


Druggable Molecular Networks in BRCA1/BRCA2-Mutated Breast Cancer

 ,
,  and
and 
Simple Summary
Abstract

1. Introduction
1.1. Breast Cancer: Overview
1.2. Genetic Causes of BC
2. Materials and Methods
3. Relationship Between BRCA1/2 Mutations, Gene Expression, and Biological Processes
3.1. BRCA Mutations Interfere with Homologous Recombination and Cell Cycle
3.2. BRCA1/2 Modulate Transcription of Specific Genes
3.3. Differences Between BRCA1 and BRCA2 Mutations in Trascriptional Regulation and Dysregulation of Cell Cycle
4. Targeted Therapies
4.1. Genes Involved in BC Resistance to Treatment
4.2. Synthetic Lethality: PARPi for BRCA-Mutated Tumors
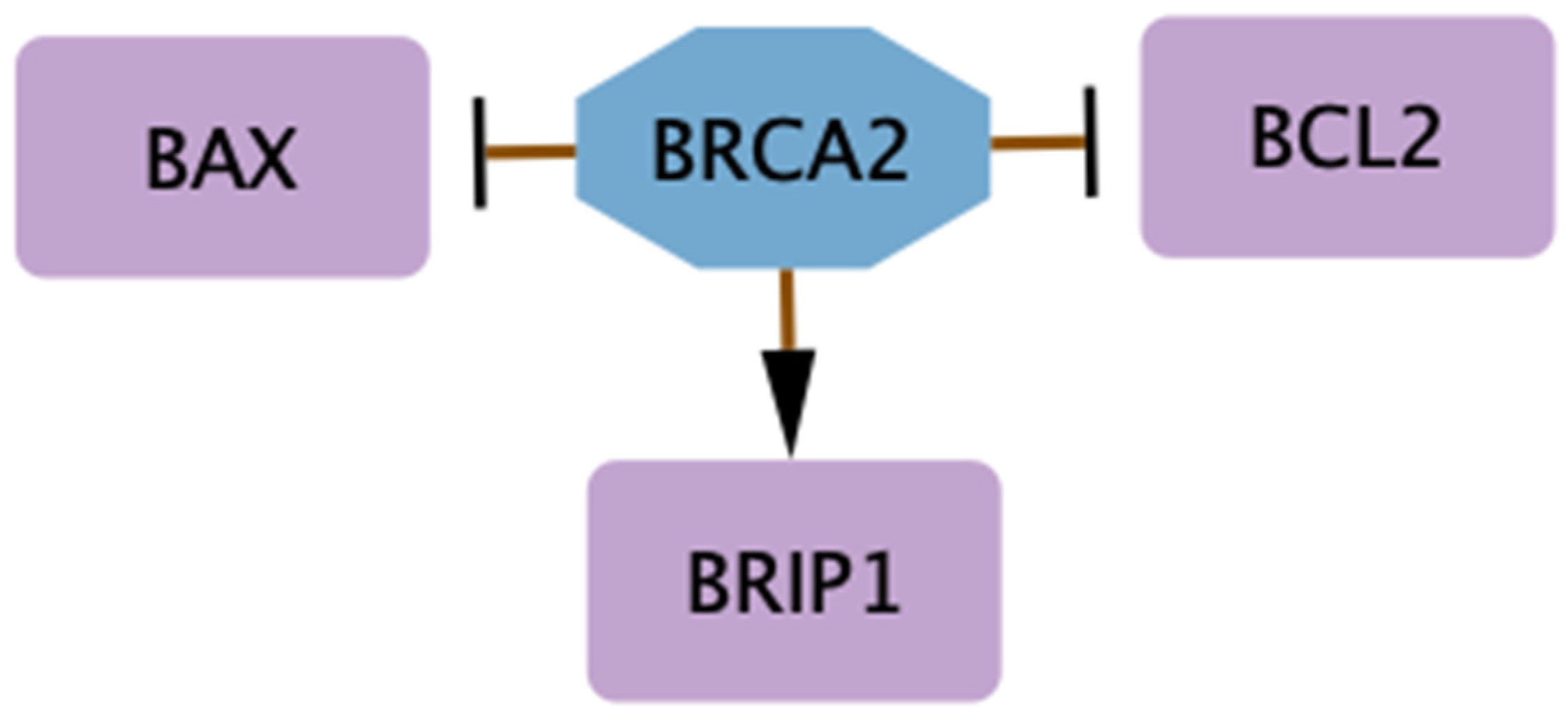
4.3. BRCA2 Deficiency, Genome Stability, and Sensitivity to Phytoestrogens and Radiation
5. Single Cell Analysis: BRCA1 and TP53 Expression in BRCA1-Mutated Models
5.1. BRCA1 Mutation and the Cellular Origins of BC: Insights from Luminal Progenitors
5.2. Aberrant Alveolar Differentiation in Luminal Progenitors Drives Early Tumorigenesis in BRCA1/p53 Models
6. Novel Therapies Suggested in the Management of BRCA1-Mutated Tumors
6.1. Efficacy of BET Inhibitors and Their Effects on BRCA1 Deficient Cells
6.2. CDDO Treatment Impacts Survivin Expression
6.3. Resveratrol Modulates BIRC5 and SIRT1 in BRCA1-Mutated Models
6.4. EZH2 Differential Expression in BRCA1-Mutated BC After DZNep Exposure
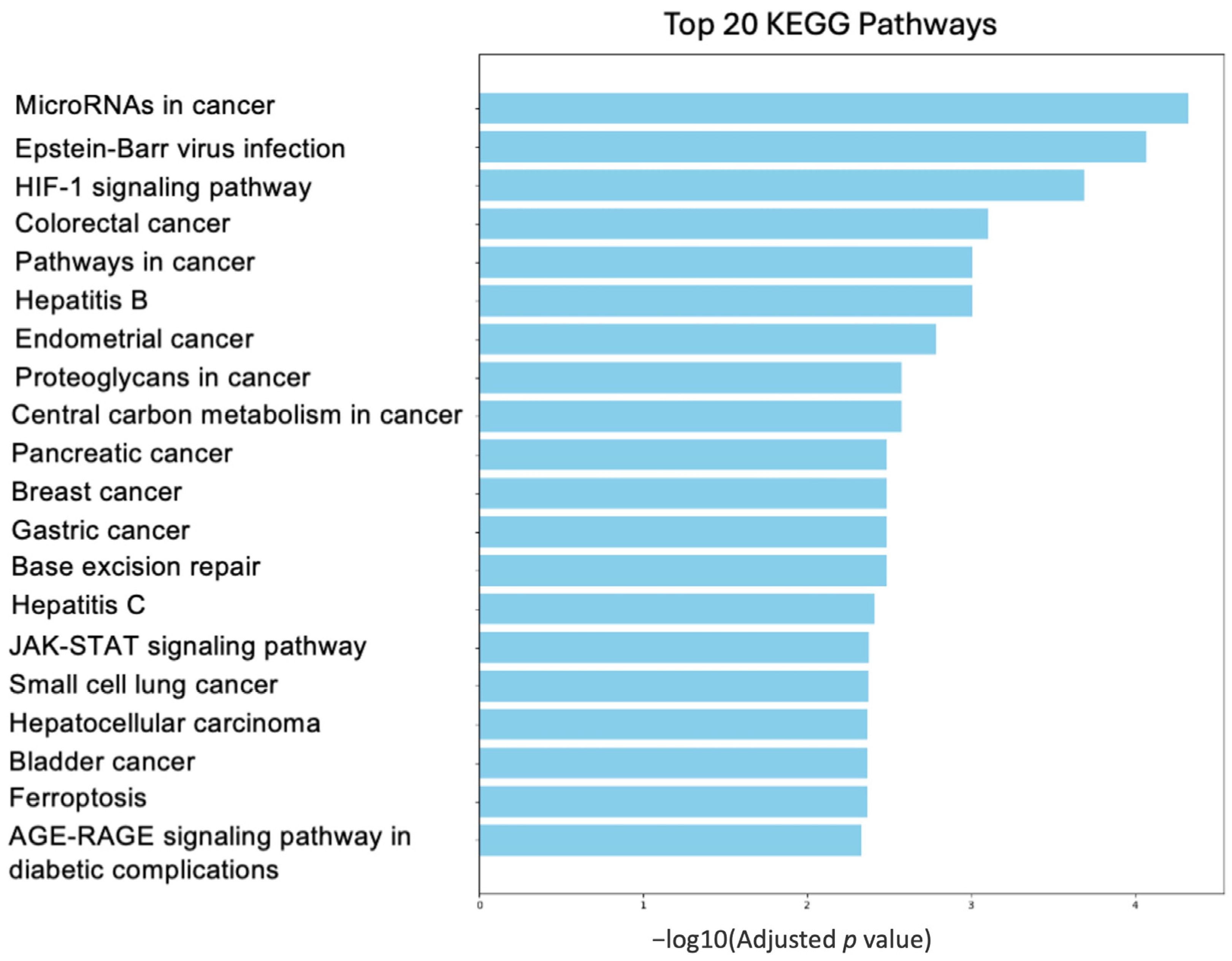
7. Pathway Involving Deregulated Genes by BRCA1
7.1. Proteoglycan Pathway
7.2. Ferroptosis Pathway
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 15 November 2024).
