
Mutual Interactions Between Microbiota and the Human Immune System During the First 1000 Days of Life

 , and
, and 
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
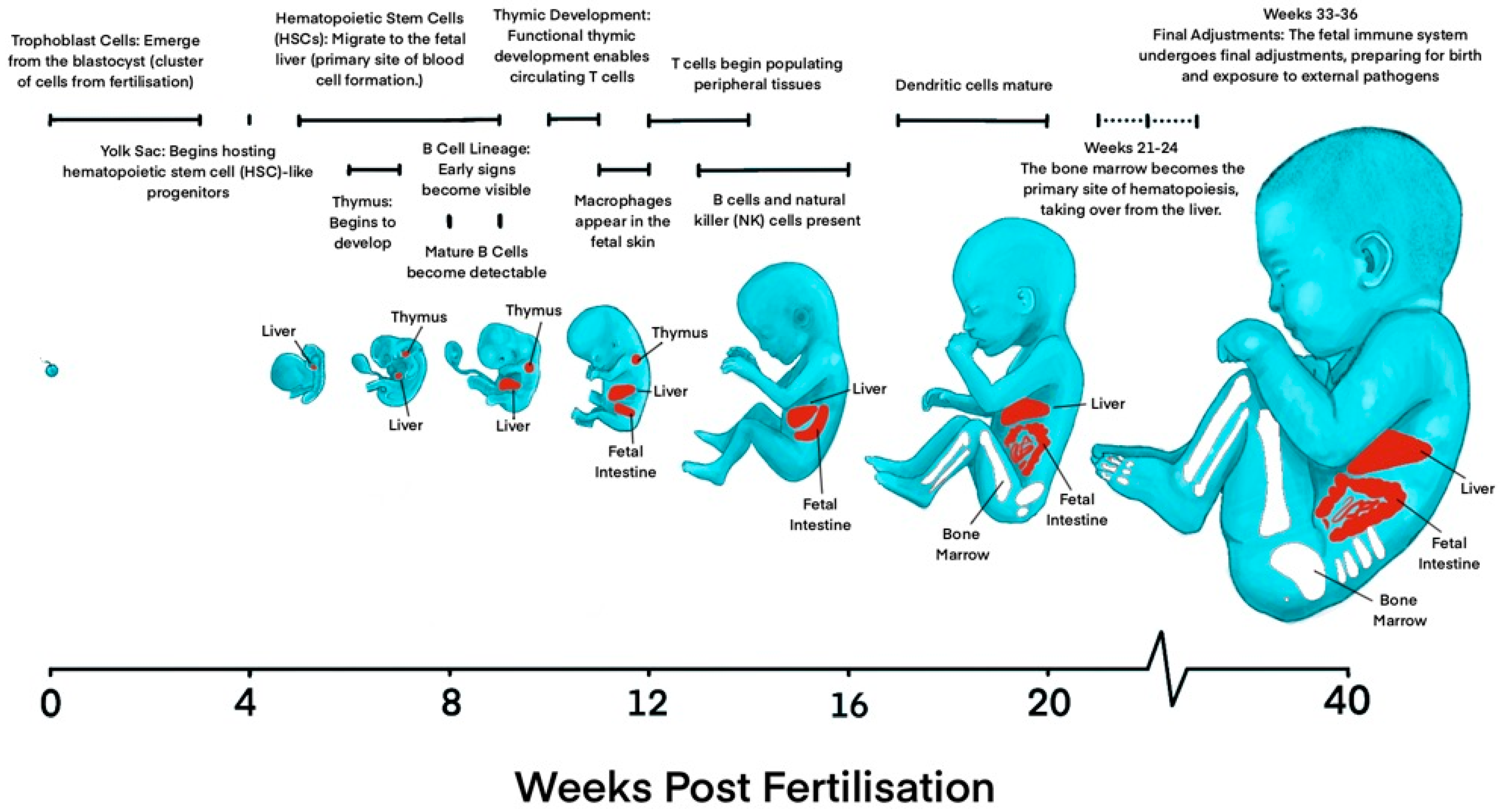
2. Pre- and Postnatal Development of the Immune System
2.1. Prenatal Development of the Immune System
2.1.1. Development of Innate Immune System
2.1.2. Development of the Humoral Immune System
2.1.3. Development of the Cellular Immune System
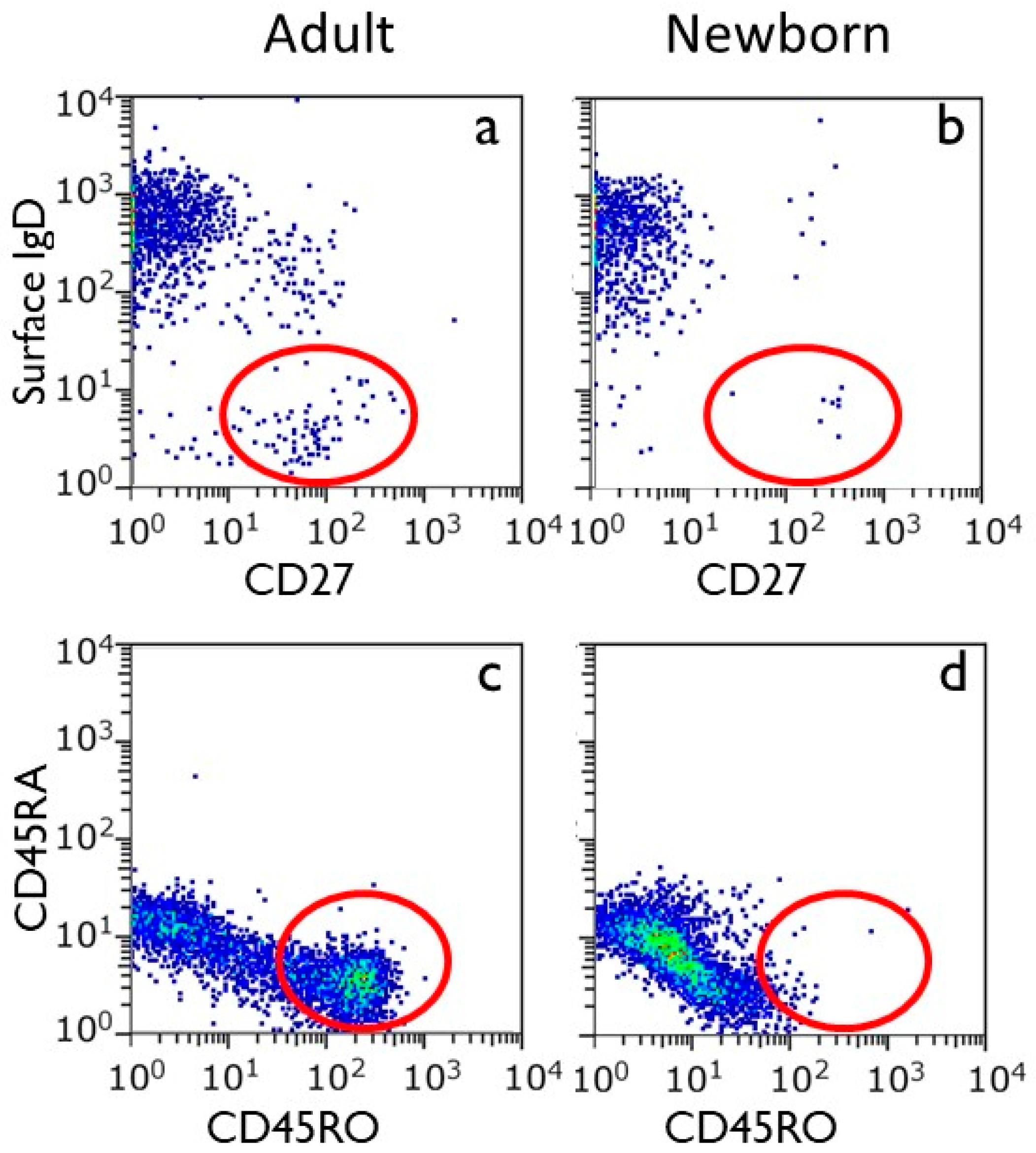
2.1.4. Development of Immunological Memory
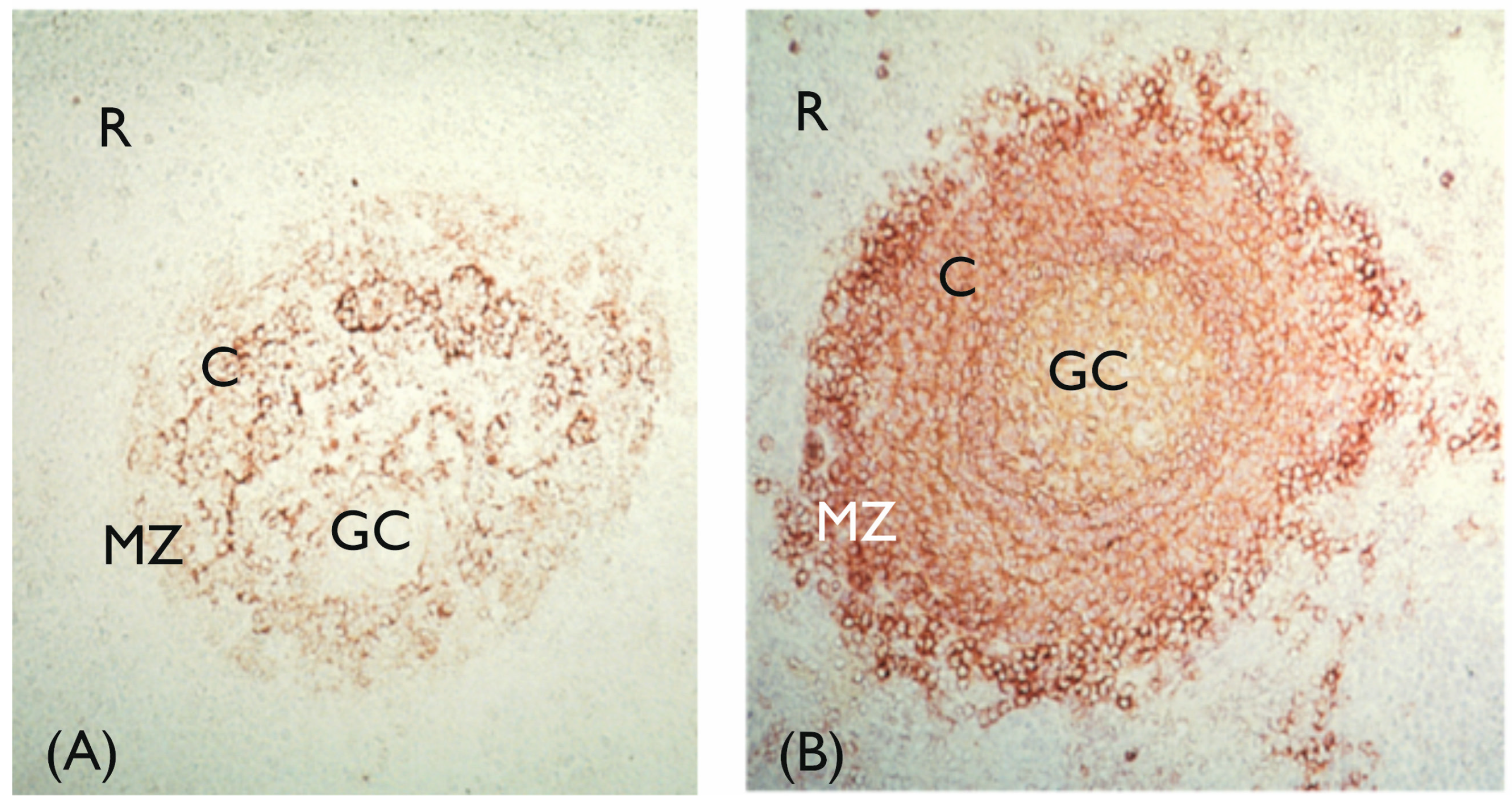
2.2. Postnatal Development of the Immune System
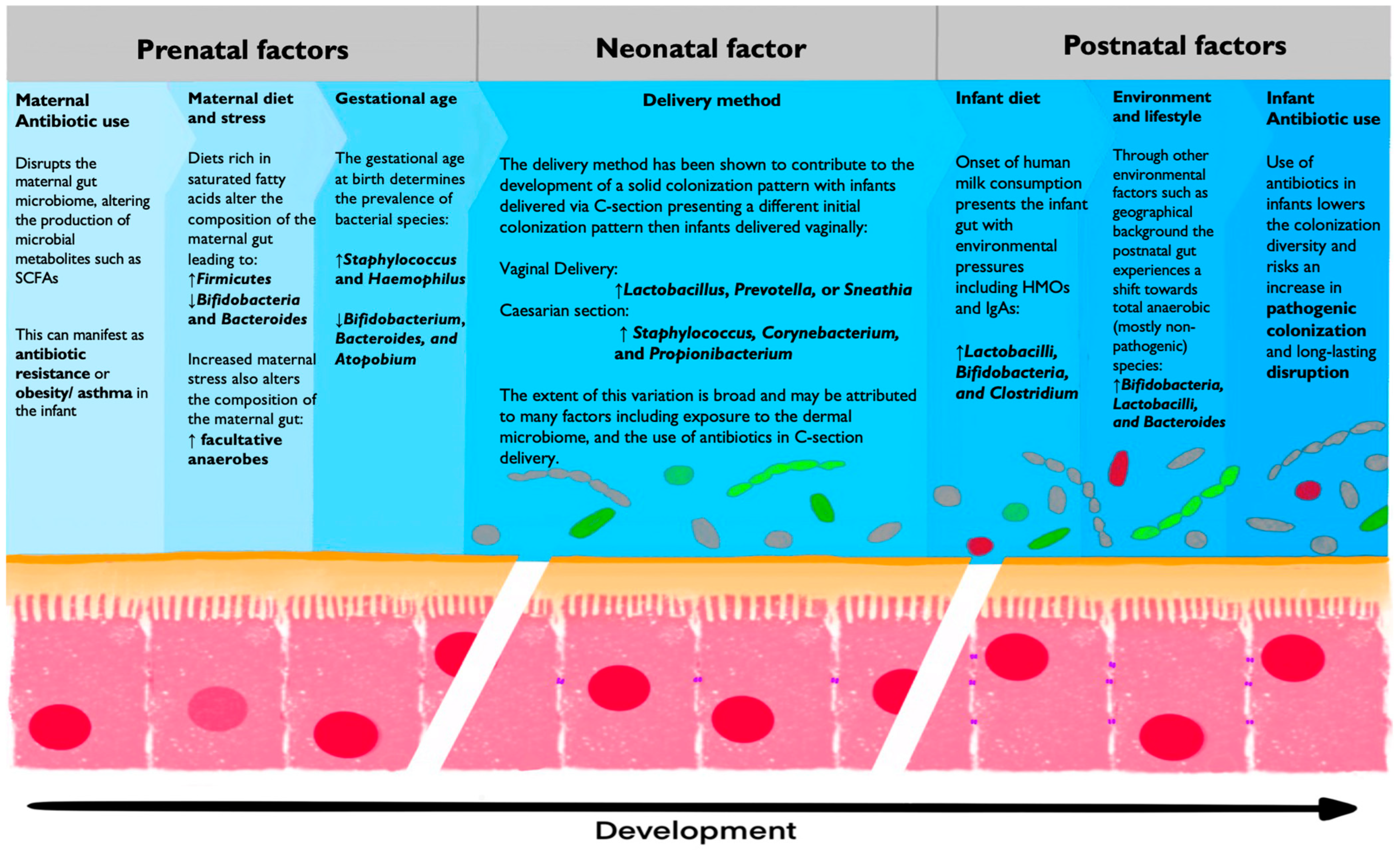
3. Development of the Infant Gut Microbiome
3.1. Maternal Microbiota
3.1.1. The Role of Maternal Microbiota During Fetal Development
3.1.2. The Composition of Maternal Microbiota Can Be Changed Through External Factors
3.2. Prenatal Bacterial Colonization
3.2.1. The Sterile Womb Hypothesis
3.2.2. The Sterility of the Placenta, Amniotic Fluid, and Fetus
3.2.3. The in Utero Hypothesis
3.3. Bacterial Colonization of Premature Children
The Development of the Gut Microbiome Is Specific to Gestational Age
3.4. Neonatal Bacterial Colonization
3.4.1. Delivery Method
3.4.2. Long-Term Development of Gut Microbiota
Bifidobacterium
Lactobacillacea
Clostridium
Bacteroides
3.4.3. First 1000 Days of Life and Nutritional Weaning
4. Cellular and Molecular Interactions Between Microbiota and the Immune System
4.1. Gastrointestinal Microbe Activity
4.1.1. Interactions at Mucosal Surfaces
4.1.2. Epithelium
4.1.3. Introduction of Milk and Its Impact on the Gut Microbiota’s Immune System
4.1.4. Introduction of Solid Food and Its Impact on the Gut Microbiota and the Immune System
4.2. Interaction Between Microbiota, Including Their Metabolites and Cells and Molecules of the Immune System
4.2.1. MAIT Cells
Anti-Inflammatory Effects of Bacterial Metabolites
Pro-Inflammatory Effects of Microbiota
4.2.2. B Cells and Immunoglobulin A
4.2.3. Dendritic Cells Connecting Gut Microbiota to Immune Systems
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sansonetti, P.J. War and peace at mucosal surfaces. Nat. Rev. Immunol. 2004, 4, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 2009, 10, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.; Boroviak, T.E. Origin and function of the yolk sac in primate embryogenesis. Nat. Commun. 2020, 11, 3760. [Google Scholar] [CrossRef]
- Enders, A.C. Implantation in the macaque: Expansion of the implantation site during the first week of implantation. Placenta 2007, 28, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Gou, F.; Zheng, Z.; Zhang, Y.; Zhang, Y.; Dong, F.; Cheng, T.; Cheng, H. Decoding human bone marrow hematopoietic stem and progenitor cells from fetal to birth. iScience 2024, 27, 110445. [Google Scholar] [CrossRef]
- Park, J.E.; Jardine, L.; Gottgens, B.; Teichmann, S.A.; Haniffa, M. Prenatal development of human immunity. Science 2020, 368, 600–603. [Google Scholar] [CrossRef]
- Popescu, D.M.; Botting, R.A.; Stephenson, E.; Green, K.; Webb, S.; Jardine, L.; Calderbank, E.F.; Polanski, K.; Goh, I.; Efremova, M.; et al. Decoding human fetal liver haematopoiesis. Nature 2019, 574, 365–371. [Google Scholar] [CrossRef]
- Goh, I.; Botting, R.A.; Rose, A.; Webb, S.; Engelbert, J.; Gitton, Y.; Stephenson, E.; Quiroga Londoño, M.; Mather, M.; Mende, N.; et al. Yolk sac cell atlas reveals multiorgan functions during human early development. Science 2023, 381, eadd7564. [Google Scholar] [CrossRef]
- Slayton, W.B.; Li, Y.; Calhoun, D.A.; Juul, S.E.; Iturraspe, J.; Braylan, R.C.; Christensen, R.D. The first-appearance of neutrophils in the human fetal bone marrow cavity. Early Hum. Dev. 1998, 53, 129–144. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef]
- Banaei-Bouchareb, L.; Peuchmaur, M.; Czernichow, P.; Polak, M. A transient microenvironment loaded mainly with macrophages in the early developing human pancreas. J. Endocrinol. 2006, 188, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Menassa, D.A.; Gomez-Nicola, D. Microglial dynamics during human brain development. Front. Immunol. 2018, 9, 1014. [Google Scholar] [CrossRef]
- Jardine, L.; Schim van der Loeff, I.; Haq, I.J.; Sproat, T.D.R. Gestational development of the human immune system. Immunol. Allergy Clin. N. Am. 2023, 43, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alsinet, C.; Primo, M.N.; Lorenzi, V.; Bello, E.; Kelava, I.; Jones, C.P.; Vilarrasa-Blasi, R.; Sancho-Serra, C.; Knights, A.J.; Park, J.E.; et al. Robust temporal map of human in vitro myelopoiesis using single-cell genomics. Nat. Commun. 2022, 13, 2885. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol. 2018, 330, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Jardine, L.; Webb, S.; Goh, I.; Quiroga Londoño, M.; Reynolds, G.; Mather, M.; Olabi, B.; Stephenson, E.; Botting, R.A.; Horsfall, D.; et al. Blood and immune development in human fetal bone marrow and Down syndrome. Nature 2021, 598, 327–331. [Google Scholar] [CrossRef]
- Schuster, C.; Vaculik, C.; Prior, M.; Fiala, C.; Mildner, M.; Eppel, W.; Stingl, G.; Elbe-Bürger, A. Phenotypic characterization of leukocytes in prenatal human dermis. J. Investig. Dermatol. 2012, 132, 2581–2592. [Google Scholar] [CrossRef]
- Msallam, R.; Balla, J.; Rathore, A.P.S.; Kared, H.; Malleret, B.; Saron, W.A.A.; Liu, Z.; Hang, J.W.; Dutertre, C.A.; Larbi, A.; et al. Fetal mast cells mediate postnatal allergic responses dependent on maternal IgE. Science 2020, 370, 941–950. [Google Scholar] [CrossRef]
- Hoorweg, K.; Narang, P.; Li, Z.; Thuery, A.; Papazian, N.; Withers, D.R.; Coles, M.C.; Cupedo, T. A stromal cell niche for human and mouse type 3 innate lymphoid cells. J. Immunol. 2015, 195, 4257–4263. [Google Scholar] [CrossRef]
- van de Pavert, S.A.; Mebius, R.E. New insights into the development of lymphoid tissues. Nat. Rev. Immunol. 2010, 10, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Leeansyah, E.; Loh, L.; Nixon, D.F.; Sandberg, J.K. Acquisition of innate-like microbial reactivity in mucosal tissues during human fetal MAIT-cell development. Nat. Commun. 2014, 5, 3143. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Lim, K.; Sun, D.; Pett, J.P.; Jeng, Q.; Polanski, K.; Dong, Z.; Bolt, L.; Richardson, L.; Mamanova, L.; et al. A human fetal lung cell atlas uncovers proximal-distal gradients of differentiation and key regulators of epithelial fates. Cell 2022, 185, 4841–4860.e25. [Google Scholar] [CrossRef]
- Schafflick, D.; Wolbert, J.; Heming, M.; Thomas, C.; Hartlehnert, M.; Börsch, A.L.; Ricci, A.; Martín-Salamanca, S.; Li, X.; Lu, I.N.; et al. Single-cell profiling of CNS border compartment leukocytes reveals that B cells and their progenitors reside in non-diseased meninges. Nat. Neurosci. 2021, 24, 1225–1234. [Google Scholar] [CrossRef]
