Integrated Systems Approach Reveals Sphingolipid Metabolism Pathway Dysregulation in Association with Late-Onset Alzheimer’s Disease

Abstract
:1. Introduction
2. Materials and Methods
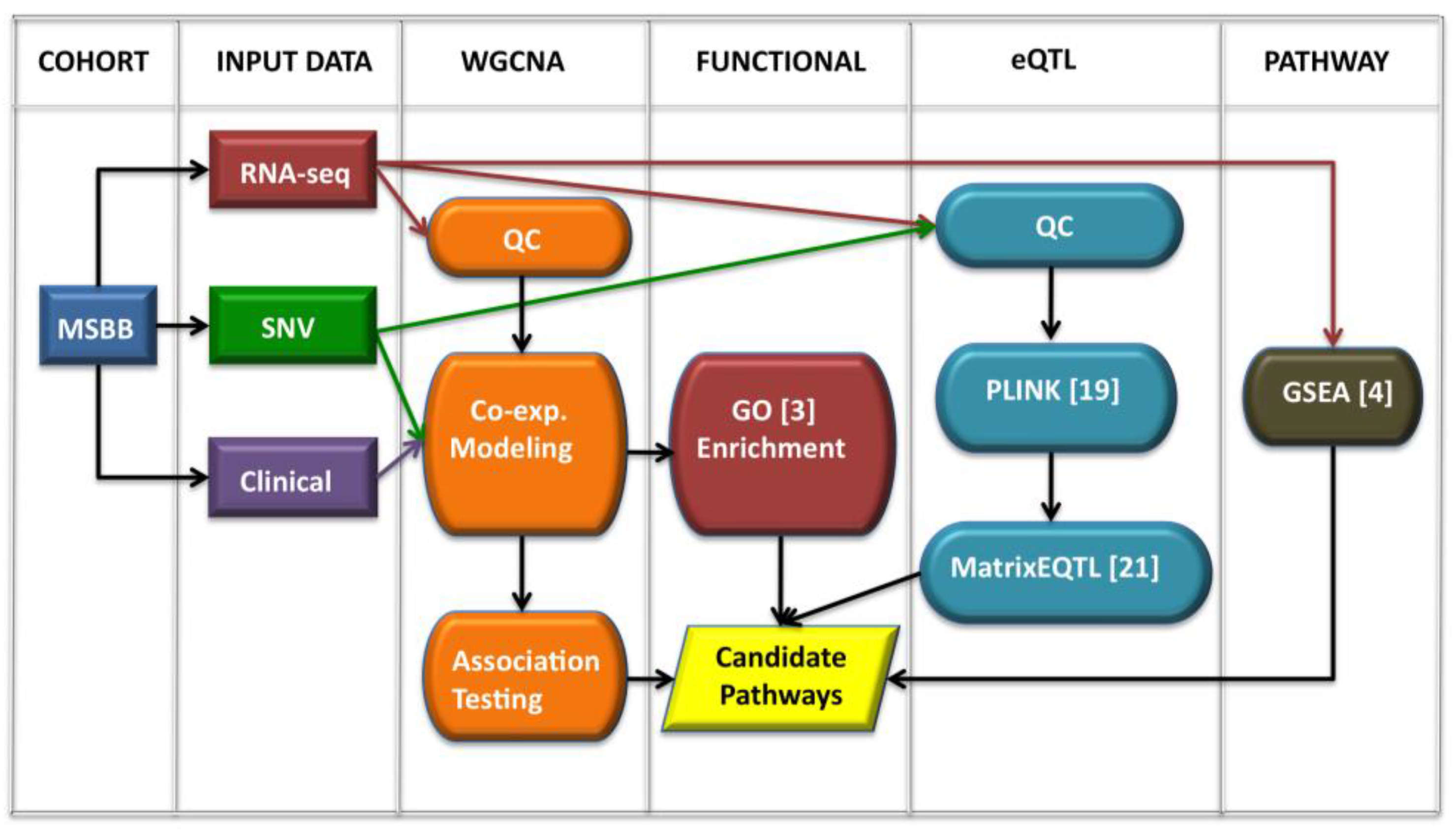
2.1. Workflow Overview
2.2. Data Description
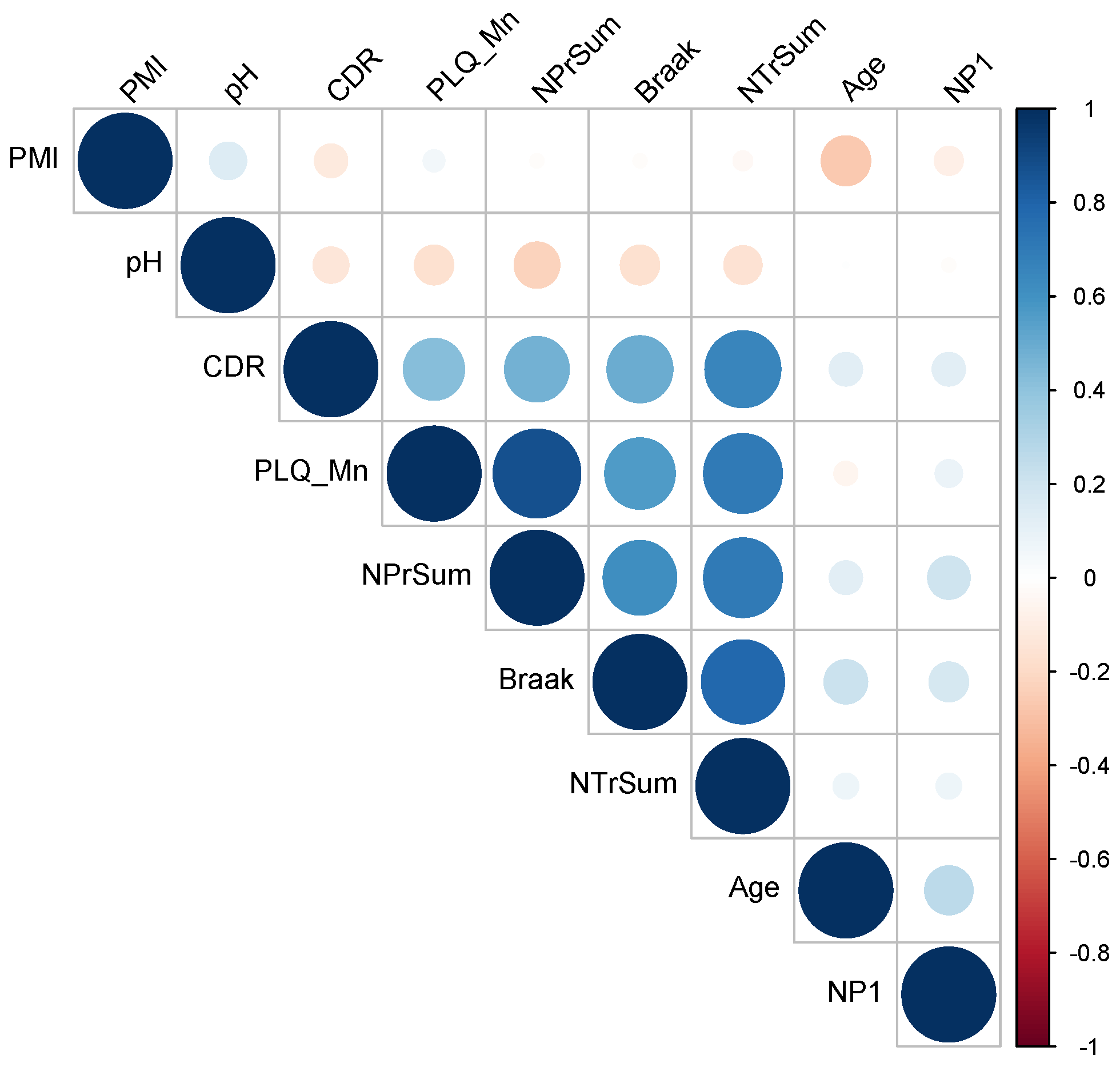
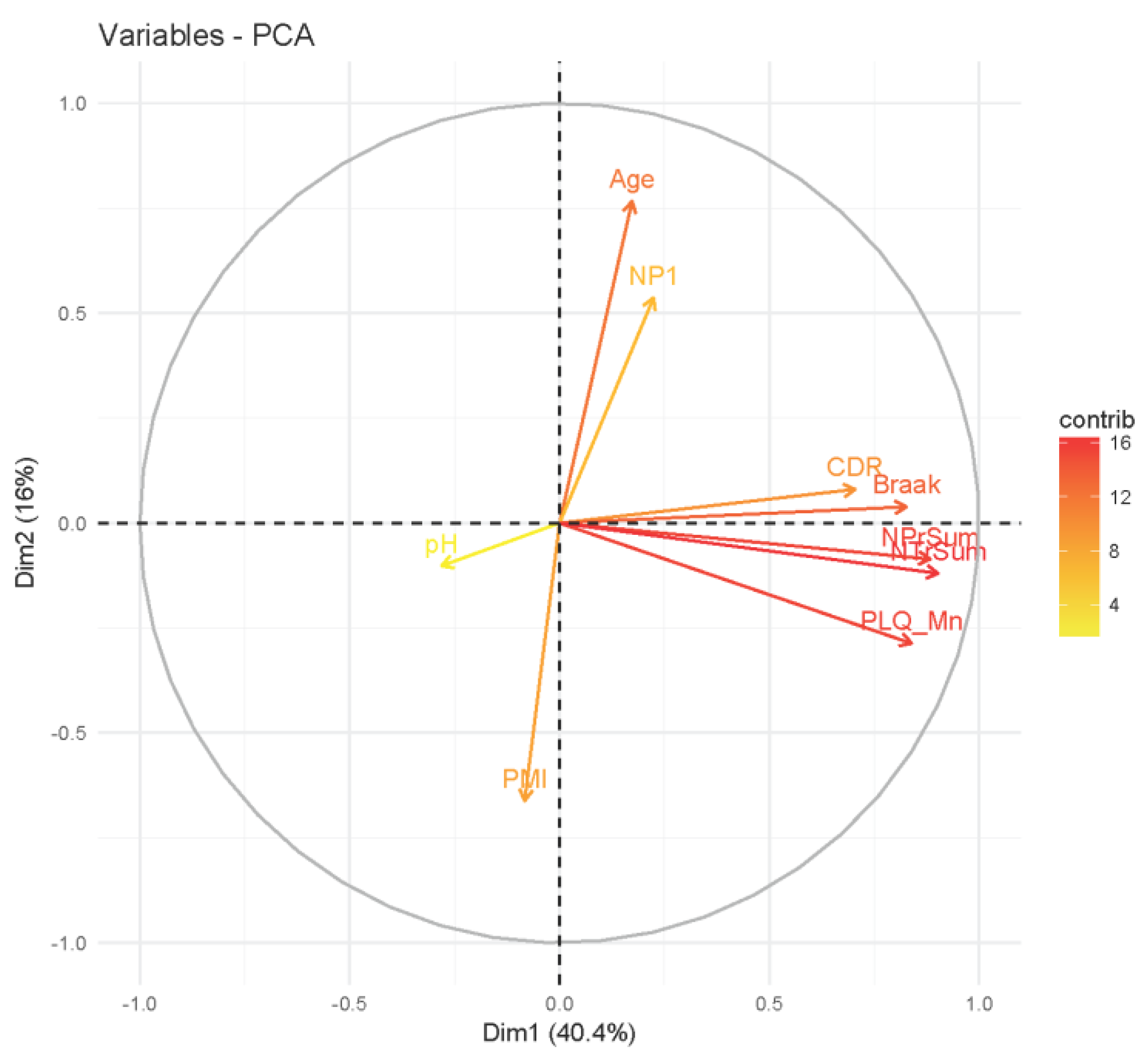
2.3. Analysis of Clinical Data
2.4. Quality Filtering of RNA-seq Data
2.5. Association Testing
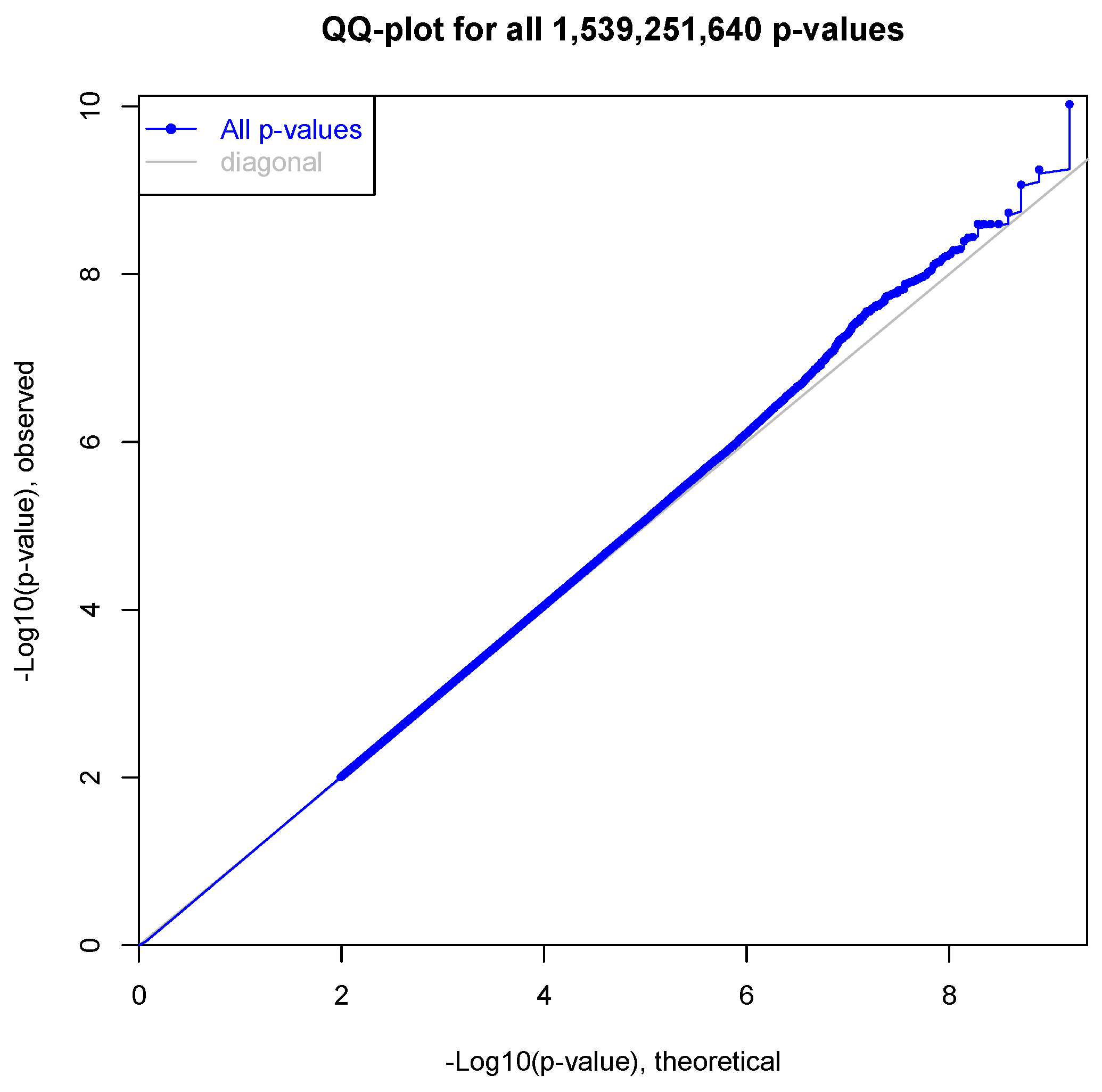
2.6. Expression Quantitative Trait Loci Analysis
2.7. Gene Set Enrichment Analysis
3. Results
3.1. Association Testing
3.2. Expression Quantitative Trait Loci
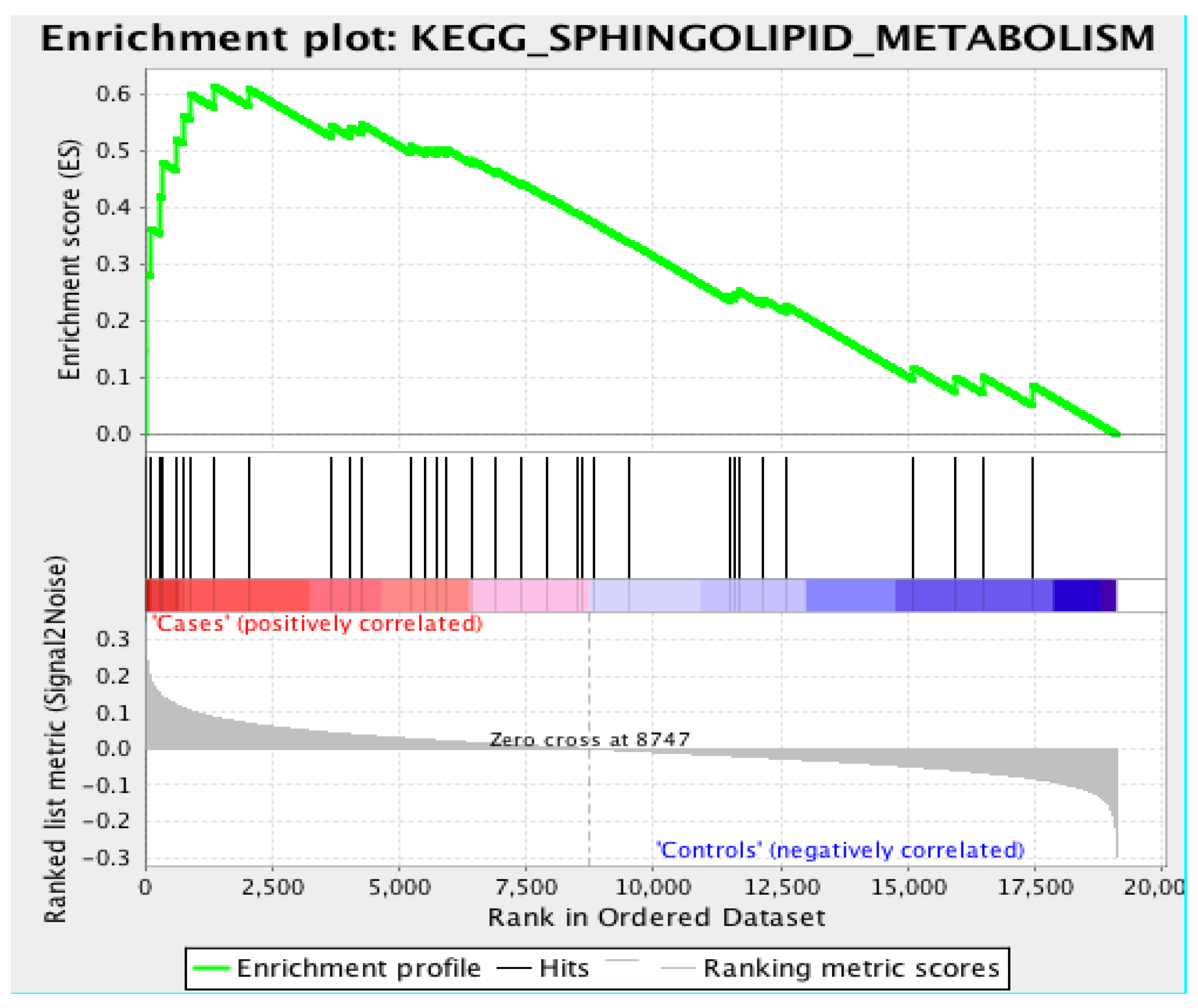
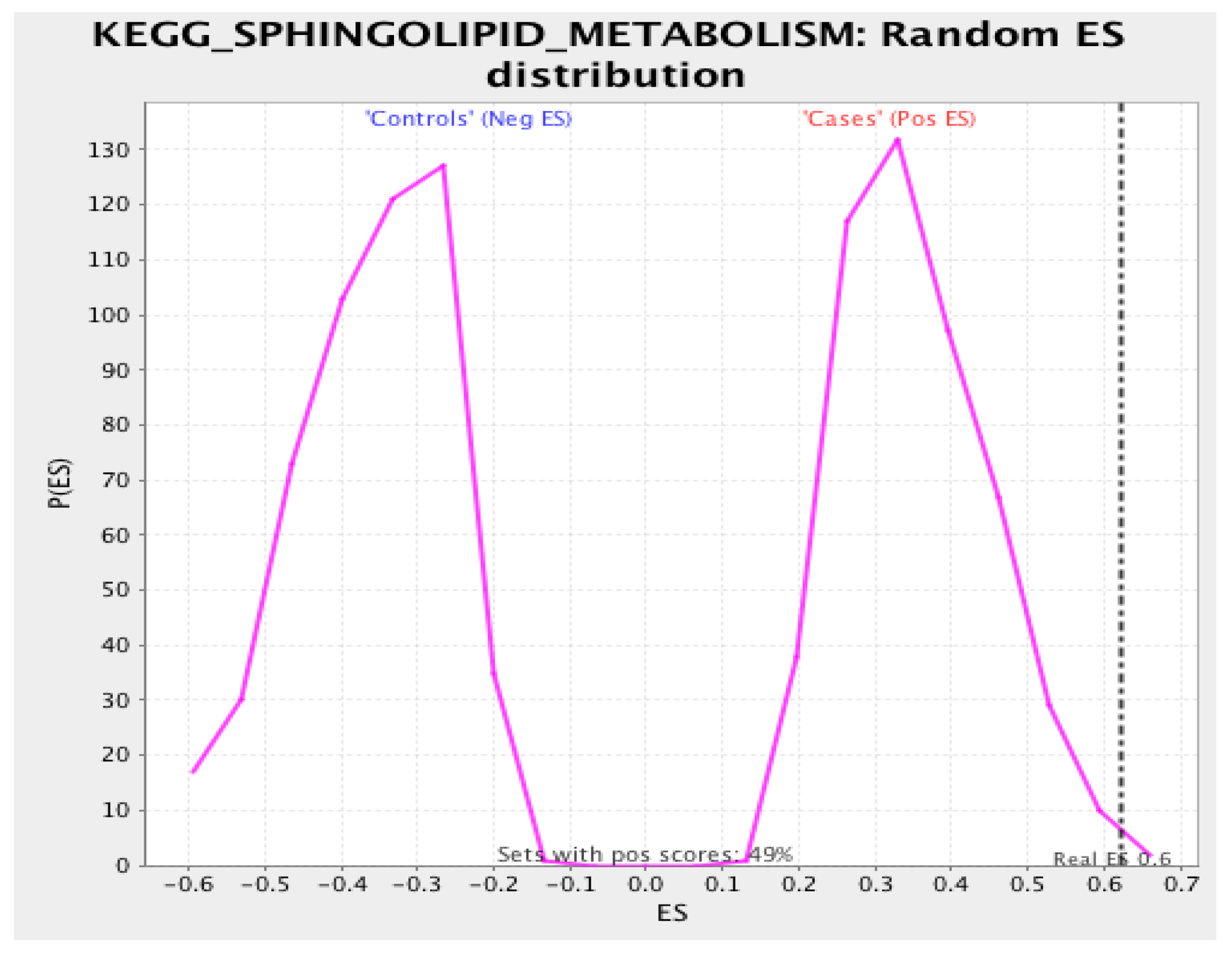
3.3. Gene Set Enrichment Analysis
4. Discussion
4.1. Biological Relevance
4.2. Sphingolipid Pathway
4.3. Myelin Maintenance
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Hurd, M.D.; Martorell, P.; Delavande, A.; Mullen, K.J.; Langa, K.M. Monetary costs of dementia in the united states. N. Engl. J. Med. 2013, 368, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. Wgcna: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Kegg as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Scheff, M.A.; Mufson, E.J. Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and alzheimer’s disease. J. Alzheimers Dis. 2011, 24, 547–557. [Google Scholar] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Lê, S.E.A. Factominer: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Benjamini, Y.A.Y.H. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- McKenzie, M.; Henders, A.K.; Caracella, A.; Wray, N.R.; Powell, J.E. Overlap of expression quantitative trait loci (eQTL) in human brain and blood. BMC Med Genom. 