- Godet, I.; Gilkes, D.M. BRCA1 and BRCA2 Mutations and Treatment Strategies for Breast Cancer. Integr. Cancer Ther. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Cserni, G.; Chmielik, E.; Cserni, B.; Tot, T. The new TNM-based staging of breast cancer. Virchows Arch. Int. J. Pathol. 2018, 472, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Orrantia-Borunda, E.; Anchondo-Nuñez, P.; Acuña-Aguilar, L.E.; Gómez-Valles, F.O.; Ramírez-Valdespino, C.A. Subtypes of breast cancer. In Breast Cancer; Mayrovitz, H.N., Ed.; Exon Publications: Brisbane, Australia, 2022; Chapter 3. [Google Scholar]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Liu, Y.-R.; Jiang, Y.-Z.; Xu, X.-E.; Yu, K.-D.; Jin, X.; Hu, X.; Zuo, W.-J.; Hao, S.; Wu, J.; Liu, G.-Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Sarhangi, N.; Hajjari, S.; Heydari, S.F.; Ganjizadeh, M.; Rouhollah, F.; Hasanzad, M. Breast Cancer in the Era of precision Medicine. Mol. Biol. Rep. 2022, 49, 10023–10037. [Google Scholar] [CrossRef]
- Gradishar, W.; Salerno, K.E. NCCN Guidelines Update: Breast Cancer. J. Natl. Compr. Cancer Netw. 2016, 14, 641–644. [Google Scholar] [CrossRef]
- Krishnamurti, U.; Silverman, J.F. HER2 in Breast Cancer: A Review and Update. Adv. Anat. Pathol. 2014, 21, 100–107. [Google Scholar] [CrossRef]
- Korde, L.A.; Somerfield, M.R.; Carey, L.A.; Crews, J.R.; Denduluri, N.; Hwang, E.S.; Khan, S.A.; Loibl, S.; Morris, E.A.; Perez, A.; et al. Neoadjuvant Chemotherapy, Endocrine Therapy, and Targeted Therapy for Breast Cancer: ASCO Guideline. J. Clin. Oncol. 2021, 39, 1485–1505. [Google Scholar] [CrossRef]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 610303. [Google Scholar] [CrossRef] [PubMed]
- De Santis, P.; Perrone, M.; Guarini, C.; Santoro, A.N.; Laface, C.; Carrozzo, D.; Oliva, G.R.; Fedele, P. Early-stage triple negative breast cancer: The therapeutic role of immunotherapy and the prognostic value of pathological complete response. Explor. Target. Antitumor Ther. 2024, 5, 232–250. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Li, Y.; Tan, Y.; Fu, S.; Tang, F.; Deng, X. Immune Checkpoint Inhibition for Triple-Negative Breast Cancer: Current Landscape and Future Perspectives. Front. Oncol. 2021, 11, 648139. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Speers, C.; Nyati, S. Present and Future of Immunotherapy for Triple-Negative Breast Cancer. Cancers 2024, 16, 3250. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 Expression in Triple-Negative Breast Cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Peleg Hasson, S.; Menes, T.; Sonnenblick, A. Comparison of Patient Susceptibility Genes Across Breast Cancer: Implications for Prognosis and Therapeutic Outcomes. Pharmacogenom. Pers. Med. 2020, 13, 227–238. [Google Scholar] [CrossRef]
- Marvali, C.; Datta, A.; Lee, S.C. Role of p53 in breast cancer progression: An insight into p53 targeted therapy. Theranostics 2023, 13, 1421–1442. [Google Scholar] [CrossRef]
- Rocca, V.; Blandino, G.; D’Antona, L.; Iuliano, R.; Di Agostino, S. Li-Fraumeni syndrome: Mutation of TP53 is a biomarker of hereditary predisposition to tumor: New insights and advances in the treatment. Cancers 2022, 14, 3664. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Kenzelmann Broz, D.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.K.; Levine, A.J. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc. Natl. Acad. Sci. USA 1996, 93, 15335–15340. [Google Scholar] [CrossRef]
- Berger, M.; Vogt Sionov, R.; Levine, A.J.; Haupt, Y. A role for the polyproline domain of p53 in its regulation by Mdm2. J. Biol. Chem. 2001, 276, 3785–3790. [Google Scholar] [CrossRef]
- el-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Kandil, A.N. Conformational shifts propagate from the oligomerization domain of p53 to its tetrameric DNA binding domain and restore DNA binding to select p53 mutants. EMBO J. 1993, 12, 5057–5064. [Google Scholar] [CrossRef]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Ponti, G.; De Angelis, C.; Ponti, R.; Pongetti, L.; Losi, L.; Sticchi, A.; Tomasi, A.; Ozben, T. Hereditary breast and ovarian cancer: From genes to molecular targeted therapies. Crit. Rev. Clin. Lab. Sci. 2023, 60, 640–650. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Marks, J.H.; Mandell, J.B.; New York Breast Cancer Study Group. Breast and ovarian cancer risk due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Moreno, M.; Oliveira, J.S.; Brianese, R.C.; de Castro, D.G.; Sanches, S.M.; Torrezan, G.T.; Santiago, K.M.; De Brot, M.; Cordeiro de Lima, V.C.; Baroni Alves Makdissi, F.; et al. Risk of Metastasis in BRCA2 Carriers Diagnosed with Triple-Negative Breast Cancer. Cancer Med. 2023, 12, 16129–16141. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-Cancer Risk in Families with Mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Tischkowitz, M.; Xia, B. PALB2/FANCN: Recombining Cancer and Fanconi Anemia. Cancer Res. 2010, 70, 7353–7359. [Google Scholar] [CrossRef]
- Catucci, I.; Milgrom, R.; Kushnir, A.; Laitman, Y.; Paluch-Shimon, S.; Volorio, S.; Ficarazzi, F.; Bernard, L.; Radice, P.; Friedman, E. Germline Mutations in BRIP1 and PALB2 in Jewish High Cancer Risk Families. Fam. Cancer 2012, 11, 483–491. [Google Scholar] [CrossRef]
- Yoshida, K.; Miki, Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004, 95, 866–871. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, D. Repair Pathway Choice for Double-Strand Breaks. Essays Biochem. 2020, 64, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Yarden, R.I.; Metsuyanim, S.; Pickholtz, I.; Shabbeer, S.; Tellio, H.; Papa, M.Z. BRCA1-Dependent Chk1 Phosphorylation Triggers Partial Chromatin Disassociation of Phosphorylated Chk1 and Facilitates S-Phase Cell Cycle Arrest. Int. J. Biochem. Cell Biol. 2012, 44, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.J.; Soares, D.G.; Bouzid, H.; Henriques, J.A.P.; Larsen, A.K.; Escargueil, A.E. BRCA2 Is Needed for Both Repair and Cell Cycle Arrest in Mammalian Cells Exposed to S23906, an Anticancer Monofunctional DNA Binder. Cell Cycle 2015, 14, 2080–2090. [Google Scholar] [CrossRef]