- Rechavi, E.; Lev, A.; Lee, Y.N.; Simon, A.J.; Yinon, Y.; Lipitz, S.; Amariglio, N.; Weisz, B.; Notarangelo, L.D.; Somech, R. Timely and spatially regulated maturation of B and T cell repertoire during human fetal development. Sci. Transl. Med. 2015, 7, 276ra225. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, M.A.; Loh, L.; Marquardt, N.; Kekäläinen, E.; Berglin, L.; Björkström, N.K.; Westgren, M.; Nixon, D.F.; Michaëlsson, J. Differentiation and functional regulation of human fetal NK cells. J. Clin. Investig. 2013, 123, 3889–3901. [Google Scholar] [CrossRef]
- Sagebiel, A.F.; Steinert, F.; Lunemann, S.; Körner, C.; Schreurs, R.; Altfeld, M.; Perez, D.; Reinshagen, K.; Bunders, M.J. Tissue-resident Eomes(+) NK cells are the major innate lymphoid cell population in human infant intestine. Nat. Commun. 2019, 10, 975. [Google Scholar] [CrossRef]
- Angelo, L.S.; Bimler, L.H.; Nikzad, R.; Aviles-Padilla, K.; Paust, S. CXCR6(+) NK cells in human fetal liver and spleen possess unique phenotypic and functional capabilities. Front. Immunol. 2019, 10, 469. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Heinly, C.S. Early human T cell development: Analysis of the human thymus at the time of initial entry of hematopoietic stem cells into the fetal thymic microenvironment. J. Exp. Med. 1995, 181, 1445–1458. [Google Scholar] [CrossRef]
- Suo, C.; Dann, E.; Goh, I.; Jardine, L.; Kleshchevnikov, V.; Park, J.E.; Botting, R.A.; Stephenson, E.; Engelbert, J.; Tuong, Z.K.; et al. Mapping the developing human immune system across organs. Science 2022, 376, eabo0510. [Google Scholar] [CrossRef]
- Wang, H.; Zúñiga-Pflücker, J.C. ThymicmMicroenvironment: Interactions between innate immune cells and developing thymocytes. Front. Immunol. 2022, 13, 885280. [Google Scholar] [CrossRef]
- Park, J.E.; Botting, R.A.; Domínguez Conde, C.; Popescu, D.M.; Lavaert, M.; Kunz, D.J.; Goh, I.; Stephenson, E.; Ragazzini, R.; Tuck, E.; et al. A cell atlas of human thymic development defines T cell repertoire formation. Science 2020, 367, eaay3224. [Google Scholar] [CrossRef]
- Zeng, Y.; Liu, C.; Gong, Y.; Bai, Z.; Hou, S.; He, J.; Bian, Z.; Li, Z.; Ni, Y.; Yan, J.; et al. Single-cell RNA sequencing resolves spatiotemporal development of pre-thymic lymphoid progenitors and thymus organogenesis in human embryos. Immunity 2019, 51, 930–948.e6. [Google Scholar] [CrossRef]
- Haynes, B.F.; Martin, M.E.; Kay, H.H.; Kurtzberg, J. Early events in human T cell ontogeny. Phenotypic characterization and immunohistologic localization of T cell precursors in early human fetal tissues. J. Exp. Med. 1988, 168, 1061–1080. [Google Scholar] [CrossRef] [PubMed]
- Rackaityte, E.; Halkias, J. Mechanisms of fetal T cell tolerance and immune regulation. Front. Immunol. 2020, 11, 588. [Google Scholar] [CrossRef] [PubMed]
- Ignacio, A.; Czyz, S.; McCoy, K.D. Early life microbiome influences on development of the mucosal innate immune system. Semin. Immunol. 2024, 73, 101885. [Google Scholar] [CrossRef]
- Mold, J.E.; Venkatasubrahmanyam, S.; Burt, T.D.; Michaëlsson, J.; Rivera, J.M.; Galkina, S.A.; Weinberg, K.; Stoddart, C.A.; McCune, J.M. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science 2010, 330, 1695–1699. [Google Scholar] [CrossRef]
- Hayakawa, S.; Ohno, N.; Okada, S.; Kobayashi, M. Significant augmentation of regulatory T cell numbers occurs during the early neonatal period. Clin. Exp. Immunol. 2017, 190, 268–279. [Google Scholar] [CrossRef]
- McDavid, A.; Laniewski, N.; Grier, A.; Gill, A.L.; Kessler, H.A.; Huyck, H.; Carbonell, E.; Holden-Wiltse, J.; Bandyopadhyay, S.; Carnahan, J.; et al. Aberrant newborn T cell and microbiota developmental trajectories predict respiratory compromise during infancy. iScience 2022, 25, 104007. [Google Scholar] [CrossRef]
- Smith, N.L.; Patel, R.K.; Reynaldi, A.; Grenier, J.K.; Wang, J.; Watson, N.B.; Nzingha, K.; Yee Mon, K.J.; Peng, S.A.; Grimson, A.; et al. Developmental origin governs CD8(+) T cell fate decisions during infection. Cell 2018, 174, 117–130.e14. [Google Scholar] [CrossRef]
- McGovern, N.; Shin, A.; Low, G.; Low, D.; Duan, K.; Yao, L.J.; Msallam, R.; Low, I.; Shadan, N.B.; Sumatoh, H.R.; et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature 2017, 546, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Feyaerts, D.; Urbschat, C.; Gaudillière, B.; Stelzer, I.A. Establishment of tissue-resident immune populations in the fetus. Semin. Immunopathol. 2022, 44, 747–766. [Google Scholar] [CrossRef] [PubMed]
- Olin, A.; Henckel, E.; Chen, Y.; Lakshmikanth, T.; Pou, C.; Mikes, J.; Gustafsson, A.; Bernhardsson, A.K.; Zhang, C.; Bohlin, K.; et al. Stereotypic immune system development in newborn children. Cell 2018, 174, 1277–1292.e14. [Google Scholar] [CrossRef]
- Schreurs, R.R.C.E.; Baumdick, M.E.; Sagebiel, A.F.; Kaufmann, M.; Mokry, M.; Klarenbeek, P.L.; Schaltenberg, N.; Steinert, F.L.; van Rijn, J.M.; Drewniak, A.; et al. Human fetal TNF-α-cytokine-producing CD4(+) effector memory T cells promote intestinal development and mediate inflammation early in life. Immunity 2019, 50, 462–476.e8. [Google Scholar] [CrossRef]
- Zhang, X.; Mozeleski, B.; Lemoine, S.; Dériaud, E.; Lim, A.; Zhivaki, D.; Azria, E.; Le Ray, C.; Roguet, G.; Launay, O.; et al. CD4 T cells with effector memory phenotype and function develop in the sterile environment of the fetus. Sci. Transl. Med. 2014, 6, 238ra272. [Google Scholar] [CrossRef]
- Li, N.; van Unen, V.; Guo, N.; Abdelaal, T.; Somarakis, A.; Eggermont, J.; Mahfouz, A.; Chuva de Sousa Lopes, S.M.; Lelieveldt, B.P.F.; Koning, F. Early-life compartmentalization of immune cells in human fetal tissues revealed by high-dimensional mass cytometry. Front. Immunol. 2019, 10, 1932. [Google Scholar] [CrossRef]
- Stras, S.F.; Werner, L.; Toothaker, J.M.; Olaloye, O.O.; Oldham, A.L.; McCourt, C.C.; Lee, Y.N.; Rechavi, E.; Shouval, D.S.; Konnikova, L. Maturation of the human intestinal immune system occurs early in fetal development. Dev. Cell 2019, 51, 357–373.e5. [Google Scholar] [CrossRef]
- van den Berg, J.P.; Westerbeek, E.A.; van der Klis, F.R.; Berbers, G.A.; van Elburg, R.M. Transplacental transport of IgG antibodies to preterm infants: A review of the literature. Early Hum. Dev. 2011, 87, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Hong, R. Transient hypogammaglobulinemia of infancy. J. Pediatr. 1978, 92, 521–522. [Google Scholar] [CrossRef]
- Benediktsdóttir, B. Upper airway infections in preschool children-frequency and risk factors. Scand. J. Prim. Health Care 1993, 11, 197–201. [Google Scholar] [CrossRef]