2014, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, A.; Trabzuni, D.; Guelfi, S.; Varghese, V.; Smith, C.; Walker, R.; De, T.; UK Brain Expression Consortium; North American Brain Expression Consortium; Coin, L.; et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 2014, 17, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S.; Boks, M.P.; Fuller, T.F.; Strengman, E.; Janson, E.; de Kovel, C.G.; Ori, A.P.; Vi, N.; Mulder, F.; Blom, J.D.; et al. A gene co-expression network in whole blood of schizophrenia patients is independent of antipsychotic-use and enriched for brain-expressed genes. PLoS ONE 2012, 7, e39498. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset al.zheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Hardy, G.H. Mendelian proportions in a mixed population. Science 1908, 28, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, A.A. Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012, 28, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Cool, B.H.; Chan, G.C.; Lee, L.; Oshima, J.; Martin, G.M.; Hu, Q. A flanking gene problem leads to the discovery of a gprc5b splice variant predominantly expressed in c57bl/6j mouse brain and in maturing neurons. PLoS ONE 2010, 5, e10351. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Mou, X.; Liu, Q.; Chen, Z.; Vanderburg, C.R.; Rogers, J.T.; Huang, X. Independent component analysis of alzheimer’s DNA microarray gene expression data. Mol. Neurodegener. 2009, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashby, E.L.; Kehoe, P.G.; Love, S. Kallikrein-related peptidase 6 in Alzheimer’s disease and vascular dementia. Brain Res. 2010, 1363, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zarghooni, M.; Soosaipillai, A.; Grass, L.; Scorilas, A.; Mirazimi, N.; Diamandis, E.P. Decreased concentration of human kallikrein 6 in brain extracts of alzheimer’s disease patients. Clin. Biochem. 2002, 35, 225–231. [Google Scholar] [CrossRef]