- Marmorstein, L.Y.; Kinev, A.V.; Chan, G.K.; Bochar, D.A.; Beniya, H.; Epstein, J.A.; Yen, T.J.; Shiekhattar, R. A Human BRCA2 Complex Containing a Structural DNA Binding Component Influences Cell Cycle Progression. Cell 2001, 104, 247–257. [Google Scholar] [CrossRef]
- Yarden, R.I.; Pardo-Reoyo, S.; Sgagias, M.; Cowan, K.H.; Brody, L.C. BRCA1 Regulates the G2/M Checkpoint by Activating Chk1 Kinase upon DNA Damage. Nat. Genet. 2002, 30, 285–289. [Google Scholar] [CrossRef]
- Hutchins, J.R.A.; Clarke, P.R. Many Fingers on the Mitotic Trigger: Post-Translational Regulation of the Cdc25C Phosphatase. Cell Cycle 2004, 3, 41–45. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, R.S.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional Interactions between BRCA1 and the Checkpoint Kinase ATR during Genotoxic Stress. Genes Dev. 2000, 14, 2989–3002. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.X. BRCA1: Cell Cycle Checkpoint, Genetic Instability, DNA Damage Response and Cancer Evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef]
- Deng, C.X.; Brodie, S.G. Roles of BRCA1 and Its Interacting Proteins. Bioessays 2000, 22, 728–737. [Google Scholar] [CrossRef]
- Bièche, I.; Lidereau, R. Genetic Alterations in Breast Cancer. Genes Chromosomes Cancer 1995, 14, 227–251. [Google Scholar] [CrossRef]
- Guy, C.T.; Webster, M.A.; Schaller, M.; Parsons, T.J.; Cardiff, R.D.; Muller, W.J. Expression of the Neu Protooncogene in the Mammary Epithelium of Transgenic Mice Induces Metastatic Disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10578–10582. [Google Scholar] [CrossRef]
- Opitz, O.G.; Nakagawa, H.; Rustgi, A.K. Cyclin D1 Transgenic Mouse Models. Tumor Models in Cancer Research. In Cancer Drug Discovery and Development; Teicher, B.A., Ed.; Humana Press: Totowa, NJ, USA, 2002; pp. 223–230. [Google Scholar] [CrossRef]
- Brodie, S.G.; Xu, X.; Qiao, W.; Li, W.M.; Cao, L.; Deng, C.X. Multiple Genetic Changes Are Associated with Mammary Tumorigenesis in Brca1 Conditional Knockout Mice. Oncogene 2001, 20, 7514–7523. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Muller, W.J. Transgenic Mouse Models of Human Breast Cancer. Oncogene 2000, 19, 6130–6137. [Google Scholar] [CrossRef]
- Leder, A.; Pattengale, P.K.; Kuo, A.; Stewart, T.A.; Leder, P. Consequences of Widespread Deregulation of the C-Myc Gene in Transgenic Mice: Multiple Neoplasms and Normal Development. Cell 1986, 45, 485–495. [Google Scholar] [CrossRef]
- Zhang, H.; Somasundaram, K.; Peng, Y.; Tian, H.; Zhang, H.; Bi, D.; Weber, B.L.; El-Deiry, W.S. BRCA1 Physically Associates with P53 and Stimulates Its Transcriptional Activity. Oncogene 1998, 16, 1713–1721. [Google Scholar] [CrossRef]
- Chai, Y.L.; Cui, J.; Shao, N.; Shyam, E.; Reddy, P.; Rao, V.N. The Second BRCT Domain of BRCA1 Proteins Interacts with P53 and Stimulates Transcription from the p21WAF1/CIP1 Promoter. Oncogene 1999, 18, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, K.; Zhang, H.; Zeng, Y.X.; Houvras, Y.; Peng, Y.; Zhang, H.; Wu, G.S.; Licht, J.D.; Weber, B.L.; El-Deiry, W.S. Arrest of the Cell Cycle by the Tumour-Suppressor BRCA1 Requires the CDK-Inhibitor p21WAF1/CiP1. Nature 1997, 389, 187–190. [Google Scholar] [CrossRef]
- Shen, S.X.; Weaver, Z.; Xu, X.; Li, C.; Weinstein, M.; Chen, L.; Guan, X.Y.; Ried, T.; Deng, C.X. A Targeted Disruption of the Murine Brca1 Gene Causes Gamma-Irradiation Hypersensitivity and Genetic Instability. Oncogene 1998, 17, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- De Brakeleer, S.; De Grève, J.; Desmedt, C.; Joris, S.; Sotiriou, C.; Piccart, M.; Pauwels, I.; Teugels, E. Frequent incidence of BARD1-truncating mutations in germline DNA from triple-negative breast cancer patients. Clin. Genet. 2016, 89, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Xu, A.; Gingold, J.; Strong, L.C.; Zhao, R.; Lee, D.F. Li-Fraumeni Syndrome Disease Model: A Platform to Develop Precision Cancer Therapy Targeting Oncogenic p53. Trends Pharmacol. Sci. 2017, 38, 908–927. [Google Scholar] [CrossRef]
- Xu, X.; Qiao, W.; Linke, S.P.; Cao, L.; Li, W.M.; Furth, P.A.; Harris, C.C.; Deng, C.X. Genetic Interactions between Tumor Suppressors Brca1 and P53 in Apoptosis, Cell Cycle and Tumorigenesis. Nat. Genet. 2001, 28, 266–271. [Google Scholar] [CrossRef]
- Murray, M.M.; Mullan, P.B.; Harkin, D.P. Role Played by BRCA1 in Transcriptional Regulation in Response to Therapy. Biochem. Soc. Trans. 2007, 35, 1342–1346. [Google Scholar] [CrossRef]
- Preobrazhenska, O.; Yakymovych, M.; Kanamoto, T.; Yakymovych, I.; Stoika, R.; Heldin, C.-H.; Souchelnytskyi, S. BRCA2 and Smad3 Synergize in Regulation of Gene Transcription. Oncogene 2002, 21, 5660–5664. [Google Scholar] [CrossRef]
- Landberg, G. Multiparameter Analyses of Cell Cycle Regulatory Proteins in Human Breast Cancer: A Key to Definition of Separate Pathways in Tumorigenesis. Adv. Cancer Res. 2002, 84, 35–56. [Google Scholar] [CrossRef]
- Armes, J.E.; Trute, L.; White, D.; Southey, M.C.; Hammet, F.; Tesoriero, A.; Hutchins, A.M.; Dite, G.S.; McCredie, M.R.; Giles, G.G.; et al. Distinct Molecular Pathogeneses of Early-Onset Breast Cancers in BRCA1 and BRCA2 Mutation Carriers: A Population-Based Study. Cancer Res. 1999, 59, 2011–2017. [Google Scholar]
- Palacios, J.; Honrado, E.; Osorio, A.; Cazorla, A.; Sarrió, D.; Barroso, A.; Rodríguez, S.; Cigudosa, J.C.; Diez, O.; Alonso, C. Phenotypic Characterization of BRCA1 and BRCA2 Tumors Based in a Tissue Microarray Study with 37 Immunohistochemical Markers. Breast Cancer Res. Treat. 2005, 90, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Hedenfalk, I.; Duggan, D.; Chen, Y.; Radmacher, M.; Bittner, M.; Simon, R.; Meltzer, P.; Gusterson, B.; Esteller, M.; Kallioniemi, O.P.; et al. Gene-Expression Profiles in Hereditary Breast Cancer. N. Engl. J. Med. 2001, 344, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, Which Encodes a BRCA2-Interacting Protein, Is a Breast Cancer Susceptibility Gene. Nat. Genet. 2007, 39, 165–167. [Google Scholar] [CrossRef]
- Casadei, S.; Norquist, B.M.; Walsh, T.; Stray, S.; Mandell, J.B.; Lee, M.K.; Stamatoyannopoulos, J.A.; King, M.C. Contribution of Inherited Mutations in the BRCA2-Interacting Protein PALB2 to Familial Breast Cancer. Cancer Res. 2011, 71, 2222–2229. [Google Scholar] [CrossRef]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic Mutations in PALB2 Cause Fanconi Anemia Subtype FA-N and Predispose to Childhood Cancer. Nat. Genet. 2007, 39, 162–164. [Google Scholar] [CrossRef]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated with Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef]
