, B.S.; Sharma, M.; Malik, R.; Prajapati, V.K.; Murugesan, S. Molecular Docking and Molecular Dynamics Simulation Based Approach to Explore the Dual Inhibitor Against HIV-1 Reverse Transcriptase and Integrase. Comb. Chem. High Throughput Screen. 2017, 20, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.; Darden, T.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Götz, A.W.; Homeyer, N.; et al. Amber 16; University of California: San Francisco, CA, USA, 2016. [Google Scholar] [CrossRef]
- Brice, A.R.; Dominy, B.N. Examining Electrostatic Influences on Base-Flipping: A Comparison of TIP3P and GB Solvent Models. Commun. Comput. Phys. 2013, 13, 223–237. [Google Scholar] [CrossRef]
- Uberuaga, B.P.; Anghel, M.; Voter, A.F. Synchronization of Trajectories in Canonical Molecular-Dynamics Simulations: Observation, Explanation, and Exploitation. J. Chem. Phys. 2004, 120, 6363–6374. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Zhang, Y.; Feller, S.E.; Brooks, B.R.; Pastor, R.W. Computer Simulation of Liquid/Liquid Interfaces. I. Theory and Application to Octane/Water. J. Chem. Phys. 1995, 103, 10252–10266. [Google Scholar] [CrossRef]
- Carugo, O. How Root-Mean-Square Distance (r.m.s.d.) Values Depend on the Resolution of Protein Structures That Are Compared. J. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]
- Rappuoli, R. Reverse Vaccinology, a Genome-Based Approach to Vaccine Development. Vaccine 2001, 19, 2688–2691. [Google Scholar] [CrossRef]
- Das, N.C.; Ray, A.S.; Bayry, J.; Mukherjeee, S. Therapeutic Efficacy of Anti-Bestrophin Antibodies against Experimental Filariasis: Immunological, Immune-Informatics and Immune Simulation Investigations. Antibodies 2021, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Gorai, S.; Das, N.C.; Gupta, P.S.S.; Panda, S.K.; Rana, M.K.; Mukherjee, S. Designing Efficient Multi-Epitope Peptide-Based Vaccine by Targeting the Antioxidant Thioredoxin of Bancroftian Filarial Parasite. Infect. Genet. Evol. 2022, 98, 105237. [Google Scholar] [CrossRef]
- Carreira-Rosario, A.; Bhargava, V.; Hillebrand, J.; Kollipara, R.K.; Ramaswami, M.; Buszczak, M. Repression of Pumilio Protein Expression by Rbfox1 Promotes Germ Cell Differentiation. Dev. Cell 2016, 36, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl Huber, S.; van Beek, J.; de Jonge, J.; Luytjes, W.; van Baarle, D. T Cell Responses to Viral Infections—Opportunities for Peptide Vaccination. Front. Immunol. 2014, 5, 171. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T-and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef]
- Murdock, J.L.; Núñez, G. TLR4: The Winding Road to the Discovery of the LPS Receptor. J. Immunol. 2016, 197, 2561–2562. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Epitopes | Residue No. | MHC Binding Affinity | Rescale Binding Affinity | C-Terminal Cleavage Affinity | Transport Affinity | Combined Score | MHC-I Binding | Antigenicity | Toxicity (<0) (Non-Toxic) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | KLKMKGLTY | 576 | 0.1418 | 0.6022 | 0.9640 | 2.8820 | 0.8909 | Yes | 1.45 | −0.77 |
| 2 | VTDLRNCSW | 994 | 0.1987 | 0.8437 | 0.9180 | 0.7970 | 1.0212 | Yes | 1.37 | −0.73 |
| 3 | GTDCWYAME | 1103 | 0.2409 | 1.0229 | 0.0282 | −1.9320 | 0.9305 | Yes | 1.16 | −0.50 |
| 4 | WTSKGTITV | 1677 | 0.2342 | 0.9944 | 0.9409 | 0.2320 | 1.1471 | Yes | 1.26 | −0.95 |
| 5 | WSEWTNIDI | 2286 | 0.1964 | 0.8337 | 0.5968 | 0.4510 | 0.9458 | Yes | 1.61 | −1.21 |
| 6 | VTSLGWNLI | 2627 | 0.1649 | 0.7002 | 0.8243 | 0.6070 | 0.8542 | Yes | 1.33 | −1.32 |
| 7 | FLEFEALGF | 2995 | 0.1492 | 0.6333 | 0.3797 | 2.4470 | 0.8126 | Yes | 1.77 | −0.94 |
| 8 | GVEGISLNY | 3018 | 0.3920 | 1.6642 | 0.9715 | 2.8940 | 1.9546 | Yes | 1.40 | −1.21 |
