
Genome-Wide Scans for Selection Signatures in Haimen Goats Reveal Candidate Genes Associated with Growth Traits
 , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Measurement of Phenotype
2.2. Genome Sequencing, Variant Calling, and Annotation
2.3. Identification of Selection Signatures
2.4. Primer Design and Amplification
2.5. SNP/Indel Verification and Correlation Analysis
2.6. HE and ORO Staining
2.7. Western Blotting Assay
2.8. Statistical Analysis
3. Results
3.1. Sequencing and Variation Calling
3.2. Population Genetic Structure
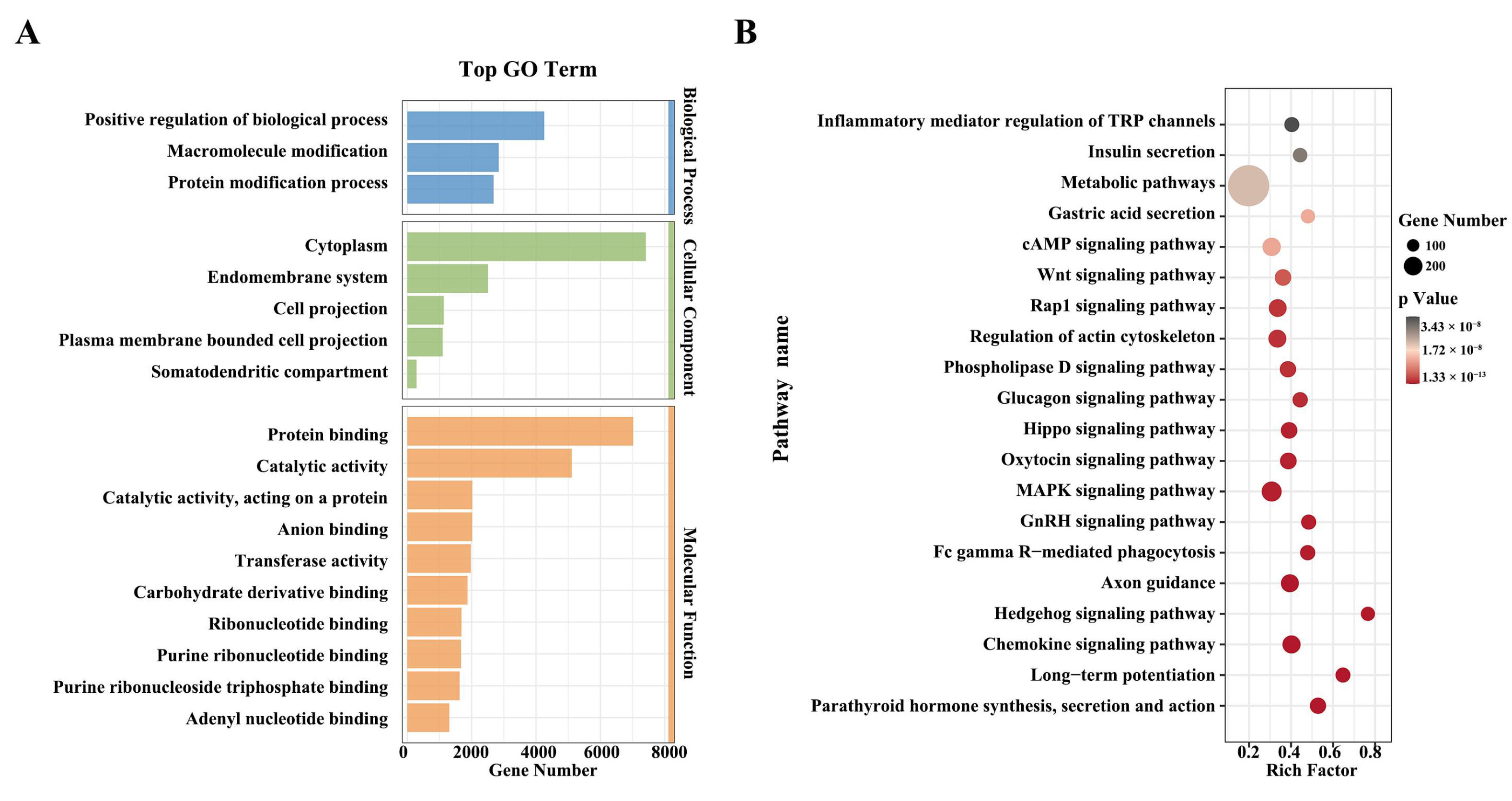
3.3. Functional Enrichment Analysis of Selected Candidate Regions
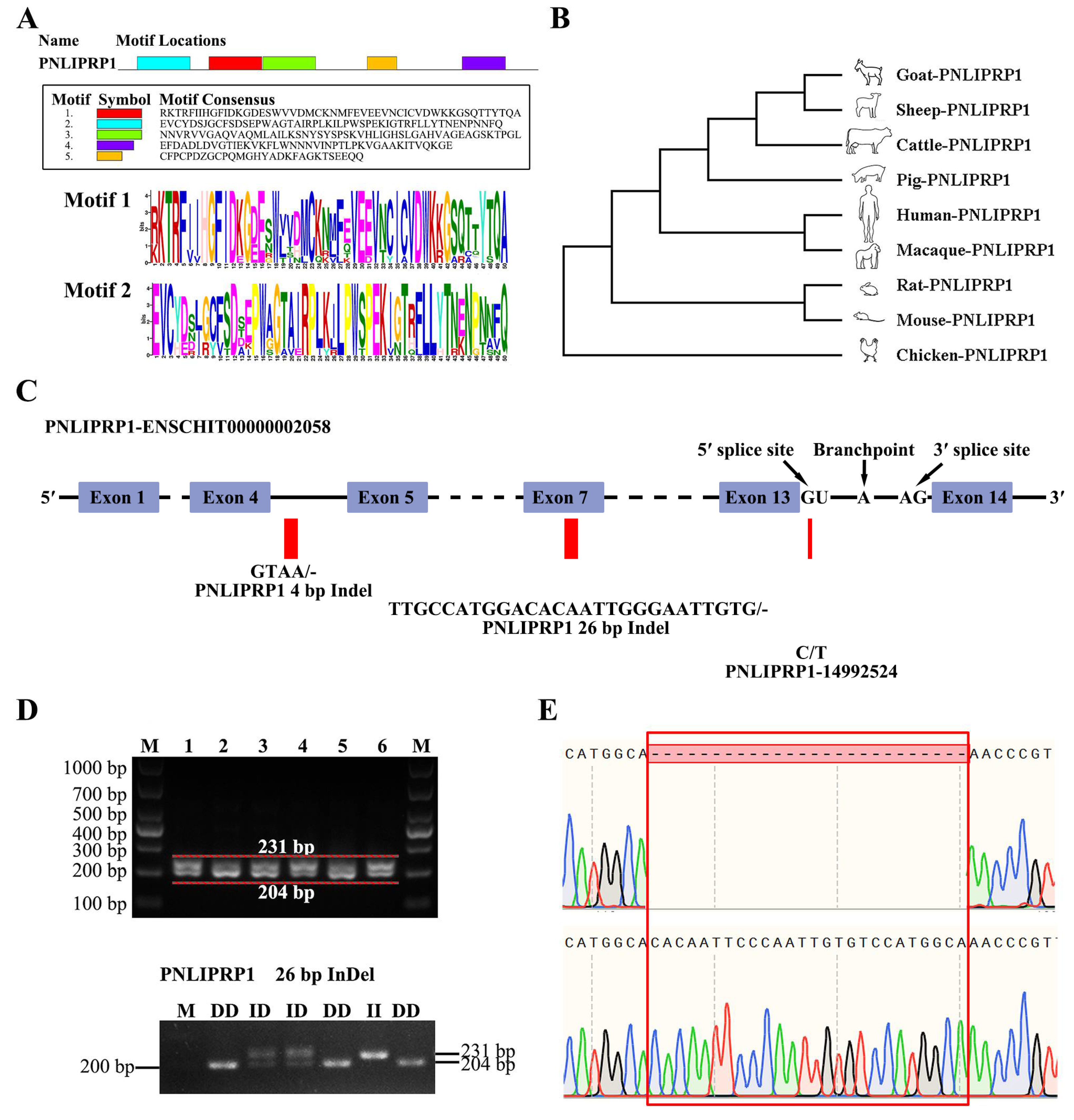
3.4. Identification and Analysis of the PNLIPRP1 Gene
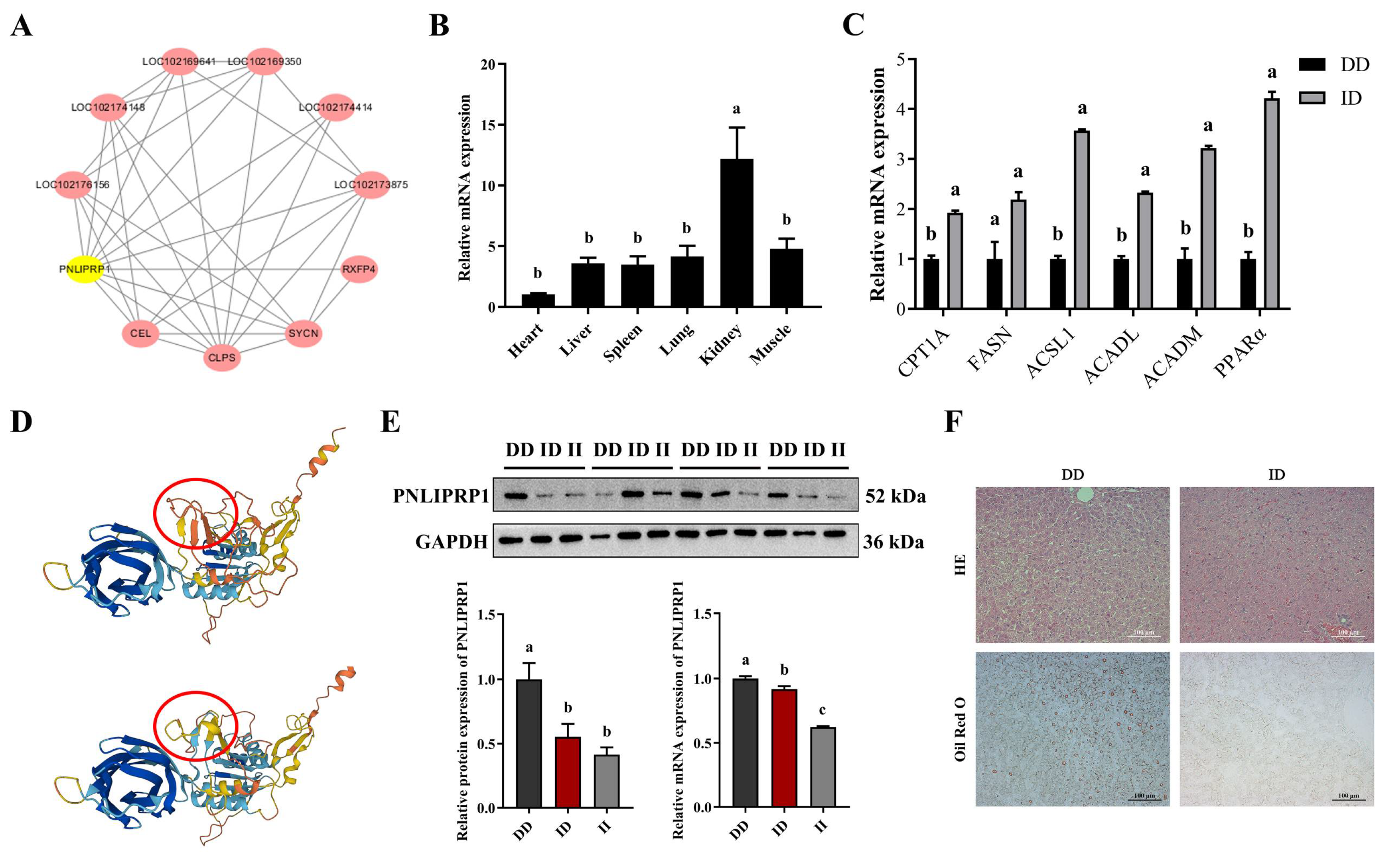
3.5. Effect of PNLIPRP1 Gene Polymorphism on Goat Growth Traits
3.6. Impact of PNLIPRP1 on Hepatic Fat Deposition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corva, P.M.; Medrano, J.F. Quantitative trait loci (QTLs) mapping for growth traits in the mouse: A review. Genet. Sel. Evol. 2001, 33, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Guo, J.; Li, L.; Zhong, T.; Wang, L.; Zhan, S.; Lu, J.; Wang, D.; Dai, D.; Liu, G.E.; et al. Genetic Diversity and Selection Signatures in Jianchang Black Goats Revealed by Whole-Genome Sequencing Data. Animals 2022, 12, 2365. [Google Scholar] [CrossRef] [PubMed]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Ankersmit-Udy, A.; Turner, S.-A.; Scholtens, M.; Littlejohn, M.D.; Lopez-Villalobos, N.; Proser, C.G.; Snell, R.G.; Lehnert, K. A Capra hircus chromosome 19 locus linked to milk production influences mammary conformation. J. Anim. Sci. Biotechnol. 2022, 13, 4–13. [Google Scholar] [CrossRef]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef]
- Cadzow, M.; Boocock, J.; Nguyen, H.T.; Wilcox, P.; Merriman, T.R.; Black, M.A. A bioinformatics workflow for detecting signatures of selection in genomic data. Front. Genet. 2014, 5, 293–300. [Google Scholar] [CrossRef]