- Tung, N.; Domchek, S.M.; Stadler, Z.; Nathanson, K.L.; Couch, F.; Garber, J.E.; Offit, K.; Robson, M.E. Counselling Framework for Moderate-Penetrance Cancer-Susceptibility Mutations. Nat. Rev. Clin. Oncol. 2016, 13, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Aas, T.; Børresen, A.L.; Geisler, S.; Smith-Sørensen, B.; Johnsen, H.; Varhaug, J.E.; Akslen, L.A.; Lønning, P.E. Specific p53 mutations are associated with de Novo resistance to doxorubicin in breast cancer patients. Nat. Med. 1996, 2, 811–814. [Google Scholar] [CrossRef]
- Chrisanthar, R.; Knappskog, S.; Løkkevik, E.; Anker, G.; Østenstad, B.; Lundgren, S.; Berge, E.O.; Risberg, T.; Mjaaland, I.; Maehle, L.; et al. CHEK2 mutations affecting kinase activity together with mutations in TP53 indicate a functional pathway associated with resistance to epirubicin in primary breast cancer. PLoS ONE 2008, 3, e3062. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, J.; Gu, R.; Shi, Y.; Hu, H.; Liu, J.; Huang, J.; Zhong, C.; Zhou, W.; Yang, Y.; et al. Comprehensive analysis of transcriptomics and genetic alterations identifies potential mechanisms underlying anthracycline therapy resistance in breast cancer. Biomolecules 2022, 12, 1834. [Google Scholar] [CrossRef]
- Bekele, R.T.; Venkatraman, G.; Liu, R.Z.; Tang, X.; Mi, S.; Benesch, M.G.; Mackey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.; et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: Implications for tamoxifen therapy and resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Clarke, P.A.; Parmar, J.; Hu, R.; Schwartz-Roberts, J.L.; Abu-Asab, M.; Wärri, A.; Baumann, W.T.; Clarke, R. Knockdown of estrogen receptor-α induces autophagy and inhibits antiestrogen-mediated unfolded protein response activation, promoting ROS-induced breast cancer cell death. FASEB J. 2014, 28, 3891–3905. [Google Scholar] [CrossRef]
- Kurimchak, A.M.; Herrera-Montávez, C.; Montserrat-Sangrà, S.; Araiza-Olivera, D.; Hu, J.; Neumann-Domer, R.; Kuruvilla, M.; Bellacosa, A.; Testa, J.R.; Jin, J.; et al. The Drug Efflux Pump MDR1 Promotes Intrinsic and Acquired Resistance to PROTACs in Cancer Cells. Sci. Signal 2022, 15, eabn2707. [Google Scholar] [CrossRef]
- Pulliam, N.; Tang, J.; Wang, W.; Fang, F.; Sood, R.; O’Hagan, H.M.; Miller, K.D.; Clarke, R.; Nephew, K.P. Poly-ADP-ribosylation of estrogen receptor-alpha by PARP1 mediates antiestrogen resistance in human breast cancer cells. Cancers 2019, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; De Angelis, C.; Brown, M.; Schiff, R. The evolving role of the estrogen receptor mutations in endocrine therapy-resistant breast cancer. Curr. Oncol. Rep. 2017, 19, 35. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 Mutations in Hormone-Resistant Metastatic Breast Cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 Ligand-Binding Domain Mutations in Hormone-Resistant Breast Cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Toi, M.; Neven, P.; Sohn, J.; Grischke, E.-M.; Llombart-Cussac, A.; Soliman, H.; Wang, H.; Wijayawardana, S.; Jansen, V.M.; et al. Clinical Significance of PIK3CA and ESR1 Mutations in Circulating Tumor DNA: Analysis from the MONARCH 2 Study of Abemaciclib plus Fulvestrant. Clin. Cancer Res. 2022, 28, 4587. [Google Scholar] [CrossRef]
- Clusan, L.; Le Goff, P.; Flouriot, G.; Pakdel, F. A Closer Look at Estrogen Receptor Mutations in Breast Cancer and Their Implications for Estrogen and Antiestrogen Responses. Int. J. Mol. Sci. 2021, 22, 756. [Google Scholar] [CrossRef]
- Araki, K.; Miyoshi, Y. Mechanism of Resistance to Endocrine Therapy in Breast Cancer: The Important Role of PI3K/Akt/mTOR in Estrogen Receptor-Positive, HER2-Negative Breast Cancer. Breast Cancer 2018, 25, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Nunnery, S.E.; Mayer, I.A. Targeting the PI3K/AKT/mTOR Pathway in Hormone-Positive Breast Cancer. Drugs 2020, 80, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Presti, D.; Quaquarini, E. The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2- Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers 2019, 11, 1242. [Google Scholar] [CrossRef]
- Singh, H.; Kang, A.; Bloudek, L.; Hsu, L.I.; Corinna Palanca-Wessels, M.; Stecher, M.; Siadak, M.; Ng, K. Systematic Literature Review and Meta-Analysis of HER2 Amplification, Overexpression, and Positivity in Colorectal Cancer. JNCI Cancer Spectr. 2024, 8, pkad082. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, H.; Yu, X.; Qin, J.J. Drug-Resistant HER2-Positive Breast Cancer: Molecular Mechanisms and Overcoming Strategies. Front. Pharmacol. 2022, 13, 1012552. [Google Scholar] [CrossRef] [PubMed]
- Mouabbi, J.A.; Singareeka Raghavendra, A.; Bassett, R.L.; Hassan, A.; Tripathy, D.; Layman, R.M. Survival Outcomes in Patients With Hormone Receptor-Positive Metastatic Breast Cancer With Low or No ERBB2 Expression Treated With Targeted Therapies Plus Endocrine Therapy. JAMA Netw. Open 2023, 6, e2313017. [Google Scholar] [CrossRef]
- Bose, R.; Ma, C.X. Breast Cancer, HER2 Mutations, and Overcoming Drug Resistance. N. Engl. J. Med. 2021, 385, 1241–1243. [Google Scholar] [CrossRef]
- Tufail, M.; Hu, J.J.; Liang, J.; He, C.Y.; Wan, W.D.; Huang, Y.Q.; Jiang, C.H.; Wu, H.; Li, N. Hallmarks of Cancer Resistance. iScience 2024, 27, 109979. [Google Scholar] [CrossRef]
- Teh, J.L.F.; Aplin, A.E. Arrested Developments: CDK4/6 Inhibitor Resistance and Alterations in the Tumor Immune Microenvironment. Clin. Cancer Res. 2019, 25, 921–927. [Google Scholar] [CrossRef]
