Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death


Abstract
1. Introduction
2. Channelopathies
2.1. Long QT Syndrome
2.2. Brugada Syndrome
2.3. Catecholaminergic Polymorphic Ventricular Tachycardia
2.4. Short QT Syndrome
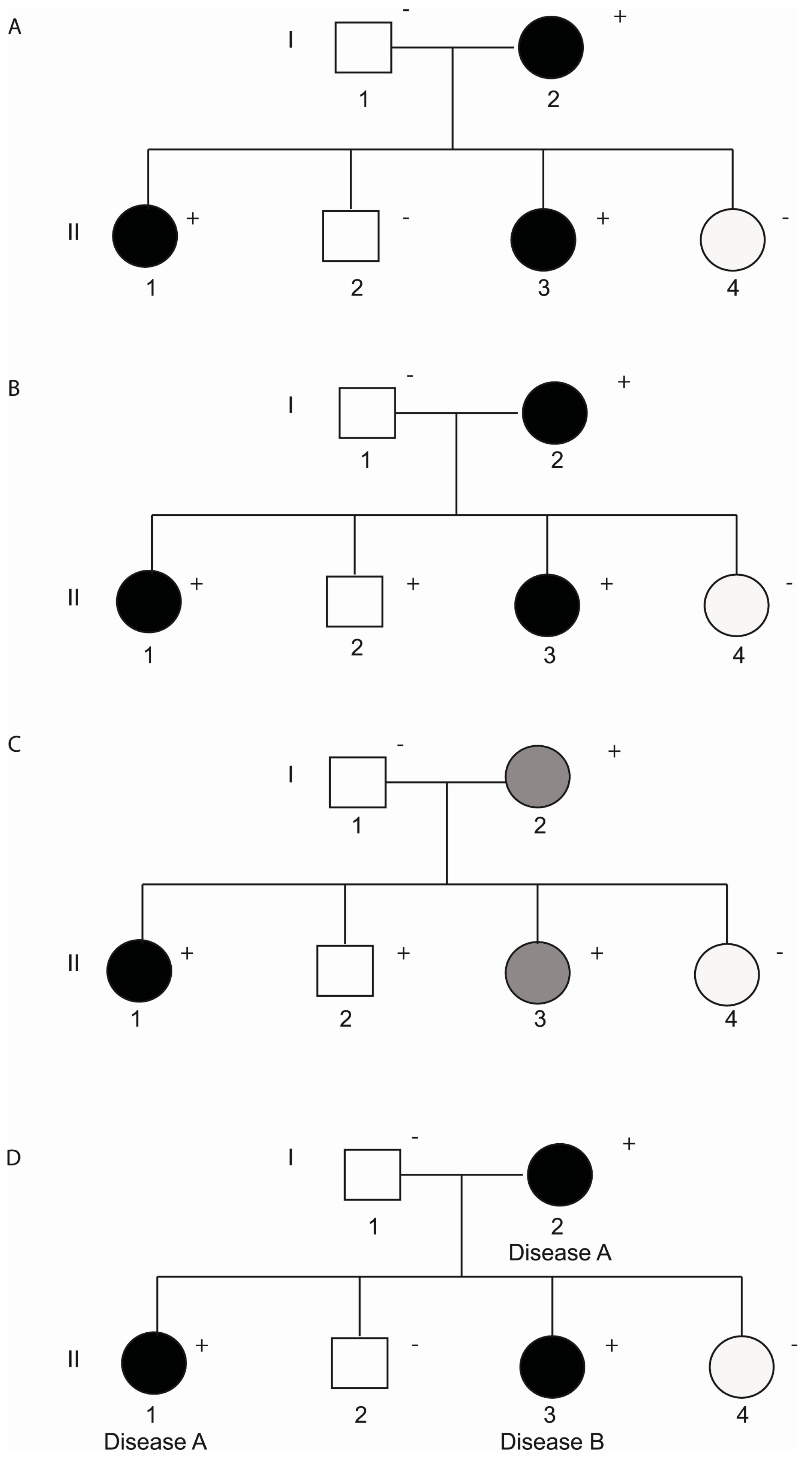
3. Incomplete Penetrance and Variable Expressivity
3.1. Non-Genetic Modifiers
3.2. Genetic Modifiers
3.2.1. Coding Variants
3.2.2. Non-Coding Variants
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bayes de Luna, A.; Elosua, R. Sudden death. Rev. Esp. Cardiol. 2012, 65, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Marsman, R.F.; Tan, H.L.; Bezzina, C.R. Genetics of sudden cardiac death caused by ventricular arrhythmias. Nat. Rev. Cardiol. 2014, 11, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.; Sinner, M.F.; Siebermair, J.; Raufhake, C.; Beckmann, B.M.; Veith, S.; Duvel, D.; Steinbeck, G.; Kaab, S. Incidence of sudden cardiac death in Germany: Results from an emergency medical service registry in Lower Saxony. Europace 2014, 16, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Brugada, R.; Brugada, J. Genetics of channelopathies associated with sudden cardiac death. Glob. Cardiol. Sci. Pract. 2015, 2015, 39. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.; Wilde, A.A. Inherited ion channel diseases: A brief review. Europace 2015. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef] [PubMed]
- Mizusawa, Y. Recent advances in genetic testing and counseling for inherited arrhythmias. J. Arrhythm. 2016, 32, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Falgueras, A.; Sarquella-Brugada, G.; Brugada, J.; Brugada, R.; Campuzano, O. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Marcou, C.A.; Tester, D.J. Personalized medicine: Genetic diagnosis for inherited cardiomyopathies/channelopathies. Rev. Esp. Cardiol. 2013, 66, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Holst, A.G.; Jensen, H.K.; Eschen, O.; Henriksen, F.L.; Kanters, J.; Bundgaard, H.; Svendsen, J.H.; Haunso, S.; Tfelt-Hansen, J. Low disease prevalence and inappropriate implantable cardioverter defibrillator shock rate in Brugada syndrome: A nationwide study. Europace 2012, 14, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K. Sudden unexplained death syndrome in Southeast Asia. Am. J. Cardiol. 1997, 79, 10–11. [Google Scholar] [CrossRef]
- Wanguemert, F.; Bosch Calero, C.; Perez, C.; Campuzano, O.; Beltran-Alvarez, P.; Scornik, F.S.; Iglesias, A.; Berne, P.; Allegue, C.; Ruiz Hernandez, P.M.; et al. Clinical and molecular characterization of a cardiac ryanodine receptor founder mutation causing catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2015, 12, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.; van der Werf, C.; Wilde, A.A. Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. J. 2016, 80, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Roux-Buisson, N.; Cacheux, M.; Fourest-Lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012, 21, 2759–2767. [Google Scholar] [CrossRef] [PubMed]