- Rijkers, G.T.; Sanders, E.A.; Breukels, M.A.; Zegers, B.J. Infant B cell responses to polysaccharide determinants. Vaccine 1998, 16, 1396–1400. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Toebes, E.A.; Zegers, B.J.; Rijkers, G.T. Role of CR2 in the human adult and neonatal in vitro antibody response to type 4 pneumococcal polysaccharide. Cell. Immunol. 1992, 143, 11–22. [Google Scholar] [CrossRef]
- Griffioen, A.W.; Franklin, S.W.; Zegers, B.J.; Rijkers, G.T. Expression and functional characteristics of the complement receptor type 2 on adult and neonatal B lymphocytes. Clin. Immunol. Immunopathol. 1993, 69, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Peset Llopis, M.J.; Harms, G.; Hardonk, M.J.; Timens, W. Human immune response to pneumococcal polysaccharides: Complement-mediated localization preferentially on CD21-positive splenic marginal zone B cells and follicular dendritic cells. J. Allergy Clin. Immunol. 1996, 97, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Timens, W.; Boes, A.; Rozeboom-Uiterwijk, T.; Poppema, S. Immaturity of the human splenic marginal zone in infancy. Possible contribution to the deficient infant immune response. J. Immunol. 1989, 143, 3200–3206. [Google Scholar] [CrossRef]
- Rijkers, G.; Dekker, K.; Berbers, G. Oswald Avery: Pioneer of bacterial vaccines and the first to discover the function of DNA. Cureus 2024, 16, e71465. [Google Scholar] [CrossRef]
- Goldman, A.S.; Schmalstieg, F.C. Karl Otto Landsteiner (1868–1943). Physician-biochemist-immunologist. J. Med. Biogr. 2019, 27, 67–75. [Google Scholar] [CrossRef]
- Zorn, E. ABO antibody development, a new look at a very old question. Transplantation 2023, 107, 2311–2312. [Google Scholar] [CrossRef]
- Adalsteinsson, S. Possible changes in the frequency of the human ABO blood groups in Iceland due to smallpox epidemics selection. Ann. Hum. Genet. 1985, 49, 275–281. [Google Scholar] [CrossRef]
- Comans-Bitter, W.M.; de Groot, R.; van den Beemd, R.; Neijens, H.J.; Hop, W.C.; Groeneveld, K.; Hooijkaas, H.; van Dongen, J.J. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J. Pediatr. 1997, 130, 388–393. [Google Scholar] [CrossRef]
- Blanco, E.; Pérez-Andrés, M.; Arriba-Méndez, S.; Contreras-Sanfeliciano, T.; Criado, I.; Pelak, O.; Serra-Caetano, A.; Romero, A.; Puig, N.; Remesal, A.; et al. Age-associated distribution of normal B-cell and plasma cell subsets in peripheral blood. J. Allergy Clin. Immunol. 2018, 141, 2208–2219.e16. [Google Scholar] [CrossRef]
- Irjala, K.; Koskinen, P.; Icen, A.; Palosuo, T. Reference intervals for immunoglobulins IgA, IgG and IgM in serum in adults and in children aged 6 months to 14 years. Scand. J. Clin. Lab. Investig. 1990, 50, 573–577. [Google Scholar] [CrossRef]
- Grunewald, O.; Lopez, B.; Brabant, S.; Rogeau, S.; Deschildre, A.; Phrommavanh, V.; Lefort, M.; Moitrot, E.; Gyselinckx, D.; Deleplancque, A.S.; et al. Immunoglobulin G (IgG) and IgG subclass reference intervals in children, using Optilite® reagents. Clin. Chem. Lab. Med. 2018, 56, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Nikita, L.; Galuppi, M.; Lamont, R.F.; Chaemsaithong, P.; Miranda, J.; et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2014, 2, 4. [Google Scholar] [CrossRef]
- Altemani, F.; Barrett, H.L.; Gomez-Arango, L.; Josh, P.; David McIntyre, H.; Callaway, L.K.; Morrison, M.; Tyson, G.W.; Dekker Nitert, M. Pregnant women who develop preeclampsia have lower abundance of the butyrate-producer Coprococcus in their gut microbiota. Pregnancy Hypertens. 2021, 23, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cai, X.; Fei, W.; Ye, Y.; Zhao, M.; Zheng, C. The role of short-chain fatty acids in immunity, inflammation and metabolism. Crit. Rev. Food Sci. Nutr. 2022, 62, 1–12. [Google Scholar] [CrossRef]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.; Yim, Y.S.; Ha, S.; Atarashi, K.; Tan, T.G.; Longman, R.S.; Honda, K.; Littman, D.R.; Choi, G.B.; et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 2017, 549, 528–532. [Google Scholar] [CrossRef]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra265. [Google Scholar] [CrossRef]
- Marques, A.H.; O’Connor, T.G.; Roth, C.; Susser, E.; Bjørke-Monsen, A.L. The influence of maternal prenatal and early childhood nutrition and maternal prenatal stress on offspring immune system development and neurodevelopmental disorders. Front. Neurosci. 2013, 7, 120. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.E.; O’Brien, E.C.; Moore, R.L.; Byrne, D.F.; Geraghty, A.A.; Saldova, R.; Murphy, E.F.; Van Sinderen, D.; Cotter, P.D.; McAuliffe, F.M. The association between the maternal diet and the maternal and infant gut microbiome: A systematic review. Br. J. Nutr. 2023, 129, 1491–1499. [Google Scholar] [CrossRef]
- Chu, D.M.; Antony, K.M.; Ma, J.; Prince, A.L.; Showalter, L.; Moller, M.; Aagaard, K.M. The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Med. 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.N.; McKenzie, C.I.; Shen, S.; Stanley, D.; Macia, L.; Mason, L.J.; Roberts, L.K.; Wong, C.H.; Shim, R.; Robert, R.; et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat. Commun. 2015, 6, 7320. [Google Scholar] [CrossRef]
- Chu, D.M.; Meyer, K.M.; Prince, A.L.; Aagaard, K.M. Impact of maternal nutrition in pregnancy and lactation on offspring gut microbial composition and function. Gut Microbes 2016, 7, 459–470. [Google Scholar] [CrossRef]
- Frias, A.E.; Morgan, T.K.; Evans, A.E.; Rasanen, J.; Oh, K.Y.; Thornburg, K.L.; Grove, K.L. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 2011, 152, 2456–2464. [Google Scholar] [CrossRef]
- Myles, I.A.; Fontecilla, N.M.; Janelsins, B.M.; Vithayathil, P.J.; Segre, J.A.; Datta, S.K. Parental dietary fat intake alters offspring microbiome and immunity. J. Immunol. 2013, 191, 3200–3209. [Google Scholar] [CrossRef]
- Pronovost, G.N.; Yu, K.B.; Coley-O’Rourke, E.J.L.; Telang, S.S.; Chen, A.S.; Vuong, H.E.; Williams, D.W.; Chandra, A.; Rendon, T.K.; Paramo, J.; et al. The maternal microbiome promotes placental development in mice. Sci. Adv. 2023, 9, eadk1887. [Google Scholar] [CrossRef]
- Lee, H.S. Impact of maternal diet on the epigenome during in utero life and the developmental programming of diseases in childhood and adulthood. Nutrients 2015, 7, 9492–9507. [Google Scholar] [CrossRef] [PubMed]
- Jašarević, E.; Howard, C.D.; Misic, A.M.; Beiting, D.P.; Bale, T.L. Stress during pregnancy alters temporal and spatial dynamics of the maternal and offspring microbiome in a sex-specific manner. Sci. Rep. 2017, 7, 44182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zhou, Y.; Liu, B.; Jin, Z.; Zhuang, X.; Dai, W.; Yang, Z.; Feng, X.; Zhou, Q.; Liu, Y.; et al. Perinatal antibiotic exposure affects the transmission between maternal and neonatal microbiota and is associated with early-onset sepsis. mSphere 2020, 5, e00984. [Google Scholar] [CrossRef]