- Roberto, M.; Astone, A.; Botticelli, A.; Carbognin, L.; Cassano, A.; D’Auria, G.; Fabbri, A.; Fabi, A.; Gamucci, T.; Krasniqi, E.; et al. CDK4/6 Inhibitor Treatments in Patients with Hormone Receptor Positive, Her2 Negative Advanced Breast Cancer: Potential Molecular Mechanisms, Clinical Implications and Future Perspectives. Cancers 2021, 13, 332. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. Mechanisms of PARP Inhibitor Sensitivity and Resistance. DNA Rep. 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol. Cancer 2020, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Geenen, J.J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H.M. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin. Pharmacokinet. 2018, 57, 427–437. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Sullivan, M.J.; Wynn, R.R.; Demagall, A.; Hendrix, A.S.; Sindhwani, P.; Petros, F.G.; Nadiminty, N. PARP Inhibitors Chemopotentiate and Synergize with Cisplatin to Inhibit Bladder Cancer Cell Survival and Tumor Growth. BMC Cancer 2022, 22, 312. [Google Scholar] [CrossRef]
- Horton, J.K.; Wilson, S.H. Predicting Enhanced Cell Killing through PARP Inhibition. Mol. Cancer Res. 2013, 11, 13–18. [Google Scholar] [CrossRef]
- Kedar, P.S.; Stefanick, D.F.; Horton, J.K.; Wilson, S.H. Increased PARP-1 Association with DNA in Alkylation Damaged, PARP-Inhibited Mouse Fibroblasts. Mol. Cancer Res. 2012, 10, 360–368. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Regairaz, M.; Seiler, J.A.; Agama, K.K.; Doroshow, J.H.; Pommier, Y. Poly (ADP-Ribose) Polymerase and XPF-ERCC1 Participate in Distinct Pathways for the Repair of Topoisomerase I-Induced DNA Damage in Mammalian Cells. Nucleic Acids Res. 2011, 39, 3607–3620. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to Therapy Caused by Intragenic Deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Barge, A.; Parris, C.N. Combination strategies with PARP inhibitors in BRCA-mutated triple-negative breast cancer: Overcoming resistance mechanisms. Oncogene 2025, 44, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e23. [Google Scholar] [CrossRef] [PubMed]
- Dilmac, S.; Ozpolat, B. Mechanisms of PARP-Inhibitor-Resistance in BRCA-Mutated Breast Cancer and New Therapeutic Approaches. Cancers 2023, 15, 3642. [Google Scholar] [CrossRef]
- Giudice, E.; Gentile, M.; Salutari, V.; Ricci, C.; Musacchio, L.; Carbone, M.V.; Ghizzoni, V.; Camarda, F.; Tronconi, F.; Nero, C.; et al. PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers 2022, 14, 1420. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary Mutations as a Mechanism of Cisplatin Resistance in BRCA2-Mutated Cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Zheng, L.; Li, S.; Boyer, T.G.; Lee, W.H. Lessons Learned from BRCA1 and BRCA2. Oncogene 2000, 19, 6159–6175. [Google Scholar] [CrossRef]
- Collins, N.; McManus, R.; Wooster, R.; Mangion, J.; Seal, S.; Lakhani, S.R.; Ormiston, W.; Daly, P.A.; Ford, D.; Easton, D.F. Consistent Loss of the Wild Type Allele in Breast Cancers from a Family Linked to the BRCA2 Gene on Chromosome 13q12-13. Oncogene 1995, 10, 1673–1675. [Google Scholar]
- Gudmundsson, J.; Johannesdottir, G.; Bergthorsson, J.T.; Arason, A.; Ingvarsson, S.; Egilsson, V.; Barkardottir, R.B. Different Tumor Types from BRCA2 Carriers Show Wild-Type Chromosome Deletions on 13q12-Q13. Cancer Res. 1995, 55, 4830–4832. [Google Scholar]
- Sharan, S.K.; Bradley, A. Murine Brca2: Sequence, Map Position, and Expression Pattern. Genomics 1997, 40, 234–241. [Google Scholar] [CrossRef]
- Connor, F.; Bertwistle, D.; Mee, P.J.; Ross, G.M.; Swift, S.; Grigorieva, E.; Tybulewicz, V.L.; Ashworth, A. Tumorigenesis and a DNA Repair Defect in Mice with a Truncating Brca2 Mutation. Nat. Genet. 1997, 17, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.S.; Thistlethwaite, F.C.; Patel, K.J.; Yu, V.P.; Lee, H.; Venkitaraman, A.R.; Abel, K.J.; Carlton, M.B.; Hunter, S.M.; Colledge, W.H.; et al. Thymic Lymphomas in Mice with a Truncating Mutation in Brca2. Cancer Res. 1998, 58, 1338–1343. [Google Scholar] [PubMed]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic Lethality and Radiation Hypersensitivity Mediated by Rad51 in Mice Lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.K.; Misra, S.; Khedkar, S.V.; Hamilton, N.; Irvin-Wilson, C.; Sharan, C.; Sealy, L.; Chaudhuri, G. Regulation of BRCA2 gene expression by the SLUG repressor protein in human breast cells. J. Biol. Chem. 2005, 280, 17163–17171. [Google Scholar] [CrossRef]
- Bernard-Gallon, D.J.; Satih, S.; Chalabi, N.; Rabiau, N.; Bosviel, R.; Fontana, L.; Bignon, Y.J. Phytoestrogens Regulate the Expression of Genes Involved in Different Biological Processes in BRCA2 Knocked down MCF-7, MDA-MB-231 and MCF-10a Cell Lines. Oncol. Rep. 2010, 23, 647–653. [Google Scholar] [CrossRef]
- Katheeja, M.N.; Das, S.P.; Das, R.; Laha, S. BRCA1 interactors, RAD50 and BRIP1, as prognostic markers for triple-negative breast cancer severity. Front. Genet. 2023, 14, 1035052. [Google Scholar] [CrossRef]
- Castro, M.A.; Dalmolin, R.J.; Moreira, J.C.; Mombach, J.C.; de Almeida, R.M. Evolutionary origins of human apoptosis and genome-stability gene networks. Nucleic Acids Res. 2008, 36, 6269–6283. [Google Scholar] [CrossRef]
- Cimmino, A.; Fasciglione, G.F.; Gioia, M.; Marini, S.; Ciaccio, C. Multi-Anticancer Activities of Phytoestrogens in Human Osteosarcoma. Int. J. Mol. Sci. 2023, 24, 13344. [Google Scholar] [CrossRef]
- Hu, L.; Su, L.; Cheng, H.; Mo, C.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Single-Cell RNA Sequencing Reveals the Cellular Origin and Evolution of Breast Cancer in BRCA1 Mutation Carriers. Cancer Res. 2021, 81, 2600–2611. [Google Scholar] [CrossRef]
- Buckley, N.E.; Mullan, P.B. BRCA1-Conductor of the Breast Stem Cell Orchestra: The Role of BRCA1 in Mammary Gland Development and Identification of Cell of Origin of BRCA1 Mutant Breast Cancer. Stem Cell Rev. Rep. 2012, 8, 982–993. [Google Scholar] [CrossRef]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant Luminal Progenitors as the Candidate Target Population for Basal Tumor Development in BRCA1 Mutation Carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic Predisposition Directs Breast Cancer Phenotype by Dictating Progenitor Cell Fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 Basal-like Breast Cancers Originate from Luminal Epithelial Progenitors and Not from Basal Stem Cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Sau, A.; Lau, R.; Cabrita, M.A.; Nolan, E.; Crooks, P.A.; Visvader, J.E.; Pratt, M.A.C. Persistent Activation of NF-κB in BRCA1-Deficient Mammary Progenitors Drives Aberrant Proliferation and Accumulation of DNA Damage. Cell Stem Cell 2016, 19, 52–65. [Google Scholar] [CrossRef]