- Cao, X.; Ling, C.; Liu, Y.; Gu, Y.; Huang, J.; Sun, W. Pleiotropic Gene HMGA2 Regulates Myoblast Proliferation and Affects Body Size of Sheep. Animals 2024, 14, 2721. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, X.; Zhao, Z.; Li, J.; Albrecht, E.; Schering, L.; Maak, S.; Yang, R. Polymorphisms of the ASIP gene and the haplotype are associated with fat deposition traits and fatty acid composition in Chinese Simmental steers. Arch. Anim. Breed. 2019, 62, 135–142. [Google Scholar] [CrossRef]
- Rubin, C.-J.; Megens, H.-J.; Barrio, A.M.; Maqbool, K.; Sayyab, S.; Schwochow, D.; Wang, C.; Carlborg, Ö.; Jern, P.; Jørgensen, C.B.; et al. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA 2012, 109, 19529–19536. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, W.; Liu, C.; He, Y.; Zhang, H.; Xu, L.; Yang, B.; Zhao, Y.; Ma, Y.; Chu, M.; et al. Genome-Wide Selective Analysis of Boer Goat to Investigate the Dynamic Heredity Evolution under Different Stages. Animals 2022, 12, 1356. [Google Scholar] [CrossRef]
- Lestari, D.A.; Oikawa, T.; Sutopo, S.; Purbowati, E.; Setiaji, A.; Kurnianto, E. Effect of insulin-like growth factor 1 gene on growth traits of Kejobong goat and its growth analysis. Vet. World 2020, 13, 127–133. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kendig, K.I.; Baheti, S.; Bockol, M.A.; Drucker, T.M.; Hart, S.N.; Heldenbrand, J.R.; Hernaez, M.; Hudson, M.E.; Kalmbach, M.T.; Klee, E.W.; et al. Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy. Front. Genet. 2019, 10, 736–742. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2014, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g: Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, M.; Qian, J.; Xu, Y.; Li, B.; Le, G.; Xie, Y. Dietary Methionine Restriction Promotes Fat Browning and Attenuates Hepatic Lipid Accumulation in High-Choline-Fed Mice Associated with the Improvement of Thyroid Function. J. Agric. Food Chem. 2023, 71, 1447–1463. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Wei, C.; Wang, H.; Liu, G.; Wu, M.; Cao, J.; Liu, Z.; Liu, R.; Zhao, F.; Zhang, L.; Lu, J.; et al. Genome-wide analysis reveals population structure and selection in Chinese indigenous sheep breeds. BMC Genom. 2015, 16, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Vlaic, B.A.; Vlaic, A.; Russo, I.-R.; Colli, L.; Bruford, M.W.; Odagiu, A.; Orozco-terWengel, P. Analysis of Genetic Diversity in Romanian Carpatina Goats Using SNP Genotyping Data. Animals 2024, 14, 560. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Chen, D.; Guo, P.; Hu, L.; Zheng, X.; Cheng, J.; Chen, H. Ecogroups and maternal haplogroups reveal the ancestral origin of native Chinese goat populations based on the variation of mtDNA D-loop sequences. Ecol. Evol. 2023, 13, e10382. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tao, H.; Li, P.; Li, L.; Zhong, T.; Wang, L.; Ma, J.; Chen, X.; Song, T.; Zhang, H. Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci. Rep. 2018, 8, 10405–10415. [Google Scholar] [CrossRef]
- Li, X.; Kumar, A.; Carmeliet, P. Metabolic Pathways Fueling the Endothelial Cell Drive. Annu. Rev. Physiol. 2019, 81, 483–503. [Google Scholar] [CrossRef]
- Ma, Z.; Jiang, K.; Wang, D.; Wang, Z.; Gu, Z.; Li, G.; Jiang, R.; Tian, Y.; Kang, X.; Li, H.; et al. Comparative analysis of hypothalamus transcriptome between laying hens with different egg-laying rates. Poult. Sci. 2021, 100, 101110–101122. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, J.; Zhou, J.; Zhang, Y.; Qiao, M.; Sun, H.; Li, Z.; Li, L.; Chen, N.; Oyelami, F.O.; et al. Integration of ATAC-seq and RNA-seq analysis identifies key genes affecting intramuscular fat content in pigs. Front. Nutr. 2022, 9, 1016956–1016967. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Zhou, G.; Guo, J.; Yan, H.; Niu, Y.; Li, Y.; Yuan, C.; Geng, R.; Lan, X.; et al. Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits. Sci. Rep. 2016, 6, 38932–38941. [Google Scholar] [CrossRef]