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; Van Cauwenberghe, C.; Kolen, K.V.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P.; et al. Increased expression of bin1 mediates alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 2013, 18, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I.; Demirci, F.Y.; Wang, X.; Minster, R.L.; Carrasquillo, M.M.; Pankratz, V.S.; Younkin, S.G.; Saykin, A.J. Alzheimer’s Disease Neuroimaging, Initiative; Jun, G.; et al. Genome-wide association study of alzheimer’s disease. Transl. Psychiatry 2012, 2, e117. [Google Scholar] [CrossRef] [PubMed]
- Barrachina, M.; Maes, T.; Buesa, C.; Ferrer, I. Lysosome-associated membrane protein 1 (lamp-1) in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2006, 32, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Ogino, K.; Shin, R.W.; Kitamoto, T.; Kikuchi, T.; Shimizu, N. Age-dependent increase in lysosome-associated membrane protein 1 and early-onset behavioral deficits in appsl transgenic mouse model of alzheimer’s disease. Neurosci. Lett. 2010, 469, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Friedman, B.A.; Larson, J.L.; Lauffer, B.E.; Goldstein, L.D.; Appling, L.L.; Borneo, J.; Poon, C.; Ho, T.; Cai, F.; et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 2016, 7, 11295. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Pan, X.L.; Wang, Y.; Tang, H.D.; Deng, Y.L.; Ren, R.J.; Xu, W.; Ma, J.F.; Wang, G.; Chen, S.D. A single nucleotide polymorphism in lrp2 is associated with susceptibility to Alzheimer’s disease in the chinese population. Clin. Chim. Acta 