| S. No. | HLA | Epitopes | Percentile Rank | Antigenicity | Allergenicity | Toxicity (<0) (Non-Toxic) |
|---|---|---|---|---|---|---|
| 1 | HLA-DRB5*01:01 | MEILWHAMVGTARSP | 0.68 | 0.49 | −1.74 | −1.21 |
| 2 | HLA-DRB3*01:01 | HRDWFNDLALPWKHE | 0.65 | 0.86 | −0.44 | −0.59 |
| 3 | HLA-DRB1*15:01 | AERLVEFGVPHAVKM | 0.74 | 0.48 | −0.66 | −1.49 |
| 4 | HLA-DRB1*07:01 | VLHTMWHVTRGAAIF | 0.89 | 0.53 | −0.56 | −0.95 |
| 5 | HLA-DRB3*01:01 | GELVLDTGRRIGAIP | 0.59 | 0.63 | −0.79 | −1.32 |
| S. No. | B Cell Epitopes Sequences | Binding Score | Allergenicity | Antigenicity | Toxicity (<0) (Non-Toxic) |
|---|---|---|---|---|---|
| 1 | AARCPAMGPATLDEEHQSGT | 0.96 | −1.32 | 0.85 | −0.65 |
| 2 | KISWKSWGQSMIWSVPEAPR | 0.924 | −0.61 | 0.48 | −1.32 |
| 3 | SPGLLWGHRQVGVGFGSKGV | 0.963 | −0.44 | 1.48 | −1.29 |
| 4 | SGKRVRFHSPAVGDQQTGNA | 0.771 | −0.76 | 0.75 | −0.71 |
| 5 | MTATPPGKSEPFPESNGAIT | 0.999 | −0.48 | 0.77 | −1.19 |
| 6 | VMCDIGESSPDAAIEGERTR | 0.962 | −0.93 | 0.63 | −0.49 |
| S. No. | Features | Assessment | Remarks |
|---|---|---|---|
| 1 | Amino acids No. | 375 | Suitable |
| 2 | Chemical formula | C1821H2817N517O508S19 | - |
| 3 | Molecular weight | 40.6 KDa | Average |
| 4 | Theoretical PI | 9.56 | Slightly basic |
| 7 | Instability Index (II) | 35.27 | Stable |
| 8 | Aliphatic index (AI) | 65.12 | Thermostable |
| 9 | GRAVY (Grand Average-of hydropathicity) | −0.394 | Hydrophilic |
| S. No. | Chain A | Chain B | Distance | ||
|---|---|---|---|---|---|
| Residues Name | Residues Number | Residues Name | Residues Number | ||
| 1 | GLU | 42 | ARG | 12 | 2.84 |
| 2 | GLU | 42 | ARG | 12 | 2.69 |
| 3 | GLU | 42 | CYS | 11 | 2.92 |
| 4 | ARG | 264 | THR | 88 | 2.69 |
| 5 | ARG | 264 | THR | 88 | 2.85 |
| 6 | LYS | 362 | TRP | 84 | 2.69 |
| 7 | LYS | 402 | SER | 140 | 2.67 |
| 8 | ASN | 409 | TYR | 59 | 2.74 |
| 9 | GLN | 423 | ASN | 142 | 2.74 |
| 10 | GLU | 425 | SER | 140 | 2.96 |
| 11 | GLU | 425 | ASN | 142 | 3.04 |
| 12 | ASN | 433 | ARG | 67 | 3.03 |
| 13 | ASN | 433 | VAL | 63 | 2.72 |
| 14 | LEU | 434 | ARG | 67 | 2.50 |
| 15 | LYS | 435 | LEU | 66 | 2.80 |
| 16 | ASN | 448 | ASN | 142 | 2.67 |
| 17 | ASP | 453 | TRP | 116 | 2.98 |
| 18 | HIS | 458 | ARG | 67 | 2.62 |
| 19 | HIS | 458 | ARG | 67 | 2.55 |
| 20 | HIS | 458 | VAL | 63 | 2.94 |
| 21 | LYS | 477 | GLY | 115 | 2.69 |
| 22 | THR | 499 | TYR | 122 | 3.17 |
| 23 | GLN | 505 | THR | 112 | 2.82 |
| 24 | GLN | 523 | TYR | 122 | 2.88 |
| Salt Bridges between TLR4 and vaccine Molecules | |||||
| 1 | GLU | 42 | ARG | 12 | 2.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alnuqaydan, A.M.; Eisa, A.A. Targeting Polyprotein to Design Potential Multiepitope Vaccine against Omsk Hemorrhagic Fever Virus (OHFV) by Evaluating Allergenicity, Antigenicity, and Toxicity Using Immunoinformatic Approaches. Biology 2024, 13, 738. https://doi.org/10.3390/biology13090738
Alnuqaydan AM, Eisa AA. Targeting Polyprotein to Design Potential Multiepitope Vaccine against Omsk Hemorrhagic Fever Virus (OHFV) by Evaluating Allergenicity, Antigenicity, and Toxicity Using Immunoinformatic Approaches. Biology. 2024; 13(9):738. https://doi.org/10.3390/biology13090738
Chicago/Turabian StyleAlnuqaydan, Abdullah M., and Alaa Abdulaziz Eisa. 2024. "Targeting Polyprotein to Design Potential Multiepitope Vaccine against Omsk Hemorrhagic Fever Virus (OHFV) by Evaluating Allergenicity, Antigenicity, and Toxicity Using Immunoinformatic Approaches" Biology 13, no. 9: 738. https://doi.org/10.3390/biology13090738
APA StyleAlnuqaydan, A. M., & Eisa, A. A. (2024). Targeting Polyprotein to Design Potential Multiepitope Vaccine against Omsk Hemorrhagic Fever Virus (OHFV) by Evaluating Allergenicity, Antigenicity, and Toxicity Using Immunoinformatic Approaches. Biology, 13(9), 738. https://doi.org/10.3390/biology13090738