- Stokholm, J.; Schjørring, S.; Eskildsen, C.E.; Pedersen, L.; Bischoff, A.L.; Følsgaard, N.; Carson, C.G.; Chawes, B.L.; Bønnelykke, K.; Mølgaard, A.; et al. Antibiotic use during pregnancy alters the commensal vaginal microbiota. Clin. Microbiol. Infect. 2014, 20, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Tormo-Badia, N.; Håkansson, Å.; Vasudevan, K.; Molin, G.; Ahrné, S.; Cilio, C.M. Antibiotic treatment of pregnant non-obese diabetic mice leads to altered gut microbiota and intestinal immunological changes in the offspring. Scand. J. Immunol. 2014, 80, 250–260. [Google Scholar] [CrossRef]
- Madany, A.M.; Hughes, H.K.; Ashwood, P. Antibiotic treatment during pregnancy alters offspring gut microbiota in a sex-dependent manner. Biomedicines 2022, 10, 1042. [Google Scholar] [CrossRef]
- Lawn, J.E.; Cousens, S.; Zupan, J. 4 million neonatal deaths: When? Where? Why? Lancet 2005, 365, 891–900. [Google Scholar] [CrossRef]
- Goldenberg, R.L.; Hauth, J.C.; Andrews, W.W. Intrauterine infection and preterm delivery. N. Engl. J. Med. 2000, 342, 1500–1507. [Google Scholar] [CrossRef]
- Perez-Muñoz, M.E.; Arrieta, M.C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036. [Google Scholar] [CrossRef]
- Dos Santos, S.J.; Pakzad, Z.; Elwood, C.N.; Albert, A.Y.K.; Gantt, S.; Manges, A.R.; Dumonceaux, T.J.; Maan, E.J.; Hill, J.E.; Money, D.M. Early neonatal meconium does not have a demonstrable microbiota determined through use of robust negative controls with cpn60-based microbiome profiling. Microbiol. Spectr. 2021, 9, e0006721. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, K.M.; Gerlach, M.J.; Adam, T.; Heimesaat, M.M.; Rossi, L.; Surette, M.G.; Sloboda, D.M.; Braun, T. Fetal meconium does not have a detectable microbiota before birth. Nat. Microbiol. 2021, 6, 865–873. [Google Scholar] [CrossRef]
- Shi, Y.C.; Guo, H.; Chen, J.; Sun, G.; Ren, R.R.; Guo, M.Z.; Peng, L.H.; Yang, Y.S. Initial meconium microbiome in Chinese neonates delivered naturally or by cesarean section. Sci. Rep. 2018, 8, 3255. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W.M.; Brace, R.A. Amniotic fluid volume and normal flows to and from the amniotic cavity. Semin. Perinatol. 1993, 17, 150–157. [Google Scholar]
- Jiménez, E.; Fernández, L.; Marín, M.L.; Martín, R.; Odriozola, J.M.; Nueno-Palop, C.; Narbad, A.; Olivares, M.; Xaus, J.; Rodríguez, J.M. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr. Microbiol. 2005, 51, 270–274. [Google Scholar] [CrossRef]
- Jiménez, E.; Marín, M.L.; Martín, R.; Odriozola, J.M.; Olivares, M.; Xaus, J.; Fernández, L.; Rodríguez, J.M. Is meconium from healthy newborns actually sterile? Res. Microbiol. 2008, 159, 187–193. [Google Scholar] [CrossRef]
- Moles, L.; Gómez, M.; Heilig, H.; Bustos, G.; Fuentes, S.; de Vos, W.; Fernández, L.; Rodríguez, J.M.; Jiménez, E. Bacterial diversity in meconium of preterm neonates and evolution of their fecal microbiota during the first month of life. PLoS ONE 2013, 8, e66986. [Google Scholar] [CrossRef]
- Hansen, R.; Scott, K.P.; Khan, S.; Martin, J.C.; Berry, S.H.; Stevenson, M.; Okpapi, A.; Munro, M.J.; Hold, G.L. First-pass meconium samples from healthy term vaginally-delivered neonates: An analysis of the microbiota. PLoS ONE 2015, 10, e0133320. [Google Scholar] [CrossRef]
- Hill, C.J.; Lynch, D.B.; Murphy, K.; Ulaszewska, M.; Jeffery, I.B.; O’Shea, C.A.; Watkins, C.; Dempsey, E.; Mattivi, F.; Tuohy, K.; et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome 2017, 5, 4. [Google Scholar] [CrossRef]
- Rougé, C.; Goldenberg, O.; Ferraris, L.; Berger, B.; Rochat, F.; Legrand, A.; Göbel, U.B.; Vodovar, M.; Voyer, M.; Rozé, J.C.; et al. Investigation of the intestinal microbiota in preterm infants using different methods. Anaerobe 2010, 16, 362–370. [Google Scholar] [CrossRef]
- Cong, X.; Xu, W.; Janton, S.; Henderson, W.A.; Matson, A.; McGrath, J.M.; Maas, K.; Graf, J. Gut microbiome developmental patterns in early life of preterm infants: Impacts of feeding and gender. PLoS ONE 2016, 11, e0152751. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, A.; Neveu, D.; Aujoulat, F.; Mercier, G.; Marchandin, H.; Jumas-Bilak, E.; Picaud, J.C. Dynamics and clinical evolution of bacterial gut microflora in extremely premature patients. J. Pediatr. 2011, 158, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Arboleya, S.; Binetti, A.; Salazar, N.; Fernández, N.; Solís, G.; Hernández-Barranco, A.; Margolles, A.; de Los Reyes-Gavilán, C.G.; Gueimonde, M. Establishment and development of intestinal microbiota in preterm neonates. FEMS Microbiol. Ecol. 2012, 79, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Arboleya, S.; Sánchez, B.; Milani, C.; Duranti, S.; Solís, G.; Fernández, N.; de los Reyes-Gavilán, C.G.; Ventura, M.; Margolles, A.; Gueimonde, M. Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. J. Pediatr. 2015, 166, 538–544. [Google Scholar] [CrossRef]
- Rutten, N.B.; Rijkers, G.T.; Meijssen, C.B.; Crijns, C.E.; Oudshoorn, J.H.; van der Ent, C.K.; Vlieger, A.M. Intestinal microbiota composition after antibiotic treatment in early life: The INCA study. BMC Pediatr. 2015, 15, 204. [Google Scholar] [CrossRef]
- Cooke, R.J. Improving growth in preterm infants during initial hospital stay: Principles into practice. Arch. Dis. Child. Fetal Neonatal Ed. 2016, 101, F366–F370. [Google Scholar] [CrossRef]
- Tadros, J.S.; Llerena, A.; Sarkar, A.; Johnson, R.; Miller, E.M.; Gray, H.L.; Ho, T.T.B. Postnatal growth and gut microbiota development influenced early childhood growth in preterm infants. Front. Pediatr. 2022, 10, 850629. [Google Scholar] [CrossRef]
- van den Akker, C.H.P.; van Goudoever, J.B.; Shamir, R.; Domellöf, M.; Embleton, N.D.; Hojsak, I.; Lapillonne, A.; Mihatsch, W.A.; Berni Canani, R.; Bronsky, J.; et al. Probiotics and preterm infants: A position paper by the European Society for Paediatric Gastroenterology Hepatology and Nutrition Committee on Nutrition and the European Society for Paediatric Gastroenterology Hepatology and Nutrition Working Group for Probiotics and Prebiotics. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 664–680. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef]
- Yang, R.; Gao, R.; Cui, S.; Zhong, H.; Zhang, X.; Chen, Y.; Wang, J.; Qin, H. Dynamic signatures of gut microbiota and influences of delivery and feeding modes during the first 6 months of life. Physiol. Genom. 2019, 51, 368–378. [Google Scholar] [CrossRef]
- Montoya-Williams, D.; Lemas, D.J.; Spiryda, L.; Patel, K.; Carney, O.O.; Neu, J.; Carson, T.L. The neonatal microbiome and its partial role in mediating the association between birth by Cesarean section and adverse pediatric outcomes. Neonatology 2018, 114, 103–111. [Google Scholar] [CrossRef]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef]