2011, 412, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Wavrant-De Vrieze, F.; Compton, D.; Womick, M.; Arepalli, S.; Adighibe, O.; Li, L.; Perez-Tur, J.; Hardy, J. Abca1 polymorphisms and alzheimer’s disease. Neurosci. Lett. 2007, 416, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Lalli, M.A.; Garcia, G.; Madrigal, L.; Arcos-Burgos, M.; Arcila, M.L.; Kosik, K.S.; Lopera, F. Exploratory data from complete genomes of familial alzheimer disease age-at-onset outliers. Hum. Mutat. 2012, 33, 1630–1634. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.; Song, J.H.; Tan, M.S.; Zhang, W.; Wang, Z.X.; Jiang, T.; Tan, L.; Yu, J.T. Association of single-nucleotide polymorphism in ank1 with late-onset Alzheimer’s disease in Han Chinese. Mol. Neurobiol. 2016, 53, 6476–6481. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, Y.; Shen, J.; Lv, D.; Zhang, J. Meta-analysis of bace1 gene rs638405 polymorphism and the risk of alzheimer’s disease in caucasion and asian population. Neurosci. Lett. 2016, 616, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Preda-Pais, A.; Casares, S.; Brumeanu, T.D. Analysis of lipid rafts in t cells. Mol. Immunol. 2004, 41, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Korade, Z.; Kenworthy, A.K. Lipid rafts, cholesterol, and the brain. Neuropharmacology 2008, 55, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Mangold, C.A.; Wronowski, B.; Du, M.; Masser, D.R.; Hadad, N.; Bixler, G.V.; Brucklacher, R.M.; Ford, M.M.; Sonntag, W.E.; Freeman, W.M. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J. Neuroinflamm. 