- Gussak, I.; Brugada, P.; Brugada, J.; Wright, R.S.; Kopecky, S.L.; Chaitman, B.R.; Bjerregaard, P. Idiopathic short QT interval: A new clinical syndrome? Cardiology 2000, 94, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; O’Rourke, S.; Ng, K.; Miceli, C.; Borio, G.; Curcio, A.; Esposito, F.; Napolitano, C.; Priori, S.G. The usual suspects in sudden cardiac death of the young: A focus on inherited arrhythmogenic diseases. Expert Rev. Cardiovasc. Ther. 2014, 12, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013, 15, 1389–1406. [Google Scholar] [CrossRef] [PubMed]
- Veltmann, C.; Barajas-Martinez, H.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Pfeiffer, R.; Caceres, G.; Burashnikov, E.; Antzelevitch, C.; Hu, D. Further Insights in the Most Common SCN5A Mutation Causing Overlapping Phenotype of Long QT Syndrome, Brugada Syndrome, and Conduction Defect. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ackerman, M.J. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl. Res. 2013, 161, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, M.G. Diagnostic dilemmas: Overlapping features of brugada syndrome and arrhythmogenic right ventricular cardiomyopathy. Front. Physiol. 2012, 3, 144. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Schwartz, P.J.; Bloise, R.; Crotti, L.; Ronchetti, E. The elusive link between LQT3 and Brugada syndrome: The role of flecainide challenge. Circulation 2000, 102, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Brignole, M.; Giordano, U.; Giovannini, T.; Menozzi, C.; Bloise, R.; et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation 2000, 102, 2509–2515. [Google Scholar] [CrossRef] [PubMed]
- Postma, A.V.; Denjoy, I.; Kamblock, J.; Alders, M.; Lupoglazoff, J.M.; Vaksmann, G.; Dubosq-Bidot, L.; Sebillon, P.; Mannens, M.M.; Guicheney, P.; et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 2005, 42, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, C.; Nederend, I.; Hofman, N.; van Geloven, N.; Ebink, C.; Frohn-Mulder, I.M.; Alings, A.M.; Bosker, H.A.; Bracke, F.A.; van den Heuvel, F.; et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: Disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ. Arrhythm. Electrophysiol. 2012, 5, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Sarkozy, A.; Mont, L.; Henkens, S.; Berruezo, A.; Tamborero, D.; Arzamendi, D.; Berne, P.; Brugada, R.; Brugada, P.; et al. Gender differences in clinical manifestations of Brugada syndrome. J. Am. Coll. Cardiol. 2008, 52, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Kanthan, A.; Monteforte, N.; Memmi, M.; Bloise, R.; Novelli, V.; Miceli, C.; O’Rourke, S.; Borio, G.; Zienciuk-Krajka, A.; et al. Novel insight into the natural history of short QT syndrome. J. Am. Coll. Cardiol. 