- Kim, E.S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.; Sayre, B.; Mwacharo, J.M.; Rothschild, M.F. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity 2015, 116, 255–264. [Google Scholar] [CrossRef]
- Yang, J.; Wang, D.-F.; Huang, J.-H.; Zhu, Q.-H.; Luo, L.-Y.; Lu, R.; Xie, X.-L.; Salehian-Dehkordi, H.; Esmailizadeh, A.; Liu, G.E.; et al. Structural variant landscapes reveal convergent signatures of evolution in sheep and goats. Genome Biol. 2024, 25, 148–193. [Google Scholar] [CrossRef]
- Li, X.; Su, R.; Wan, W.; Zhang, W.; Jiang, H.; Qiao, X.; Fan, Y.; Zhang, Y.; Wang, R.; Liu, Z.; et al. Identification of selection signals by large-scale whole-genome resequencing of cashmere goats. Sci. Rep. 2017, 7, 15142–15151. [Google Scholar] [CrossRef] [PubMed]
- Madhusoodan, A.P.; Sejian, V.; Afsal, A.; Bagath, M.; Krishnan, G.; Savitha, S.T.; Rashamol, V.P.; Devaraj, C.; Bhatta, R. Differential expression patterns of candidate genes pertaining to productive and immune functions in hepatic tissue of heat-stressed Salem Black goats. Biol. Rhythm. Res. 2019, 52, 809–820. [Google Scholar] [CrossRef]
- Gao, J.; Lyu, Y.; Zhang, D.; Reddi, K.K.; Sun, F.; Yi, J.; Liu, C.; Li, H.; Yao, H.; Dai, J.; et al. Genomic Characteristics and Selection Signatures in Indigenous Chongming White Goat (Capra hircus). Front. Genet. 2020, 11, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, G.; Jiang, Y.; Tang, J.; Fan, Y.; Ren, J. Genomic insights into the conservation and population genetics of two Chinese native goat breeds. J. Anim. Sci. 2022, 100, 1–17. [Google Scholar] [CrossRef]
- Brooker, R.M.; Feeney, W.E. Animal domesticators. Curr. Biol. 2019, 29, R1168–R1169. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Lam, S.M.; Chen, L.; Gao, Y.; Wang, W.; Xu, Y.; Tan, T.; Yu, H.; Zhang, M.; et al. Low-input lipidomics reveals lipid metabolism remodelling during early mammalian embryo development. Nat. Cell Biol. 2024, 26, 278–293. [Google Scholar] [CrossRef]