2017, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Blom, T.; Li, Z.; Bittman, R.; Somerharju, P.; Ikonen, E. Tracking sphingosine metabolism and transport in sphingolipidoses: Npc1 deficiency as a test case. Traffic 2012, 13, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Kagedal, K.; Kim, W.S.; Appelqvist, H.; Chan, S.; Cheng, D.; Agholme, L.; Barnham, K.; McCann, H.; Halliday, G.; Garner, B. Increased expression of the lysosomal cholesterol transporter npc1 in alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Marks, D.L.; Park, W.D.; Wheatley, C.L.; Puri, V.; O’Brien, J.F.; Kraft, D.L.; Lundquist, P.A.; Patterson, M.C.; Pagano, R.E.; et al. Niemann-pick c variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of npc1. Am. J. Hum. Genet. 2001, 68, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Lieberman, A.P. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 2013, 9, e1003462. [Google Scholar] [CrossRef] [PubMed]
- Becker, I.; Wang-Eckhardt, L.; Yaghootfam, A.; Gieselmann, V.; Eckhardt, M. Differential expression of (dihydro)ceramide synthases in mouse brain: Oligodendrocyte-specific expression of cers2/lass2. Histochem. Cell Biol. 2008, 129, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Bosio, A.; Binczek, E.; Stoffel, W. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 13280–13285. [Google Scholar] [CrossRef] [PubMed]
- Hooijmans, C.R.; Pasker-de Jong, P.C.; de Vries, R.B.; Ritskes-Hoitinga, M. The effects of long-term omega-3 fatty acid supplementation on cognition and alzheimer’s pathology in animal models of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimers Dis. 2012, 28, 191–209. [Google Scholar] [PubMed]