- Korpela, K.; de Vos, W.M. Early life colonization of the human gut: Microbes matter everywhere. Curr. Opin. Microbiol. 2018, 44, 70–78. [Google Scholar] [CrossRef]
- Sandall, J.; Tribe, R.M.; Avery, L.; Mola, G.; Visser, G.H.; Homer, C.S.; Gibbons, D.; Kelly, N.M.; Kennedy, H.P.; Kidanto, H.; et al. Short-term and long-term effects of caesarean section on the health of women and children. Lancet 2018, 392, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Słabuszewska-Jóźwiak, A.; Szymański, J.K.; Ciebiera, M.; Sarecka-Hujar, B.; Jakiel, G. Pediatrics consequences of caesarean section-A systematic review and meta-analysis. Int. J. Environ. Res. Public Health 2020, 17, 8031. [Google Scholar] [CrossRef] [PubMed]
- Hyde, M.J.; Modi, N. The long-term effects of birth by caesarean section: The case for a randomised controlled trial. Early Hum. Dev. 2012, 88, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.L. Caesarean delivery, immune function and inflammation in early life among Ecuadorian infants and young children. J. Dev. Orig. Health Dis. 2019, 10, 555–562. [Google Scholar] [CrossRef]
- Keag, O.E.; Norman, J.E.; Stock, S.J. Long-term risks and benefits associated with cesarean delivery for mother, baby, and subsequent pregnancies: Systematic review and meta-analysis. PLoS Med. 2018, 15, e1002494. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, J.; Bai, S.; Du, S.; Yang, X.; Wang, Z. Short-term and long-term effects of cesarean section on asthma and wheezing: A cohort study and meta-analysis. Respir. Med. 2023, 215, 107300. [Google Scholar] [CrossRef]
- Neu, J.; Rushing, J. Cesarean versus vaginal delivery: Long-term infant outcomes and the hygiene hypothesis. Clin. Perinatol. 2011, 38, 321–331. [Google Scholar] [CrossRef]
- Chua, W.C.; Chen, Y.L.; Yen, C.F.; Chen, H.L. Long-term health outcomes of children born by cesarean section: A nationwide population-based retrospective cohort study in Taiwan. J. Formos. Med. Assoc. 2024, 1, 00444. [Google Scholar] [CrossRef] [PubMed]
- Stinson, L.F.; Payne, M.S.; Keelan, J.A. A critical review of the bacterial baptism hypothesis and the impact of Cesarean delivery on the infant microbiome. Front. Med. 2018, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- Wernroth, M.L.; Peura, S.; Hedman, A.M.; Hetty, S.; Vicenzi, S.; Kennedy, B.; Fall, K.; Svennblad, B.; Andolf, E.; Pershagen, G.; et al. Development of gut microbiota during the first 2 years of life. Sci. Rep. 2022, 12, 9080. [Google Scholar] [CrossRef] [PubMed]
- Barker-Tejeda, T.C.; Zubeldia-Varela, E.; Macías-Camero, A.; Alonso, L.; Martín-Antoniano, I.A.; Rey-Stolle, M.F.; Mera-Berriatua, L.; Bazire, R.; Cabrera-Freitag, P.; Shanmuganathan, M.; et al. Comparative characterization of the infant gut microbiome and their maternal lineage by a multi-omics approach. Nat. Commun. 2024, 15, 3004. [Google Scholar] [CrossRef]
- Sakanaka, M.; Gotoh, A.; Yoshida, K.; Odamaki, T.; Koguchi, H.; Xiao, J.Z.; Kitaoka, M.; Katayama, T. Varied pathways of infant gut-associated Bifidobacterium to assimilate human milk oligosaccharides: Prevalence of the gene set and its correlation with Bifidobacteria-rich microbiota formation. Nutrients 2019, 12, 71. [Google Scholar] [CrossRef]
- Alcon-Giner, C.; Dalby, M.J.; Caim, S.; Ketskemety, J.; Shaw, A.; Sim, K.; Lawson, M.A.E.; Kiu, R.; Leclaire, C.; Chalklen, L.; et al. Microbiota supplementation with Bifidobacterium and Lactobacillus modifies the preterm infant gut microbiota and metabolome: An observational study. Cell Rep. Med. 2020, 1, 100077. [Google Scholar] [CrossRef]
- Jamyuang, C.; Phoonlapdacha, P.; Chongviriyaphan, N.; Chanput, W.; Nitisinprasert, S.; Nakphaichit, M. Characterization and probiotic properties of Lactobacilli from human breast milk. 3 Biotech 2019, 9, 398. [Google Scholar] [CrossRef]
- Al Radaideh, A.J.; Badran, E.F.; Shehabi, A.A. Diversity of toxin genotypes and antimicrobial susceptibility of Clostridium perfringens isolates from feces of infants. Germs 2019, 9, 28–34. [Google Scholar] [CrossRef]
- Kim, Y.G.; Sakamoto, K.; Seo, S.U.; Pickard, J.M.; Gillilland, M.G., 3rd; Pudlo, N.A.; Hoostal, M.; Li, X.; Wang, T.D.; Feehley, T.; et al. Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science 2017, 356, 315–319. [Google Scholar] [CrossRef]
- Gregory, K.E.; LaPlante, R.D.; Shan, G.; Kumar, D.V.; Gregas, M. Mode of birth influences preterm infant intestinal colonization with bacteroides over the early neonatal period. Adv. Neonatal Care 2015, 15, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Shimojo, N.; Tajiri, Y.; Kumemura, M.; Kohno, Y. A quantitative and relative increase in intestinal bacteroides in allergic infants in rural Japan. Asian Pac. J. Allergy Immunol. 2008, 26, 113–119. [Google Scholar] [PubMed]
- Biagioli, V.; Volpedo, G.; Riva, A.; Mainardi, P.; Striano, P. From birth to weaning: A window of opportunity for microbiota. Nutrients 2024, 16, 272. [Google Scholar] [CrossRef]
- Schwab, C. The development of human gut microbiota fermentation capacity during the first year of life. Microb. Biotechnol. 2022, 15, 2865–2874. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- van Hoffen, E.; Ruiter, B.; Faber, J.; M’Rabet, L.; Knol, E.F.; Stahl, B.; Arslanoglu, S.; Moro, G.; Boehm, G.; Garssen, J. A specific mixture of short-chain galacto-oligosaccharides and long-chain fructo-oligosaccharides induces a beneficial immunoglobulin profile in infants at high risk for allergy. Allergy 2009, 64, 484–487. [Google Scholar] [CrossRef]
- Zuurveld, M.; Ayechu-Muruzabal, V.; Folkerts, G.; Garssen, J.; Van’t Land, B.; Willemsen, L.E.M. Specific human milk oligosaccharides differentially promote Th1 and regulatory responses in a CpG-activated epithelial/immune cell coculture. Biomolecules 2023, 13, 263. [Google Scholar] [CrossRef]
- Zuurveld, M.; Kiliaan, P.C.J.; van Grinsven, S.E.L.; Folkerts, G.; Garssen, J.; Van’t Land, B.; Willemsen, L.E.M. Ovalbumin-induced epithelial activation directs monocyte-derived dendritic cells to instruct type 2 inflammation in T cells which is differentially modulated by 2′-fucosyllactose and 3-fucosyllactose. J. Innate Immun. 2023, 15, 222–239. [Google Scholar] [CrossRef]
- Rahman, T.; Sarwar, P.F.; Potter, C.; Comstock, S.S.; Klepac-Ceraj, V. Role of human milk oligosaccharide metabolizing bacteria in the development of atopic dermatitis/eczema. Front. Pediatr. 2023, 11, 1090048. [Google Scholar] [CrossRef]