- Assareh, N.; Shahemabadi, M.; Varnosfaderani, S.R.; Jafarpour, F.; Hajian, M.; Nasr-Esfahani, M.H. Sequential IVM by CNP preincubation and cooperating of PGE2 with AREG enhances developmental competence of SCNT reconstructs in goat. Sci. Rep. 2022, 12, 4243–4254. [Google Scholar] [CrossRef]
- Maurin, L.; Marselli, L.; Boissel, M.; Ning, L.; Boutry, R.; Fernandes, J.; Suleiman, M.; Luca, C.D.; Leloire, A.; Pascat, V.; et al. PNLIPRP1 hypermethylation in exocrine pancreas links type 2 diabetes and cholesterol metabolism. Diabetes 2024, 73, 1908–1918. [Google Scholar] [CrossRef]
- van Es, M.A.; van Vught, P.W.J.; Blauw, H.M.; Franke, L.; Saris, C.G.J.; Van Den Bosch, L.; de Jong, S.W.; de Jong, V.; Baas, F.; van’t Slot, R.; et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2007, 40, 29–31. [Google Scholar] [CrossRef]
- Hansen, P.J.; Pareek, C.S.; Smoczyński, R.; Kadarmideen, H.N.; Dziuba, P.; Błaszczyk, P.; Sikora, M.; Walendzik, P.; Grzybowski, T.; Pierzchała, M.; et al. Single Nucleotide Polymorphism Discovery in Bovine Pituitary Gland Using RNA-Seq Technology. PLoS ONE 2016, 11, e0161370. [Google Scholar]
- Ouzounis, C.A.; Zhang, Y.; Zagnitko, O.; Rodionova, I.; Osterman, A.; Godzik, A. The FGGY Carbohydrate Kinase Family: Insights into the Evolution of Functional Specificities. PLoS Comput. Biol. 2011, 7, e1002318. [Google Scholar]
- Santana, M.H.A.; Utsunomiya, Y.T.; Neves, H.H.R.; Gomes, R.C.; Garcia, J.F.; Fukumasu, H.; Silva, S.L.; Leme, P.R.; Coutinho, L.L.; Eler, J.P.; et al. Genome-wide association study for feedlot average daily gain in Nellore cattle (Bos indicus). J. Anim. Breed. Genet. 2014, 131, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L.; Shi, L.; Zhang, P.; Li, Y.; Li, M.; Tian, J.; Wang, L.; Zhao, F. GWAS of Reproductive Traits in Large White Pigs on Chip and Imputed Whole-Genome Sequencing Data. Int. J. Mol. Sci. 2022, 23, 13338. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Han, B.; Li, X.; Liu, D.; Zhou, B.; Zhao, C.; Zhang, N.; Wang, L.; Pei, Q.; Zhao, K. Genetic diversity and selection of Tibetan sheep breeds revealed by whole-genome resequencing. Anim. Biosci. 2023, 36, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Cendron, F.; Sottile, G.; Niero, G.; Portolano, B.; Biscarini, F.; Cassandro, M. Genome-Wide Analyses Identifies Known and New Markers Responsible of Chicken Plumage Color. Animals 2020, 10, 493. [Google Scholar] [CrossRef]
- Light, S.; Sagit, R.; Sachenkova, O.; Ekman, D.; Elofsson, A. Protein Expansion Is Primarily due to Indels in Intrinsically Disordered Regions. Mol. Biol. Evol. 2013, 30, 2645–2653. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Autosome | Variant | Gene | Position | Reference | Alternate Base | Type |
|---|---|---|---|---|---|---|---|
| rs652752535 | 26 | SNP | PNLIPRP1 | 14992524 | C | T | Splice donor variant and Intron variant |
| rs654084728 | 19 | SNP | NCOR1 | 33085630 | G | A | Missense variant |
| DPP6 g.3352780G>A | 4 | SNP | DPP6 | 3352780 | G | A | Splice donor variant and Intron variant |
| NOTCH2 g.97297615G>C | 3 | SNP | NOTCH2 | 97297615 | G | C | Missense variant |
| rs636776710 | 3 | SNP | COL6A3 | 3897592 | T | G | Missense variant |
| rs641287990 | 3 | SNP | FGGY | 34104364 | C | T | Missense variant |
| PNLIPRP1 26 bp Indel | 26 | Indel | PNLIPRP1 | 14998429 | TTGCCATGGACACAAT TGGGAATTGTG | - | Frameshift variant |
| PNLIPRP1 4 bp Indel | 26 | Indel | PNLIPRP1 | 15000823 | GTAA | - | Splice region variant |
| Locus | Genotypic Frequencies | Allelic Frequencies | Diversity Parameters | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ho | He | Ne | PIC | Hardy-Weinberg | ||||||
| rs652752535 | TT | TC | CC | T | C | |||||
| 0.7602 (n = 447) | 0.2177 (n = 128) | 0.0221 (n = 13) | 0.8690 | 0.1310 | 0.7724 | 0.2276 | 1.2947 | 0.2017 | 0.2905 | |
| PNLIPRP1 26 bp Indel | DD | ID | II | D | I | |||||
| 0.6034 (n = 353) | 0.3504 (n = 205) | 0.0462 (n = 27) | 0.7786 | 0.2214 | 0.6553 | 0.3447 | 1.5261 | 0.2853 | 0.6892 | |