- Wang, H.; Xiang, D.; Liu, B.; He, A.; Randle, H.J.; Zhang, K.X.; Dongre, A.; Sachs, N.; Clark, A.P.; Tao, L. Inadequate DNA Damage Repair Promotes Mammary Transdifferentiation, Leading to BRCA1 Breast Cancer. Cell 2019, 178, 135–151.e19. [Google Scholar] [CrossRef]
- Villadsen, R.; Fridriksdottir, A.J.; Rønnov-Jessen, L.; Gudjonsson, T.; Rank, F.; LaBarge, M.A.; Bissell, M.J.; Petersen, O.W. Evidence for a Stem Cell Hierarchy in the Adult Human Breast. J. Cell Biol. 2007, 177, 87–101. [Google Scholar] [CrossRef]
- Bach, K.; Pensa, S.; Zarocsinceva, M.; Kania, K.; Stockis, J.; Pinaud, S.; Lazarus, K.A.; Shehata, M.; Simões, B.M.; Greenhalgh, A.R.; et al. Time-Resolved Single-Cell Analysis of Brca1 Associated Mammary Tumourigenesis Reveals Aberrant Differentiation of Luminal Progenitors. Nat. Commun. 2021, 12, 1502. [Google Scholar] [CrossRef]
- Bach, K.; Pensa, S.; Grzelak, M.; Hadfield, J.; Adams, D.J.; Marioni, J.C.; Khaled, W.T. Differentiation Dynamics of Mammary Epithelial Cells Revealed by Single-Cell RNA Sequencing. Nat. Commun. 2017, 8, 2128. [Google Scholar] [CrossRef]
- Watson, C.J.; Khaled, W.T. Mammary Development in the Embryo and Adult: A Journey of Morphogenesis and Commitment. Dev. Camb. Engl. 2008, 135, 995–1003. [Google Scholar] [CrossRef]
- Ranjan, R.; Thompson, E.A.; Yoon, K.; Smart, R.C. C/EBPalpha expression is partially regulated by C/EBPbeta in response to DNA damage and C/EBPalpha-deficient fibroblasts display an impaired G1 checkpoint. Oncogene 2009, 28, 3235–3245. [Google Scholar] [CrossRef]
- Zhang, B.; Lyu, J.; Liu, Y.; Wu, C.; Yang, E.J.; Pardeshi, L.; Tan, K.; Wong, K.H.; Chen, Q.; Xu, X.; et al. BRCA1 Deficiency Sensitizes Breast Cancer Cells to Bromodomain and Extra-Terminal Domain (BET) Inhibition. Oncogene 2018, 37, 6341–6356. [Google Scholar] [CrossRef]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Fujino, K.; Monteiro, L.J.; Gomes, A.R.; Drost, R.; Davidson-Smith, H.; Takeda, S.; Khoo, U.S.; Jonkers, J.; Sproul, D.; et al. FOXA1 repression is associated with loss of BRCA1 and increased promoter methylation and chromatin silencing in breast cancer. Oncogene 2015, 34, 5012–5024. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Deng, C.X.; Sporn, M.B.; Liby, K.T. CDDO-Imidazolide Induces DNA Damage, G2/M Arrest and Apoptosis in BRCA1-Mutated Breast Cancer Cells. Cancer Prev. Res. Phila. Pa. 2011, 4, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Zheng, Y.; Kim, H.S.; Xu, X.; Cao, L.; Luhasen, T.; Lee, M.H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-Associated Tumorigenesis. Mol. Cell 2008, 32, 11–20. [Google Scholar] [CrossRef]
- Simeone, A.M.; Deng, C.X.; Kelloff, G.J.; Steele, V.E.; Johnson, M.M.; Tari, A.M. N-(4-Hydroxyphenyl)retinamide is more potent than other phenylretinamides in inhibiting the growth of BRCA1-mutated breast cancer cells. Carcinogenesis 2005, 26, 1000–1007. [Google Scholar] [CrossRef]
- Zajac, M.; Moneo, M.V.; Carnero, A.; Benitez, J.; Martínez-Delgado, B. Mitotic catastrophe cell death induced by heat shock protein 90 inhibitor in BRCA1-deficient breast cancer cell lines. Mol. Cancer Ther. 2008, 7, 2358–2366. [Google Scholar] [CrossRef]
- Zander, S.A.; Kersbergen, A.; van der Burg, E.; de Water, N.; van Tellingen, O.; Gunnarsdottir, S.; Jaspers, J.E.; Pajic, M.; Nygren, A.O.; Jonkers, J.; et al. Sensitivity and acquired resistance of BRCA1;p53-deficient mouse mammary tumors to the topoisomerase I inhibitor topotecan. Cancer Res. 2010, 70, 1700–1710. [Google Scholar] [CrossRef]
- Pajic, M.; Iyer, J.K.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Jonkers, J.; Borst, P.; Rottenberg, S. Moderate increase in Mdr1a/1b expression causes in vivo resistance to doxorubicin in a mouse model for hereditary breast cancer. Cancer Res. 2009, 69, 6396–6404. [Google Scholar] [CrossRef]
- Burga, L.N.; Hu, H.; Juvekar, A.; Tung, N.M.; Troyan, S.L.; Hofstatter, E.W.; Wulf, G.M. Loss of BRCA1 leads to an increase in epidermal growth factor receptor expression in mammary epithelial cells, and epidermal growth factor receptor inhibition prevents estrogen receptor-negative cancers in BRCA1-mutant mice. Breast Cancer Res. 2011, 13, R30. [Google Scholar] [CrossRef]
- Puppe, J.; Drost, R.; Liu, X.; Joosse, S.A.; Evers, B.; Cornelissen-Steijger, P.; Nederlof, P.; Yu, Q.; Jonkers, J.; van Lohuizen, M.; et al. BRCA1-Deficient Mammary Tumor Cells Are Dependent on EZH2 Expression and Sensitive to Polycomb Repressive Complex 2-Inhibitor 3-Deazaneplanocin A. Breast Cancer Res. 2009, 11, R63. [Google Scholar] [CrossRef]
- Warmoes, M.; Jaspers, J.E.; Xu, G.; Sampadi, B.K.; Pham, T.V.; Knol, J.C.; Piersma, S.R.; Boven, E.; Jonkers, J.; Rottenberg, S.; et al. Proteomics of Genetically Engineered Mouse Mammary Tumors Identifies Fatty Acid Metabolism Members as Potential Predictive Markers for Cisplatin Resistance. Mol. Cell Proteom. 2013, 12, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- Mauny, A.; Faure, S.; Derbré, S. Phytoestrogens and Breast Cancer: Should French Recommendations Evolve? Cancers 2022, 14, 6163. [Google Scholar] [CrossRef] [PubMed]
- Greville, G.; Llop, E.; Howard, J.; Madden, S.F.; Perry, A.S.; Peracaula, R.; Rudd, P.M.; McCann, A.; Saldova, R. 5-AZA-dC induces epigenetic changes associated with modified glycosylation of secreted glycoproteins and increased EMT and migration in chemo-sensitive cancer cells. Clin. Epigenet 2021, 13, 34. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Zhang, Z.C.; Wu, Y.Y.; Pi, Y.N.; Lou, S.H.; Liu, T.B.; Lou, G.; Yang, C. Bromodomain and Extraterminal (BET) Proteins: Biological Functions, Diseases, and Targeted Therapy. Signal Transduct. Target. Ther. 2023, 8, 420. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Hurvitz, S.A.; Mina, L.A.; Rugo, H.S.; Lee, K.H.; Gonçalves, A.; Diab, S.; Woodward, N.; Goodwin, A.; Yerushalmi, R.; et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: Final overall survival results from the EMBRACA trial. Ann. Oncol: ESMO 2020, 31, 1526–1535. [Google Scholar] [CrossRef]
- ClinicalTrial.gov. Available online: https://clinicaltrials.gov/study/NCT05327010 (accessed on 20 February 2025).