- Qasem, W.; Azad, M.B.; Hossain, Z.; Azad, E.; Jorgensen, S.; Castillo San Juan, S.; Cai, C.; Khafipour, E.; Beta, T.; Roberts, L.J., 2nd; et al. Assessment of complementary feeding of Canadian infants: Effects on microbiome & oxidative stress, a randomized controlled trial. BMC Pediatr. 2017, 17, 54. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Dulauroy, S.; Marques, R.; Cousu, C.; Al Bounny, S.; Déjardin, F.; Sparwasser, T.; Bérard, M.; Cerf-Bensussan, N.; Eberl, G. A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity 2019, 50, 1276–1288.e5. [Google Scholar] [CrossRef]
- Dizzell, S.; Stearns, J.C.; Li, J.; van Best, N.; Bervoets, L.; Mommers, M.; Penders, J.; Morrison, K.M.; Hutton, E.K. Investigating colonization patterns of the infant gut microbiome during the introduction of solid food and weaning from breastmilk: A cohort study protocol. PLoS ONE 2021, 16, e0248924. [Google Scholar] [CrossRef] [PubMed]
- Wiertsema, S.P.; van Bergenhenegouwen, J.; Garssen, J.; Knippels, L.M.J. The interplay between the gut microbiome and the immune system in the context of infectious diseases throughout life and the role of nutrition in optimizing treatment strategies. Nutrients 2021, 13, 886. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut microbiota and immune system interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Mulligan, C.M.; Friedman, J.E. Maternal modifiers of the infant gut microbiota: Metabolic consequences. J. Endocrinol. 2017, 235, R1–R12. [Google Scholar] [CrossRef] [PubMed]
- Golubkova, A.; Hunter, C.J. Development of the neonatal intestinal barrier, microbiome, and susceptibility to NEC. Microorganisms 2023, 11, 1247. [Google Scholar] [CrossRef] [PubMed]
- Iacob, S.; Iacob, D.G.; Luminos, L.M. Intestinal microbiota as a host defense mechanism to infectious threats. Front. Microbiol. 2018, 9, 3328. [Google Scholar] [CrossRef]
- Kong, S.; Zhang, Y.H.; Zhang, W. Regulation of Intestinal Epithelial Cells Properties and Functions by Amino Acids. BioMed Res. Int. 2018, 2018, 2819154. [Google Scholar] [CrossRef]
- Lazar, V.; Ditu, L.M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.X. Microbiota-associated metabolites and related immunoregulation in colorectal cancer. Cancers 2021, 13, 4054. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Human colonic microbiota: Ecology, physiology and metabolic potential of intestinal bacteria. Scand. J. Gastroenterol. Suppl. 1997, 222, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef]
- Kim, C.H. Control of lymphocyte functions by gut microbiota-derived short-chain fatty acids. Cell. Mol. Immunol. 2021, 18, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, J.I.; Klont, E.; van Overveld, F.J.; Rijkers, G.T. The long and winding road of faecal microbiota transplants to targeted intervention for improvement of immune checkpoint inhibition therapy. Expert Rev. Anticancer. Ther. 2023, 23, 1179–1191. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef]
- Torow, N.; Marsland, B.J.; Hornef, M.W.; Gollwitzer, E.S. Neonatal mucosal immunology. Mucosal Immunol. 2017, 10, 5–17. [Google Scholar] [CrossRef]
- Ahern, P.P.; Maloy, K.J. Understanding immune-microbiota interactions in the intestine. Immunology 2020, 159, 4–14. [Google Scholar] [CrossRef]
- Dzidic, M.; Boix-Amorós, A.; Selma-Royo, M.; Mira, A.; Collado, M.C. Gut microbiota and mucosal immunity in the neonate. Med. Sci. 2018, 6, 56. [Google Scholar] [CrossRef]
- van Es, J.H.; Jay, P.; Gregorieff, A.; van Gijn, M.E.; Jonkheer, S.; Hatzis, P.; Thiele, A.; van den Born, M.; Begthel, H.; Brabletz, T.; et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat. Cell Biol. 2005, 7, 381–386. [Google Scholar] [CrossRef]
- Thompson, J.A.; Oliveira, R.A.; Xavier, K.B. Chemical conversations in the gut microbiota. Gut Microbes 2016, 7, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, H.; Oozeer, R.; Knipping, K.; Belzer, C.; Knol, J. The first thousand days—Intestinal microbiology of early life: Establishing a symbiosis. Pediatr. Allergy Immunol. 2014, 25, 428–438. [Google Scholar] [CrossRef]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Jost, T.; Lacroix, C.; Braegger, C.P.; Chassard, C. New insights in gut microbiota establishment in healthy breast fed neonates. PLoS ONE 2012, 7, e44595. [Google Scholar] [CrossRef]
- Houghteling, P.D.; Walker, W.A. Why is initial bacterial colonization of the intestine important to infants’ and children’s health? J. Pediatr. Gastroenterol. Nutr. 2015, 60, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef]
- Managlia, E.; Yan, X.; De Plaen, I.G. Intestinal epithelial barrier function and necrotizing enterocolitis. Newborn 2022, 1, 32–43. [Google Scholar] [CrossRef]
- Yang, S.; Yu, M. Role of goblet cells in intestinal barrier and mucosal immunity. J. Inflamm. Res. 2021, 14, 3171–3183. [Google Scholar] [CrossRef]
- Godl, K.; Johansson, M.E.; Lidell, M.E.; Mörgelin, M.; Karlsson, H.; Olson, F.J.; Gum, J.R., Jr.; Kim, Y.S.; Hansson, G.C. The N terminus of the MUC2 mucin forms trimers that are held together within a trypsin-resistant core fragment. J. Biol. Chem. 2002, 277, 47248–47256. [Google Scholar] [CrossRef]
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef]
- Chairatana, P.; Nolan, E.M. Defensins, lectins, mucins, and secretory immunoglobulin A: Microbe-binding biomolecules that contribute to mucosal immunity in the human gut. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 45–56. [Google Scholar] [CrossRef]
- Birchenough, G.M.; Dalgakiran, F.; Witcomb, L.A.; Johansson, M.E.; McCarthy, A.J.; Hansson, G.C.; Taylor, P.W. Postnatal development of the small intestinal mucosa drives age-dependent, regio-selective susceptibility to Escherichia coli K1 infection. Sci. Rep. 2017, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Hui, Y.; Obelitz-Ryom, K.; Brandt, A.B.; Kot, W.; Nielsen, D.S.; Thymann, T.; Sangild, P.T.; Nguyen, D.N. Neonatal gut and immune maturation is determined more by postnatal age than by postconceptional age in moderately preterm pigs. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G855–G867. [Google Scholar] [CrossRef]
- Henderickx, J.G.E.; Zwittink, R.D.; Renes, I.B.; van Lingen, R.A.; van Zoeren-Grobben, D.; Jebbink, L.J.G.; Boeren, S.; van Elburg, R.M.; Knol, J.; Belzer, C. Maturation of the preterm gastrointestinal tract can be defined by host and microbial markers for digestion and barrier defense. Sci. Rep. 2021, 11, 12808. [Google Scholar] [CrossRef]
- Hoffmann, W. Trefoil factors TFF (trefoil factor family) peptide-triggered signals promoting mucosal restitution. Cell. Mol. Life Sci. 2005, 62, 2932–2938. [Google Scholar] [CrossRef] [PubMed]