| Locus | Genotype | BW (kg) | BL (cm) | BH (cm) | CC (cm) | CD (cm) | CW (cm) |
|---|---|---|---|---|---|---|---|
| rs652752535 | TC | 10.7 ± 0.44 a | 43.0 ± 0.78 | 44.0 ± 0.71 | 50.5 ± 1.03 | 18.9 ± 0.47 | 14.2 ± 0.34 |
| TT | 9.4 ± 0.29 b | 42.9 ± 0.51 | 44.3 ± 0.46 | 48.9 ± 0.67 | 18.2 ± 0.31 | 13.9 ± 0.22 | |
| CC | 7.5 ± 1.42 b | 39.9 ± 2.51 | 40.9 ± 2.27 | 49.1 ± 3.31 | 18.3 ± 1.51 | 13.3 ± 1.10 | |
| PNLIPRP1 26 bp Indel | DD | 9.4 ± 0.55 a | 43.8 ± 1.24 a | 45.9 ± 1.06 a | 49.1 ± 1.41 a | 18.1 ± 0.57 | 13.6 ± 0.42 |
| ID | 8.3 ± 0.61 ab | 41.4 ± 1.36 b | 43.8 ± 1.16 b | 47.4 ± 1.54 ab | 17.7 ± 0.62 | 13.3 ± 0.47 | |
| II | 7.9 ± 0.98 b | 37.6 ± 2.21 b | 41.2 ± 1.89 b | 43.3 ± 2.51 b | 17.4 ± 1.01 | 13.1 ± 0.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Lu, J.; Wang, Y.; Liu, Z.; Li, D.; Deng, K.; Zhang, G.; Zhao, B.; You, P.; Fan, Y.; et al. Genome-Wide Scans for Selection Signatures in Haimen Goats Reveal Candidate Genes Associated with Growth Traits. Biology 2025, 14, 40. https://doi.org/10.3390/biology14010040
Zhang Z, Lu J, Wang Y, Liu Z, Li D, Deng K, Zhang G, Zhao B, You P, Fan Y, et al. Genome-Wide Scans for Selection Signatures in Haimen Goats Reveal Candidate Genes Associated with Growth Traits. Biology. 2025; 14(1):40. https://doi.org/10.3390/biology14010040
Chicago/Turabian StyleZhang, Zhen, Jiafeng Lu, Yifei Wang, Zhipeng Liu, Dongxu Li, Kaiping Deng, Guomin Zhang, Bingru Zhao, Peihua You, Yixuan Fan, and et al. 2025. "Genome-Wide Scans for Selection Signatures in Haimen Goats Reveal Candidate Genes Associated with Growth Traits" Biology 14, no. 1: 40. https://doi.org/10.3390/biology14010040
APA StyleZhang, Z., Lu, J., Wang, Y., Liu, Z., Li, D., Deng, K., Zhang, G., Zhao, B., You, P., Fan, Y., Wang, F., & Wang, Z. (2025). Genome-Wide Scans for Selection Signatures in Haimen Goats Reveal Candidate Genes Associated with Growth Traits. Biology, 14(1), 40. https://doi.org/10.3390/biology14010040