2014, 63, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Probst, V.; Denjoy, I.; Meregalli, P.G.; Amirault, J.C.; Sacher, F.; Mansourati, J.; Babuty, D.; Villain, E.; Victor, J.; Schott, J.J.; et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation 2007, 115, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Du, Y.; Yang, P.; Lin, S.; Xi, Y.; Yang, Z.; Ma, A. Age-dependent alterations of voltage-gated Na(+) channel isoforms in rat sinoatrial node. Mech. Ageing Dev. 2015, 152, 80–90. [Google Scholar] [CrossRef] [PubMed]
- McMillan, M.R.; Day, T.G.; Bartsota, M.; Mead-Regan, S.; Bryant, R.; Mangat, J.; Abrams, D.; Lowe, M.; Kaski, J.P. Feasibility and outcomes of ajmaline provocation testing for Brugada syndrome in children in a specialist paediatric inherited cardiovascular diseases centre. Open Heart 2014, 1, e000023. [Google Scholar] [CrossRef] [PubMed]
- Conte, G.; de Asmundis, C.; Ciconte, G.; Julia, J.; Sieira, J.; Chierchia, G.B.; Brugada, P. Follow-up from childhood to adulthood of individuals with family history of Brugada syndrome and normal electrocardiograms. JAMA 2014, 312, 2039–2041. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Novelli, V.; Francis, M.D.; Priori, S.G. Genetic modulators of the phenotype in the long QT syndrome: State of the art and clinical impact. Curr. Opin. Genet. Dev. 2015, 33, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Rook, M.B.; Groenewegen, W.A.; Herfst, L.J.; van der Wal, A.C.; Lam, J.; Jongsma, H.J.; Wilde, A.A.; Mannens, M.M. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ. Res. 2003, 92, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Risgaard, B.; Jabbari, R.; Refsgaard, L.; Holst, A.G.; Haunso, S.; Sadjadieh, A.; Winkel, B.G.; Olesen, M.S.; Tfelt-Hansen, J. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin. Genet. 2013, 84, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Refsgaard, L.; Holst, A.G.; Sadjadieh, G.; Haunso, S.; Nielsen, J.B.; Olesen, M.S. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur. J. Hum. Genet. 2012, 20, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Mademont-Soler, I.; Allegue, C.; Cesar, S.; Ferrer-Costa, C.; Coll, M.; Mates, J.; Iglesias, A.; Brugada, J.; et al. Identification of Genetic Alterations, as Causative Genetic Defects in Long QT Syndrome, Using Next Generation Sequencing Technology. PLoS ONE 2014, 9, e114894. [Google Scholar] [CrossRef] [PubMed]
- Mademont-Soler, I.; Pinsach-Abuin, M.L.; Riuro, H.; Mates, J.; Perez-Serra, A.; Coll, M.; Porres, J.M.; Del Olmo, B.; Iglesias, A.; Selga, E.; et al. Large Genomic Imbalances in Brugada Syndrome. PLoS ONE 2016, 11, e0163514. [Google Scholar] [CrossRef] [PubMed]