- Bossenmeyer-Pourié, C.; Kannan, R.; Ribieras, S.; Wendling, C.; Stoll, I.; Thim, L.; Tomasetto, C.; Rio, M.C. The trefoil factor 1 participates in gastrointestinal cell differentiation by delaying G1-S phase transition and reducing apoptosis. J. Cell Biol. 2002, 157, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Jakobsson, H.E.; Holmén-Larsson, J.; Schütte, A.; Ermund, A.; Rodríguez-Piñeiro, A.M.; Arike, L.; Wising, C.; Svensson, F.; Bäckhed, F.; et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe 2015, 18, 582–592. [Google Scholar] [CrossRef]
- Berg, D.; Clemente, J.C.; Colombel, J.F. Can inflammatory bowel disease be permanently treated with short-term interventions on the microbiome? Expert Rev. Gastroenterol. Hepatol. 2015, 9, 781–795. [Google Scholar] [CrossRef]
- Schnupf, P.; Gaboriau-Routhiau, V.; Cerf-Bensussan, N. Modulation of the gut microbiota to improve innate resistance. Curr. Opin. Immunol. 2018, 54, 137–144. [Google Scholar] [CrossRef]
- Libertucci, J.; Young, V.B. The role of the microbiota in infectious diseases. Nat. Microbiol. 2019, 4, 35–45. [Google Scholar] [CrossRef]
- Frazer, L.C.; Good, M. Intestinal epithelium in early life. Mucosal Immunol. 2022, 15, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Condino, A.A.; Barleycorn, A.A.; Lu, W.; Maheshwari, A.; Christensen, R.D.; Calhoun, D.A. Abnormal intestinal histology in neonates with congenital anomalies of the gastrointestinal tract. Biol. Neonate 2004, 85, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Kayama, H.; Okumura, R.; Takeda, K. Interaction between the microbiota, epithelia, and immune cells in the intestine. Annu. Rev. Immunol. 2020, 38, 23–48. [Google Scholar] [CrossRef]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef]
- Li, H.; Myeroff, L.; Smiraglia, D.; Romero, M.F.; Pretlow, T.P.; Kasturi, L.; Lutterbaugh, J.; Rerko, R.M.; Casey, G.; Issa, J.P.; et al. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc. Natl. Acad. Sci. USA 2003, 100, 8412–8417. [Google Scholar] [CrossRef]
- Miyauchi, S.; Gopal, E.; Fei, Y.J.; Ganapathy, V. Functional identification of SLC5A8, a tumor suppressor down-regulated in colon cancer, as a Na(+)-coupled transporter for short-chain fatty acids. J. Biol. Chem. 2004, 279, 13293–13296. [Google Scholar] [CrossRef]
- Yanase, H.; Takebe, K.; Nio-Kobayashi, J.; Takahashi-Iwanaga, H.; Iwanaga, T. Cellular expression of a sodium-dependent monocarboxylate transporter (Slc5a8) and the MCT family in the mouse kidney. Histochem. Cell Biol. 2008, 130, 957–966. [Google Scholar] [CrossRef]
- Suzuki, T.; Yoshida, S.; Hara, H. Physiological concentrations of short-chain fatty acids immediately suppress colonic epithelial permeability. Br. J. Nutr. 2008, 100, 297–305. [Google Scholar] [CrossRef]
- Kim, M.; Qie, Y.; Park, J.; Kim, C.H. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe 2016, 20, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Barreto, E.B.L.; Rattes, I.C.; da Costa, A.V.; Gama, P. Paneth cells and their multiple functions. Cell Biol. Int. 2022, 46, 701–710. [Google Scholar] [CrossRef]
- Grassi, R.; Farina, R.; Floriani, I.; Amodio, F.; Romano, S. Assessment of fetal swallowing with gray-scale and color Doppler sonography. AJR Am. J. Roentgenol. 2005, 185, 1322–1327. [Google Scholar] [CrossRef]
- Battersby, C.; Santhalingam, T.; Costeloe, K.; Modi, N. Incidence of neonatal necrotising enterocolitis in high-income countries: A systematic review. Arch. Dis. Child. Fetal Neonatal Ed. 2018, 103, F182–F189. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Kong, Y.; Kleinstein, S.H.; Subramanian, S.; Ahern, P.P.; Gordon, J.I.; Medzhitov, R. Analysis of gene-environment interactions in postnatal development of the mammalian intestine. Proc. Natl. Acad. Sci. USA 2015, 112, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.H. Revisiting the hygiene hypothesis for allergy and asthma. J. Allergy Clin. Immunol. 2015, 136, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Henrick, B.M.; Rodriguez, L.; Lakshmikanth, T.; Pou, C.; Henckel, E.; Arzoomand, A.; Olin, A.; Wang, J.; Mikes, J.; Tan, Z.; et al. Bifidobacteria-mediated immune system imprinting early in life. Cell 2021, 184, 3884–3898.e11. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- Sanidad, K.Z.; Zeng, M.Y. Neonatal gut microbiome and immunity. Curr. Opin. Microbiol. 2020, 56, 30–37. [Google Scholar] [CrossRef]
- Tejesvi, M.V.; Turunen, J.; Salmi, S.; Reunanen, J.; Paalanne, N.; Tapiainen, T. Delivery mode and perinatal antibiotics influence the infant gut bacteriome and mycobiome: A network analysis. J. Fungi 2023, 9, 718. [Google Scholar] [CrossRef]
- Torow, N.; Hornef, M.W. The neonatal window of opportunity: Setting the stage for life-long host-microbial interaction and immune homeostasis. J. Immunol. 2017, 198, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.M.; et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Braun-Fahrländer, C.; Riedler, J.; Herz, U.; Eder, W.; Waser, M.; Grize, L.; Maisch, S.; Carr, D.; Gerlach, F.; Bufe, A.; et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N. Engl. J. Med. 2002, 347, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Verhasselt, V. A newborn’s perspective on immune responses to food. Immunol. Rev. 2024, 326, 117–129. [Google Scholar] [CrossRef]
- Lawson, M.A.E.; O’Neill, I.J.; Kujawska, M.; Gowrinadh Javvadi, S.; Wijeyesekera, A.; Flegg, Z.; Chalklen, L.; Hall, L.J. Breast milk-derived human milk oligosaccharides promote Bifidobacterium interactions within a single ecosystem. ISME J. 2020, 14, 635–648. [Google Scholar] [CrossRef]
- Fassarella, M.; Blaak, E.E.; Penders, J.; Nauta, A.; Smidt, H.; Zoetendal, E.G. Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 2021, 70, 595–605. [Google Scholar] [CrossRef]
- Chong, H.Y.; Tan, L.T.; Law, J.W.; Hong, K.W.; Ratnasingam, V.; Ab Mutalib, N.S.; Lee, L.H.; Letchumanan, V. Exploring the potential of human milk and formula milk on infants’ gut and health. Nutrients 2022, 14, 3554. [Google Scholar] [CrossRef]
- Tromp, I.; Kiefte-de Jong, J.; Raat, H.; Jaddoe, V.; Franco, O.; Hofman, A.; de Jongste, J.; Moll, H. Breastfeeding and the risk of respiratory tract infections after infancy: The Generation R Study. PLoS ONE 2017, 12, e0172763. [Google Scholar] [CrossRef]
- Davis, M.Y.; Zhang, H.; Brannan, L.E.; Carman, R.J.; Boone, J.H. Rapid change of fecal microbiome and disappearance of Clostridium difficile in a colonized infant after transition from breast milk to cow milk. Microbiome 2016, 4, 53. [Google Scholar] [CrossRef]
- Hussain, T.; Murtaza, G.; Kalhoro, D.H.; Kalhoro, M.S.; Yin, Y.; Chughtai, M.I.; Tan, B.; Yaseen, A.; Rehman, Z.U. Understanding the immune system in fetal protection and maternal infections during pregnancy. J. Immunol. Res. 2022, 2022, 7567708. [Google Scholar] [CrossRef]