- Zaffaroni, N.; Pennati, M.; Daidone, M.G. Survivin as a Target for New Anticancer Interventions. J. Cell Mol. Med. 2005, 9, 360–372. [Google Scholar] [CrossRef]
- Denu, J.M. Linking Chromatin Function with Metabolic Networks: Sir2 Family of NAD(+)-Dependent Deacetylases. Trends Biochem. Sci. 2003, 28, 41–48. [Google Scholar] [CrossRef]
- Gasser, S.M.; Cockell, M.M. The Molecular Biology of the SIR Proteins. Gene 2001, 279, 1–16. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol Improves Health and Survival of Mice on a High-Calorie Diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Haigis, M.C.; Guarente, L.P. Mammalian Sirtuins-Emerging Roles in Physiology, Aging, and Calorie Restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Liu, J.C.; Shen, L.; Rau, K.M.; Kuo, H.P.; Li, Y.M.; Shi, D.; Lee, Y.C.; Chang, K.J.; Hung, M.C. Cancer-Specific Activation of the Survivin Promoter and Its Potential Use in Gene Therapy. Cancer Gene Ther. 2004, 11, 740–747. [Google Scholar] [CrossRef]
- Pietersen, A.M.; Horlings, H.M.; Hauptmann, M.; Langerød, A.; Ajouaou, A.; Cornelissen-Steijger, P.; Wessels, L.F.; Jonkers, J.; van de Vijver, M.J.; van Lohuizen, M. EZH2 and BMI1 Inversely Correlate with Prognosis and TP53 Mutation in Breast Cancer. Breast Cancer Res. 2008, 10, R109. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.A.B.; Otte, A.P.; Hayes, D.F.; et al. EZH2 Is a Marker of Aggressive Breast Cancer and Promotes Neoplastic Transformation of Breast Epithelial Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 Expression Is Associated with High Proliferation Rate and Aggressive Tumor Subgroups in Cutaneous Melanoma and Cancers of the Endometrium, Prostate, and Breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Raaphorst, F.M.; Meijer, C.J.L.M.; Fieret, E.; Blokzijl, T.; Mommers, E.; Buerger, H.; Packeisen, J.; Sewalt, R.A.B.; Otte, A.P.; van Diest, P.J. Poorly Differentiated Breast Carcinoma Is Associated with Increased Expression of the Human Polycomb Group EZH2 Gene. Neoplasia 2003, 5, 481–488. [Google Scholar] [CrossRef]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 Is Downstream of the pRB-E2F Pathway, Essential for Proliferation and Amplified in Cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed]
- Pietersen, A.M.; van Lohuizen, M. Stem Cell Regulation by Polycomb Repressors: Postponing Commitment. Curr. Opin. Cell Biol. 2008, 20, 201–207. [Google Scholar] [CrossRef]
- Sparmann, A.; van Lohuizen, M. Polycomb Silencers Control Cell Fate, Development and Cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef]
- Zeidler, M.; Varambally, S.; Cao, Q.; Chinnaiyan, A.M.; Ferguson, D.O.; Merajver, S.D.; Kleer, C.G. The Polycomb Group Protein EZH2 Impairs DNA Repair in Breast Epithelial Cells. Neoplasia 2005, 7, 1011–1019. [Google Scholar] [CrossRef]
- Collett, K.; Eide, G.E.; Arnes, J.; Stefansson, I.M.; Eide, J.; Braaten, A.; Aas, T.; Otte, A.P.; Akslen, L.A. Expression of Enhancer of Zeste Homologue 2 Is Significantly Associated with Increased Tumor Cell Proliferation and Is a Marker of Aggressive Breast Cancer. Clin. Cancer Res. 2006, 12, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ginestier, C.; Charafe-Jauffret, E.; Foco, H.; Kleer, C.G.; Merajver, S.D.; Dontu, G.; Wicha, M.S. BRCA1 Regulates Human Mammary Stem/Progenitor Cell Fate. Proc. Natl. Acad. Sci. USA 2008, 105, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-Wide Mapping of Polycomb Target Genes Unravels Their Roles in Cell Fate Transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of Developmental Regulators by Polycomb in Human Embryonic Stem Cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.M.; Tan, P.B.O.; Liu, E.T.; Yu, Q. Pharmacologic Disruption of Polycomb-Repressive Complex 2-Mediated Gene Repression Selectively Induces Apoptosis in Cancer Cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef]
- Agarwal, P.; Alzrigat, M.; Párraga, A.A.; Enroth, S.; Singh, U.; Ungerstedt, J.; Österborg, A.; Brown, P.J.; Ma, A.; Jin, J. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget 2016, 7, 6809–6823. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in Cancer Therapy: A Novel Approach to Reversing Drug Resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Adany, R.; Heimer, R.; Caterson, B.; Sorrell, J.M.; Iozzo, R.V. Altered Expression of Chondroitin Sulfate Proteoglycan in the Stroma of Human Colon Carcinoma. Hypomethylation of PG-40 Gene Correlates with Increased PG-40 Content and mRNA Levels. J. Biol. Chem. 1990, 265, 11389–11396. [Google Scholar] [CrossRef] [PubMed]
- Buraschi, S.; Pal, N.; Tyler-Rubinstein, N.; Owens, R.T.; Neill, T.; Iozzo, R.V. Decorin Antagonizes Met Receptor Activity and Down-Regulates {beta}-Catenin and Myc Levels. J. Biol. Chem. 2010, 285, 42075–42085. [Google Scholar] [CrossRef]
- Sears, R.C. The Life Cycle of C-Myc: From Synthesis to Degradation. Cell Cycle 2004, 3, 1133–1137. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in Cancer Biology, Tumour Microenvironment and Angiogenesis. J. Cell Mol. Med. 2011, 15, 1013–1031. [Google Scholar] [CrossRef] [PubMed]
- Deming, S.L.; Nass, S.J.; Dickson, R.B.; Trock, B.J. C-Myc Amplification in Breast Cancer: A Meta-Analysis of Its Occurrence and Prognostic Relevance. Br. J. Cancer 2000, 83, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.J.; Dickson, R.B. C-Myc in Breast Cancer. Endocr. Relat. Cancer 2000, 7, 143–164. [Google Scholar] [CrossRef]
- Adem, C.; Soderberg, C.L.; Hafner, K.; Reynolds, C.; Slezak, J.M.; Sinclair, C.S.; Sellers, T.A.; Schaid, D.J.; Couch, F.; Hartmann, L.C.; et al. ERBB2, TBX2, RPS6KB1, and MYC Alterations in Breast Tissues of BRCA1 and BRCA2 Mutation Carriers. Genes Chromosomes Cancer 2004, 41, 1–11. [Google Scholar] [CrossRef]
- Grushko, T.A.; Dignam, J.J.; Das, S.; Blackwood, A.M.; Perou, C.M.; Ridderstråle, K.K.; Anderson, K.N.; Wei, M.-J.; Adams, A.J.; Hagos, F.; et al. MYC Is Amplified in BRCA1-Associated Breast Cancers. Clin. Cancer Res. 2004, 10, 499–507. [Google Scholar] [CrossRef]
- Rodriguez-Pinilla, S.M.; Jones, R.L.; Lambros, M.B.K.; Arriola, E.; Savage, K.; James, M.; Pinder, S.E.; Reis-Filho, J.S. MYC Amplification in Breast Cancer: A Chromogenic in Situ Hybridisation Study. J. Clin. Pathol. 2007, 60, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Schaefer, R.M. Proteoglycans: From Structural Compounds to Signaling Molecules. Cell Tissue Res. 2010, 339, 237–246. [Google Scholar] [CrossRef]
- Weber, I.T.; Harrison, R.W.; Iozzo, R.V. Model Structure of Decorin and Implications for Collagen Fibrillogenesis. J. Biol. Chem. 1996, 271, 31767–31770. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Yamaguchi, Y. Proteoglycans as Modulators of Growth Factor Activities. Cell 1991, 64, 867–869. [Google Scholar] [CrossRef]
- Keene, D.R.; San Antonio, J.D.; Mayne, R.; McQuillan, D.J.; Sarris, G.; Santoro, S.A.; Iozzo, R.V. Decorin Binds near the C Terminus of Type I Collagen. J. Biol. Chem. 2000, 275, 21801–21804. [Google Scholar] [CrossRef]
- Vogel, K.G.; Paulsson, M.; Heinegård, D. Specific Inhibition of Type I and Type II Collagen Fibrillogenesis by the Small Proteoglycan of Tendon. Biochem. J. 1984, 223, 587–597. [Google Scholar] [CrossRef]
- Danielson, K.G.; Baribault, H.; Holmes, D.F.; Graham, H.; Kadler, K.E.; Iozzo, R.V. Targeted Disruption of Decorin Leads to Abnormal Collagen Fibril Morphology and Skin Fragility. J. Cell Biol. 1997, 136, 729–743. [Google Scholar] [CrossRef] [PubMed]