- Winther, B.; Hoem, N.; Berge, K.; Reubsaet, L. Elucidation of phosphatidylcholine composition in krill oil extracted from euphausia superba. Lipids 2011, 46, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Hama, H. Fatty acid 2-hydroxylation in mammalian sphingolipid biology. Biochim. Biophys. Acta 2010, 1801, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Sassa, T.; Kihara, A. Metabolism of very long-chain fatty acids: Genes and pathophysiology. Biomol. Ther. 2014, 22, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Luke, M.P.; LeVatte, T.L.; O’Reilly, A.M.; Smith, B.J.; Tremblay, F.; Brown, R.E.; Clarke, D.B. Effect of ncam on aged-related deterioration in vision. Neurobiol. Aging 2016, 41, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Hwang, J.H.; Tang, N.W.; Hunziker, W. Juxtanodin in retinal pigment epithelial cells: Expression and biological activities in regulating cell morphology and actin cytoskeleton organization. J. Comp. Neurol. 2018, 526, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Kouri, N.; Ross, O.A.; Dombroski, B.; Younkin, C.S.; Serie, D.J.; Soto-Ortolaza, A.; Baker, M.; Finch, N.C.; Yoon, H.; Kim, J.; et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 2015, 6, 7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, J.S.; Karch, C.M.; Fan, C.C.; Bonham, L.W.; Kouri, N.; Ross, O.A.; Rademakers, R.; Kim, J.; Wang, Y.; Hoglinger, G.U.; et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 2017, 133, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Y.; Yu, J.T.; Miao, D.; Ma, X.Y.; Wang, H.F.; Wang, W.; Tan, L. An exploratory study on stx6, mobp, mapt, and eif2ak3 and late-onset Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1519.e13–1519.e17. [Google Scholar] [CrossRef] [PubMed]