- Nishio, Y.; Makiyama, T.; Itoh, H.; Sakaguchi, T.; Ohno, S.; Gong, Y.Z.; Yamamoto, S.; Ozawa, T.; Ding, W.G.; Toyoda, F.; et al. D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. J. Am. Coll. Cardiol. 2009, 54, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013. [Google Scholar] [CrossRef]
- Amin, A.S.; Giudicessi, J.R.; Tijsen, A.J.; Spanjaart, A.M.; Reckman, Y.J.; Klemens, C.A.; Tanck, M.W.; Kapplinger, J.D.; Hofman, N.; Sinner, M.F.; et al. Variants in the 3’ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur. Heart J. 2012, 33, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Lahtinen, A.M.; Spazzolini, C.; Mastantuono, E.; Monti, M.C.; Morassutto, C.; Parati, G.; Heradien, M.; Goosen, A.; Lichtner, P.; et al. Genetic Modifiers for the Long-QT Syndrome: How Important Is the Role of Variants in the 3’ Untranslated Region of KCNQ1? Circ. Cardiovasc. Genet. 2016, 9, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Bronze-da-Rocha, E. MicroRNAs expression profiles in cardiovascular diseases. BioMed Res. Int. 2014, 2014, 985408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Non-Genetic Modifiers | |
|---|---|
| Gender | Worse prognosis in males |
| Age | Severe phenotypes in early manifestations, age-related penetrance to the ajmaline provocation test |
| Exogenous factors | Fever, excessive alcohol and large meals |
| Genetic Modifiers | |
| Coding variants—Rare Variants (MAF < 1%) | Additive effect of multiple independent mutations |
| CNVs | |
| New genes involved with the disease | |
| Coding variants—Common Variants (MAF > 1%) | Second hits (i.e., p.H558R_SCN5A; p.K897T_KCNH2; p.D85N_KCNE1 |
| Non-coding variants | 5′UTR and 3′UTR variants, microRNAs |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coll, M.; Pérez-Serra, A.; Mates, J.; Del Olmo, B.; Puigmulé, M.; Fernandez-Falgueras, A.; Iglesias, A.; Picó, F.; Lopez, L.; Brugada, R.; et al. Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death. Biology 2018, 7, 3. https://doi.org/10.3390/biology7010003
Coll M, Pérez-Serra A, Mates J, Del Olmo B, Puigmulé M, Fernandez-Falgueras A, Iglesias A, Picó F, Lopez L, Brugada R, et al. Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death. Biology. 2018; 7(1):3. https://doi.org/10.3390/biology7010003
Chicago/Turabian StyleColl, Monica, Alexandra Pérez-Serra, Jesus Mates, Bernat Del Olmo, Marta Puigmulé, Anna Fernandez-Falgueras, Anna Iglesias, Ferran Picó, Laura Lopez, Ramon Brugada, and et al. 2018. "Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death" Biology 7, no. 1: 3. https://doi.org/10.3390/biology7010003
APA StyleColl, M., Pérez-Serra, A., Mates, J., Del Olmo, B., Puigmulé, M., Fernandez-Falgueras, A., Iglesias, A., Picó, F., Lopez, L., Brugada, R., & Campuzano, O. (2018). Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death. Biology, 7(1), 3. https://doi.org/10.3390/biology7010003