- Santra, M.; Reed, C.C.; Iozzo, R.V. Decorin Binds to a Narrow Region of the Epidermal Growth Factor (EGF) Receptor, Partially Overlapping but Distinct from the EGF-Binding Epitope. J. Biol. Chem. 2002, 277, 35671–35681. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Santra, M.; Reed, C.C.; Eichstetter, I.; McQuillan, D.J.; Gross, D.; Nugent, M.A.; Hajnóczky, G.; Iozzo, R.V. Sustained Down-Regulation of the Epidermal Growth Factor Receptor by Decorin. A Mechanism for Controlling Tumor Growth in Vivo. J. Biol. Chem. 2000, 275, 32879–32887. [Google Scholar] [CrossRef]
- Santra, M.; Eichstetter, I.; Iozzo, R.V. An Anti-Oncogenic Role for Decorin. Down-Regulation of ErbB2 Leads to Growth Suppression and Cytodifferentiation of Mammary Carcinoma Cells. J. Biol. Chem. 2000, 275, 35153–35161. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Mu, T.; Zhao, L.; Zhu, D.; Zhong, X.; Li, Y.; Liang, C.; Li, W.; Zhou, Q. Stachydrine represses the proliferation and enhances cell cycle arrest and apoptosis of breast cancer cells via PLA2G2A/DCN axis. Chem. Biol. Drug Des. 2024, 103, e14429. [Google Scholar] [CrossRef]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme Oxygenase-1 Accelerates Erastin-Induced Ferroptotic Cell Death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the P62-Keap1-NRF2 Pathway Protects against Ferroptosis in Hepatocellular Carcinoma Cells. Hepatol. Baltim. Md. 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme Oxygenase-1 Mitigates Ferroptosis in Renal Proximal Tubule Cells. Am. J. Physiol. Renal Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-Targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme Oxygenase-1 Mediates BAY 11-7085 Induced Ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 Related Cytoprotection with Increased Expression Is Due to Reactive Iron. FASEB J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Wu, F.; Xiong, G.; Chen, Z.; Lei, C.; Liu, Q.; Bai, Y. SLC3A2 Inhibits Ferroptosis in Laryngeal Carcinoma via mTOR Pathway. Hereditas 2022, 159, 6. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Carlisle, A.E.; Peppers, A.; Park, S.J.; Doshi, M.B.; Spears, M.E.; Kim, D. xCT-Driven Expression of GPX4 Determines Sensitivity of Breast Cancer Cells to Ferroptosis Inducers. Antioxid. 2021, 10, 317. [Google Scholar] [CrossRef]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. IFNγ-Mediated Repression of System Xc- Drives Vulnerability to Induced Ferroptosis in Hepatocellular Carcinoma Cells. J. Leukoc. Biol. 2021, 110, 301–314. [Google Scholar] [CrossRef]
- Liao, P.; Wang, W.; Wang, W.; Kryczek, I.; Li, X.; Bian, Y.; Sell, A.; Wei, S.; Grove, S.; Johnson, J.K.; et al. CD8+ T Cells and Fatty Acids Orchestrate Tumor Ferroptosis and Immunity via ACSL4. Cancer Cell 2022, 40, 365–378.e6. [Google Scholar] [CrossRef]
- El Hout, M.; Dos Santos, L.; Hamaï, A.; Mehrpour, M. A Promising New Approach to Cancer Therapy: Targeting Iron Metabolism in Cancer Stem Cells. Semin. Cancer Biol. 2018, 53, 125–138. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis Is an Autophagic Cell Death Process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired Lysosomal Acidification Triggers Iron Deficiency and Inflammation in Vivo. eLife 2019, 8, e51031. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The Iron Exporter Ferroportin/Slc40a1 Is Essential for Iron Homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Meng, Y.; Li, D.; Yao, L.; Le, J.; Liu, Y.; Sun, Y.; Zeng, F.; Chen, X.; Deng, G. Ferroptosis in Cancer: From Molecular Mechanisms to Therapeutic Strategies. Signal Transduct. Target. Ther. 2024, 9, 55. [Google Scholar] [CrossRef]
- Philip, M.; Schietinger, A. CD8+ T Cell Differentiation and Dysfunction in Cancer. Nat. Rev. Immunol. 2022, 22, 209–223. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune Checkpoint Inhibitors in Melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T Cells Regulate Tumour Ferroptosis during Cancer Immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kroemer, G. Interferon-γ Induces Cancer Cell Ferroptosis. Cell Res. 2019, 29, 692–693. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, F.; Yin, L.; Wang, F.; Shi, H.; Zhao, Q.; Yang, F.; Chen, D.; Dong, X.; Gu, Y.; et al. The Establishment of Polypeptide PSMA-Targeted Chimeric Antigen Receptor-Engineered Natural Killer Cells for Castration-Resistant Prostate Cancer and the Induction of Ferroptosis-Related Cell Death. Cancer Commun. 2022, 42, 768–783. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C.; Schmidt, P.J. Iron Homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Ponka, P. The Molecular Mechanisms of the Metabolism and Transport of Iron in Normal and Neoplastic Cells. Biochim. Biophys. Acta 1997, 1331, 1–40. [Google Scholar] [CrossRef]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef]
- Li, G.; Lin, S.-S.; Yu, Z.-L.; Wu, X.-H.; Liu, J.-W.; Tu, G.-H.; Liu, Q.-Y.; Tang, Y.-L.; Jiang, Q.-N.; Xu, J.-H.; et al. A PARP1 PROTAC as a novel strategy against PARP inhibitor resistance via promotion of ferroptosis in p53-positive breast cancer. Biochem. Pharmacol. 2022, 206, 115329. [Google Scholar] [CrossRef]
- Wang, D.; Wei, G.; Ma, J.; Cheng, S.; Jia, L.; Song, X.; Zhang, M.; Ju, M.; Wang, L.; Zhao, L.; et al. Identification of the prognostic value of ferroptosis-related gene signature in breast cancer patients. BMC Cancer 2021, 21, 645. [Google Scholar] [CrossRef]
- Wang, N.; Gu, Y.; Li, L.; Chi, J.; Liu, X.; Xiong, Y.; Jiang, S.; Zhang, W.; Zhong, C. Identification of novel prognostic risk signature of breast cancer based on ferroptosis-related genes. Sci. Rep. 2022, 12, 13766. [Google Scholar] [CrossRef]
- Hong, T.; Lei, G.; Chen, X.; Li, H.; Zhang, X.; Wu, N.; Zhao, Y.; Zhang, Y.; Wang, J. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol. 2021, 42, 101928. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DRUGS | IC50 IN BRCA-MUTATED MODELS | DOSAGE | EFFICACY |
|---|---|---|---|
| topotecan + olaparib | topotecan 5.780934 μmol/L + olaparib 198.853952 μmol/L | n.d. | n.d. |
| 17-AAG | 0.059 ± 0.017 (μM) (MDA-MB-436) 0.014 ± 0.006 (μM) (HCC 1937) 0.013 ± 0.006 (μM) (UACC 3199) | n.d. | n.d. |
| CDDO-Im | n.d. | 1 μM | n.d. |
| resveratrol | 40 μM | n.d. | n.d. |
| DZNep | 163 nM | n.d. | n.d. |
| 4-HPR | n.d. | 2.5 μM | n.d. |
| genestein | 28 μM | n.d. | n.d. |
| daidzein | n.d. | 1.7 mg/day | CI95% daidzein intake/recurrence: 0.96 (0.52–1.76) [146] |
| BETi | 108.07 μM | n.d. | n.d. |
| 5-aza 2′deoxycytidine | n.d. | 0.1 μM 5-AZA-dC [147] | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbone, F.P.; Ancona, P.; Volinia, S.; Terrazzan, A.; Bianchi, N. Druggable Molecular Networks in BRCA1/BRCA2-Mutated Breast Cancer. Biology 2025, 14, 253. https://doi.org/10.3390/biology14030253
Carbone FP, Ancona P, Volinia S, Terrazzan A, Bianchi N. Druggable Molecular Networks in BRCA1/BRCA2-Mutated Breast Cancer. Biology. 2025; 14(3):253. https://doi.org/10.3390/biology14030253
Chicago/Turabian StyleCarbone, Francesca Pia, Pietro Ancona, Stefano Volinia, Anna Terrazzan, and Nicoletta Bianchi. 2025. "Druggable Molecular Networks in BRCA1/BRCA2-Mutated Breast Cancer" Biology 14, no. 3: 253. https://doi.org/10.3390/biology14030253
APA StyleCarbone, F. P., Ancona, P., Volinia, S., Terrazzan, A., & Bianchi, N. (2025). Druggable Molecular Networks in BRCA1/BRCA2-Mutated Breast Cancer. Biology, 14(3), 253. https://doi.org/10.3390/biology14030253