- Montague, P.; McCallion, A.S.; Davies, R.W.; Griffiths, I.R. Myelin-associated oligodendrocytic basic protein: A family of abundant CNS myelin proteins in search of a function. Dev. Neurosci. 2006, 28, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.; Wang-Eckhardt, L.; Hartmann, D.; Gieselmann, V.; Eckhardt, M. N-acetylaspartate synthase deficiency corrects the myelin phenotype in a canavan disease mouse model but does not affect survival time. J. Neurosci. 2015, 35, 14501–14516. [Google Scholar] [CrossRef] [PubMed]
- Besold, A.N.; Michel, S.L. Neural zinc finger factor/myelin transcription factor proteins: Metal binding, fold, and function. Biochemistry 2015, 54, 4443–4452. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Xing, Y.; Moore, M.W.; Zhang, J.; Han, D.; Schulte, B.A.; Dubno, J.R.; Lang, H. Sox10 expressing cells in the lateral wall of the aged mouse and human cochlea. PLoS ONE 2014, 9, e97389. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Marie, C.; Zhao, C.; Kim, B.; Wang, J.; Deng, Y.; Clavairoly, A.; Frah, M.; Wang, H.; He, X.; et al. Chd7 cooperates with sox10 and regulates the onset of cns myelination and remyelination. Nat. Neurosci. 2016, 19, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Harvald, E.B.; Olsen, A.S.; Faergeman, N.J. Autophagy in the light of sphingolipid metabolism. Apoptosis 2015, 20, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Description |
|---|---|
| CDR | Cognitive Dementia Rating
|
| Braak | Quantitative assessment of neurofibrillary tangles (NTF) based anti-tau (AD2) staining |
| NP1 | Neuropathology category |
| PLQ_Mn | Mean neocortical plaque density across 5 regions (# of plaques/mm2) |
| NTr_Sum | Sum of NFT density across 3 Brodmann areas |
| NPr_Sum | Sum of neuritic plaque density across 3 Brodmann areas |
| GENE | P.WEIGHTED | FDR | COR.WEIGHTED | #OF TRAITS |
|---|---|---|---|---|
| GLTP | <4.88e−09 | <2.22e−16 | 0.8465 | 3 |
| NPC1 | 4.88e−09 | 2.22e−16 | 0.8395 | 3 |