- Gomez de Agüero, M.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The maternal microbiota drives early postnatal innate immune development. Science 2016, 351, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, M.G.; Link, V.M.; Tamoutounour, S.; Wong, A.C.; Perez-Chaparro, P.J.; Han, S.J.; Chen, Y.E.; Li, K.; Farhat, S.; Weckel, A.; et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science 2019, 366, eaax6624. [Google Scholar] [CrossRef] [PubMed]
- Luu, M.; Weigand, K.; Wedi, F.; Breidenbend, C.; Leister, H.; Pautz, S.; Adhikary, T.; Visekruna, A. Regulation of the effector function of CD8(+) T cells by gut microbiota-derived metabolite butyrate. Sci. Rep. 2018, 8, 14430. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; Deroos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Akagbosu, B.; Tayyebi, Z.; Shibu, G.; Paucar Iza, Y.A.; Deep, D.; Parisotto, Y.F.; Fisher, L.; Pasolli, H.A.; Thevin, V.; Elmentaite, R.; et al. Novel antigen-presenting cell imparts T(reg)-dependent tolerance to gut microbiota. Nature 2022, 610, 752–760. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Troy, E.B.; Kasper, D.L. Beneficial effects of Bacteroides fragilis polysaccharides on the immune system. Front. Biosci. 2010, 15, 25–34. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef]
- Liu, C.H.; Lee, S.M.; Vanlare, J.M.; Kasper, D.L.; Mazmanian, S.K. Regulation of surface architecture by symbiotic bacteria mediates host colonization. Proc. Natl. Acad. Sci. USA 2008, 105, 3951–3956. [Google Scholar] [CrossRef]
- Tzianabos, A.O.; Onderdonk, A.B.; Rosner, B.; Cisneros, R.L.; Kasper, D.L. Structural features of polysaccharides that induce intra-abdominal abscesses. Science 1993, 262, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Tzianabos, A.O.; Wang, J.Y.; Lee, J.C. Structural rationale for the modulation of abscess formation by Staphylococcus aureus capsular polysaccharides. Proc. Natl. Acad. Sci. USA 2001, 98, 9365–9370. [Google Scholar] [CrossRef]
- Tzianabos, A.O.; Finberg, R.W.; Wang, Y.; Chan, M.; Onderdonk, A.B.; Jennings, H.J.; Kasper, D.L. T cells activated by zwitterionic molecules prevent abscesses induced by pathogenic bacteria. J. Biol. Chem. 2000, 275, 6733–6740. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, R.M.; Kasper, D.L. Immunomodelation by zwitterionic polysaccharides. In Microbial Glycobiology. Structure, Relevance and Applications; Holst, O.B.P.J., von Itzstein, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 957–980. [Google Scholar] [CrossRef]
- Jia, D.; Wang, Q.; Qi, Y.; Jiang, Y.; He, J.; Lin, Y.; Sun, Y.; Xu, J.; Chen, W.; Fan, L.; et al. Microbial metabolite enhances immunotherapy efficacy by modulating T cell stemness in pan-cancer. Cell 2024, 187, 1651–1665.e21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Jiang, M.; Zhao, J.; Song, Y.; Du, W.; Shi, J. The mechanism underlying the influence of indole-3-propionic acid: A relevance to metabolic disorders. Front. Endocrinol. 2022, 13, 841703. [Google Scholar] [CrossRef]
- Wang, H.C.; Zhou, Q.; Dragoo, J.; Klein, J.R. Most murine CD8+ intestinal intraepithelial lymphocytes are partially but not fully activated T cells. J. Immunol. 2002, 169, 4717–4722. [Google Scholar] [CrossRef]
- Moretto, M.; Weiss, L.M.; Khan, I.A. Induction of a rapid and strong antigen-specific intraepithelial lymphocyte response during oral Encephalitozoon cuniculi infection. J. Immunol. 2004, 172, 4402–4409. [Google Scholar] [CrossRef]
- Boismenu, R.; Havran, W.L. Modulation of epithelial cell growth by intraepithelial gamma delta T cells. Science 1994, 266, 1253–1255. [Google Scholar] [CrossRef]
- Crabbé, P.A.; Bazin, H.; Eyssen, H.; Heremans, J.F. The normal microbial flora as a major stimulus for proliferation of plasma cells synthesizing IgA in the gut. The germ-free intestinal tract. Int. Arch. Allergy Appl. Immunol. 1968, 34, 362–375. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Yilmaz, B.; Limenitakis, J.P.; Ganal-Vonarburg, S.C. IgA function in relation to the intestinal microbiota. Annu. Rev. Immunol. 2018, 36, 359–381. [Google Scholar] [CrossRef]
- Swiatczak, B.; Rescigno, M. How the interplay between antigen presenting cells and microbiota tunes host immune responses in the gut. Semin. Immunol. 2012, 24, 43–49. [Google Scholar] [CrossRef]
- Konieczna, P.; Groeger, D.; Ziegler, M.; Frei, R.; Ferstl, R.; Shanahan, F.; Quigley, E.M.; Kiely, B.; Akdis, C.A.; O’Mahony, L. Bifidobacterium infantis 35624 administration induces Foxp3 T regulatory cells in human peripheral blood: Potential role for myeloid and plasmacytoid dendritic cells. Gut 2012, 61, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, T.R.; Levy, O.; Montgomery, R.R.; Goriely, S. Innate immune function by Toll-like receptors: Distinct responses in newborns and the elderly. Immunity 2012, 37, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Figdor, C.G.; van Kooyk, Y.; Adema, G.J. C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev. Immunol. 2002, 2, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Stefan, K.L.; Kim, M.V.; Iwasaki, A.; Kasper, D.L. Commensal microbiota modulation of natural resistance to virus infection. Cell 2020, 183, 1312–1324.e10. [Google Scholar] [CrossRef]
- Menckeberg, C.L.; Hol, J.; Simons-Oosterhuis, Y.; Raatgeep, H.R.; de Ruiter, L.F.; Lindenbergh-Kortleve, D.J.; Korteland-van Male, A.M.; El Aidy, S.; van Lierop, P.P.; Kleerebezem, M.; et al. Human buccal epithelium acquires microbial hyporesponsiveness at birth, a role for secretory leukocyte protease inhibitor. Gut 2015, 64, 884–893. [Google Scholar] [CrossRef]
- Favier, C.F.; de Vos, W.M.; Akkermans, A.D. Development of bacterial and bifidobacterial communities in feces of newborn babies. Anaerobe 2003, 9, 219–229. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, M.H.; Ligthart, I.; Varga, S.; Lebeer, S.; van Overveld, F.J.; Rijkers, G.T. Mutual Interactions Between Microbiota and the Human Immune System During the First 1000 Days of Life. Biology 2025, 14, 299. https://doi.org/10.3390/biology14030299
Tang MH, Ligthart I, Varga S, Lebeer S, van Overveld FJ, Rijkers GT. Mutual Interactions Between Microbiota and the Human Immune System During the First 1000 Days of Life. Biology. 2025; 14(3):299. https://doi.org/10.3390/biology14030299
Chicago/Turabian StyleTang, Muy Heang, Ishbel Ligthart, Samuel Varga, Sarah Lebeer, Frans J. van Overveld, and Ger T. Rijkers. 2025. "Mutual Interactions Between Microbiota and the Human Immune System During the First 1000 Days of Life" Biology 14, no. 3: 299. https://doi.org/10.3390/biology14030299
APA StyleTang, M. H., Ligthart, I., Varga, S., Lebeer, S., van Overveld, F. J., & Rijkers, G. T. (2025). Mutual Interactions Between Microbiota and the Human Immune System During the First 1000 Days of Life. Biology, 14(3), 299. https://doi.org/10.3390/biology14030299