| CERS2 | 1.46e−08 | 2.22e−12 | 0.8307 | 3 |
| ST18 | 3.41e−08 | 3.89e−12 | 0.8255 | 2 |
| NCAM1 | 1.46e−07 | 1.02e−11 | 0.8151 | 3 |
| ASPA | 2.88e−07 | 1.54e−11 | 0.8103 | 2 |
| ELOVL1 | 4.89e−06 | 3.53e−11 | 0.7944 | 3 |
| ERMN | 1.16e−05 | 6.48e−11 | 0.7945 | 3 |
| MOBP | 3.56e−05 | 1.60e−10 | 0.7851 | 2 |
| FA2H | 1.55e−05 | 3.22e−10 | 0.7774 | 1 |
| UGT8 | 2.12e−04 | 3.54e−09 | 0.7691 | 2 |
| SOX10 | 4.01e−03 | 3.44e−07 | 0.7622 | 3 |
| Gene | rsID | Beta | p-Value |
|---|---|---|---|
| GAS7 [30] | 17339499 | −1.129 | 8.60e−10 |
| LRP2 [31] | 2075252 | −0.4257 | 3.15e−07 |
| ABCA1 [32] | 2230806 | 0.7890 | 1.20e−05 |
| EPHA10 [33] | 1212384 | −0.3230 | 1.22e−05 |
| ANK1 [34] | 61063081 | 0.2079 | 1.30e−05 |
| BACE2 [35] | 2252576 | 0.4662 | 3.00e−05 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malamon, J.S.; Kriete, A. Integrated Systems Approach Reveals Sphingolipid Metabolism Pathway Dysregulation in Association with Late-Onset Alzheimer’s Disease. Biology 2018, 7, 16. https://doi.org/10.3390/biology7010016
Malamon JS, Kriete A. Integrated Systems Approach Reveals Sphingolipid Metabolism Pathway Dysregulation in Association with Late-Onset Alzheimer’s Disease. Biology. 2018; 7(1):16. https://doi.org/10.3390/biology7010016
Chicago/Turabian StyleMalamon, John Stephen, and Andres Kriete. 2018. "Integrated Systems Approach Reveals Sphingolipid Metabolism Pathway Dysregulation in Association with Late-Onset Alzheimer’s Disease" Biology 7, no. 1: 16. https://doi.org/10.3390/biology7010016
APA StyleMalamon, J. S., & Kriete, A. (2018). Integrated Systems Approach Reveals Sphingolipid Metabolism Pathway Dysregulation in Association with Late-Onset Alzheimer’s Disease. Biology, 7(1), 16. https://doi.org/10.3390/biology7010016