Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging


{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Extrarenal Generation of 1,25(OH)2D
1.2. Excess Sun Exposure Does Not Cause Hypervitaminosis D
2. Vitamin D and Gene Regulation
2.1. Epigenetic Mechanisms Influence Cancer Genesis
2.2. Epigenetics and Molecular Genetics of Vitamin D
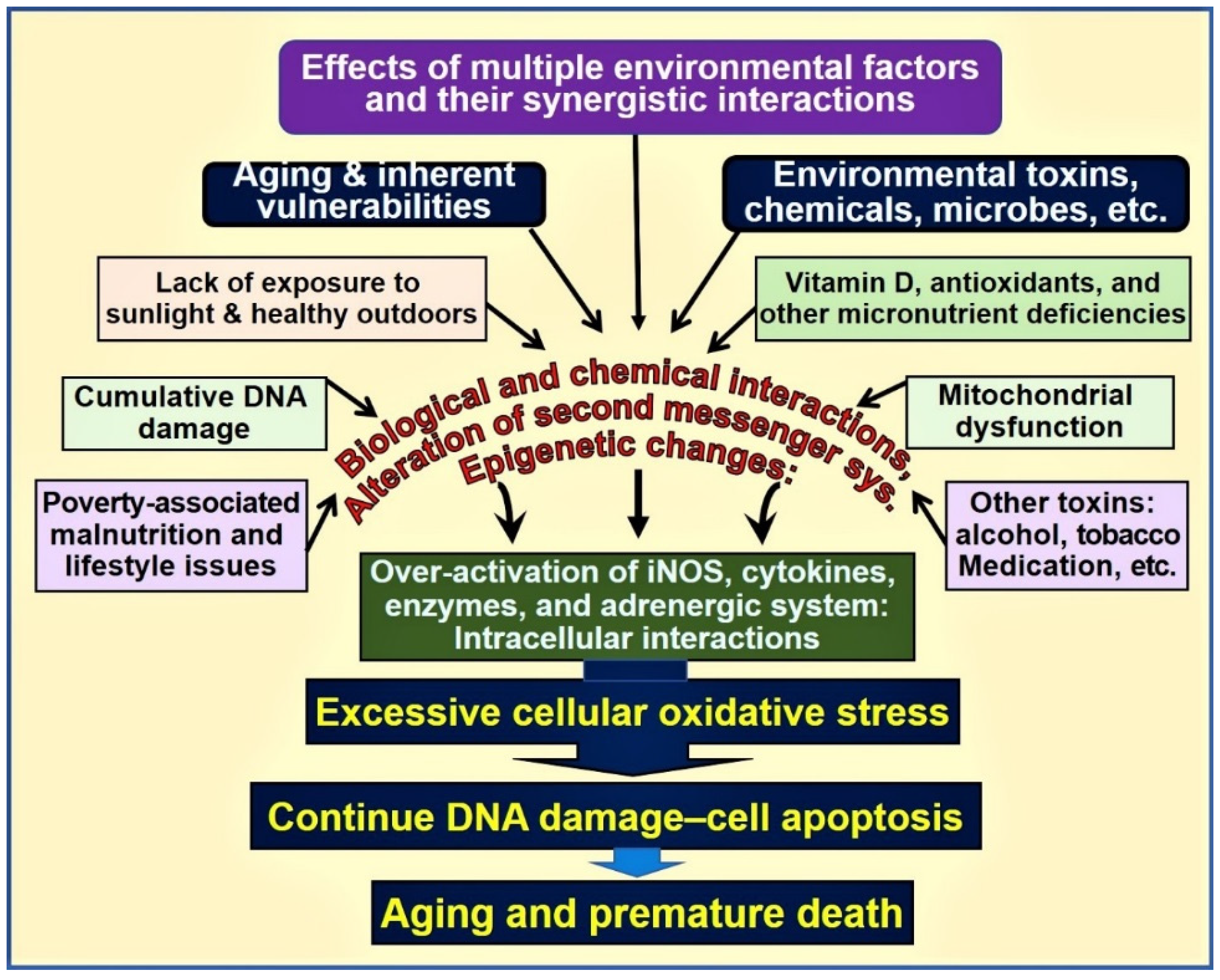
3. Vitamin D–Oxidative Stress
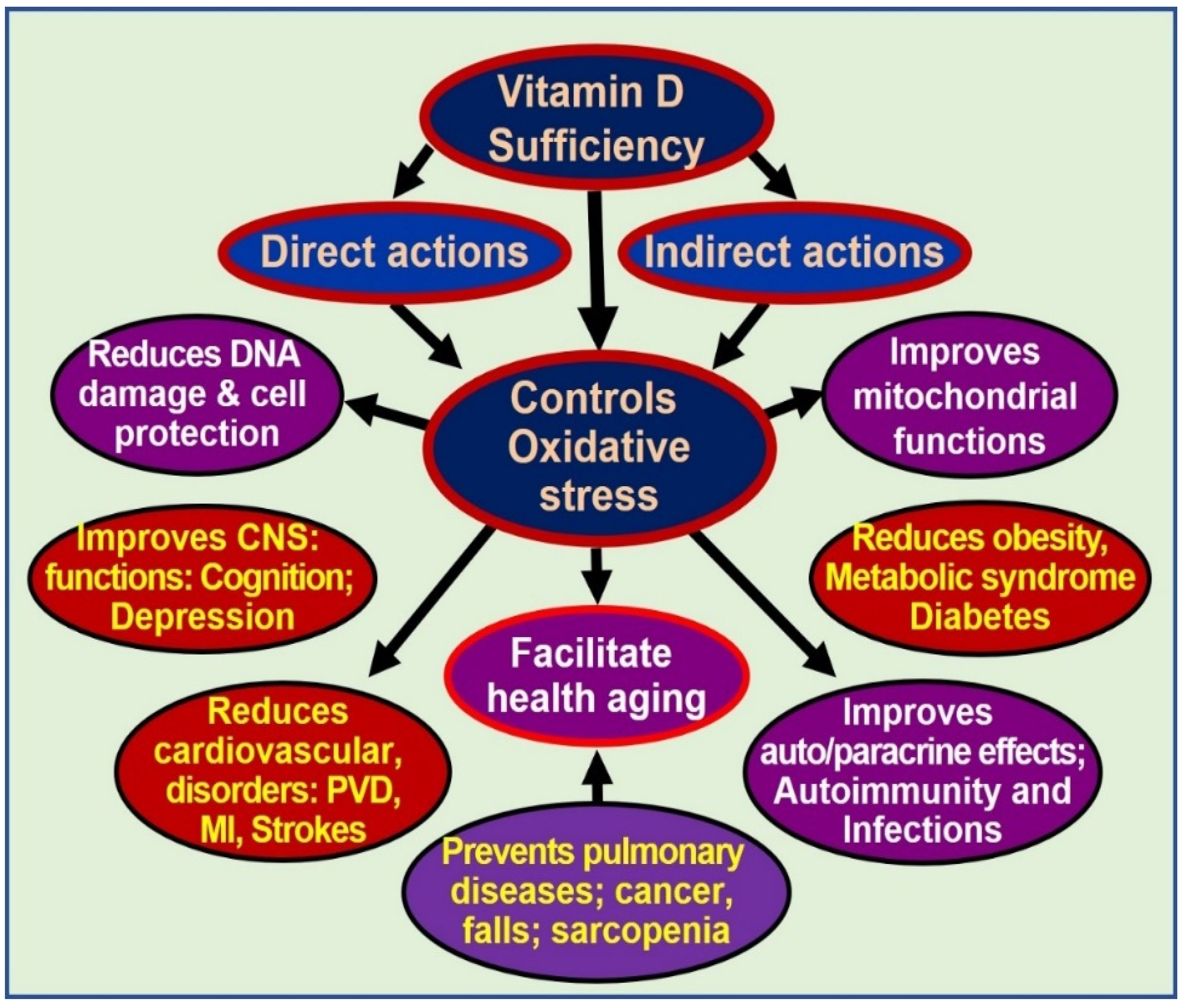
Influences of Vitamin D on Oxidative Stress
4. Role of Vitamin D in Neutralization of Toxins and Aging-Related Compounds
4.1. The Concept and the Process of Aging
4.2. Effects of Vitamin D on Apoptosis and Aging
4.3. Hypovitaminosis D Leads to Deranged Mitochondrial Respiration
4.4. Calcitriol Protects Mitochondrial Functions
5. Discussion
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1,25(OH)2D | 1,25-dihydroxyvitamin D |
| 25(OH)D | 25-hydroxy vitamin D |
| Nrf2 | Nuclear factor erythroid 2 (Nf-E2)-related factor 2 |
| PTH | Parathyroid hormone |
| ROS | Reactive oxygen species |
| UVB | Ultraviolet B |
| VDR | Vitamin D receptor |
| VDRE | Vitamin D response element |
References
- Van Schoor, N.M.; Lips, P. Worldwide vitamin D status. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Eggemoen, A.R.; Knutsen, K.V.; Dalen, I.; Jenum, A.K. Vitamin D status in recently arrived immigrants from Africa and Asia: A cross-sectional study from Norway of children, adolescents and adults. BMJ Open 2013, 3, e003293. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.F.; Gorham, E.D.; Mohr, S.B.; Garland, F.C. Vitamin D for cancer prevention: Global perspective. Ann. Epidemiol. 2009, 19, 468–483. [Google Scholar] [CrossRef]
- Hilger, J.; Friedel, A.; Herr, R.; Rausch, T.; Roos, F.; Wahl, D.A.; Pierroz, D.D.; Weber, P.; Hoffmann, K. A systematic review of vitamin D status in populations worldwide. Br. J. Nutr. 2014, 111, 23–45. [Google Scholar] [CrossRef]
- Haq, A.; Wimalawansa, S.J.; Carlberg, C. Highlights from the 5th International Conference on Vitamin D Deficiency, Nutrition and Human Health, Abu Dhabi, United Arab Emirates, March 24-25, 2016. J. Steroid Biochem. Mol. Biol. 2018, 175, 1–3. [Google Scholar] [CrossRef]
- Pludowski, P.; Holick, M.F.; Grant, W.B.; Konstantynowicz, J.; Mascarenhas, M.R.; Haq, A.; Povoroznyuk, V.; Balatska, N.; Barbosa, A.P.; Karonova, T.; et al. Vitamin D supplementation guidelines. J. Steroid Biochem. Mol. Biol. 2018, 175, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Felton, S.J.; Cooke, M.S.; Kift, R.; Berry, J.L.; Webb, A.R.; Lam, P.M.; de Gruijl, F.R.; Vail, A.; Rhodes, L.E. Concurrent beneficial (vitamin D production) and hazardous (cutaneous DNA damage) impact of repeated low-level summer sunlight exposures. Br. J. Dermatol. 2016, 175, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Thomson, C.; Tongkao-on, W.; Song, E.J.; Carter, S.E.; Dixon, K.M.; Mason, R.S. Protection from ultraviolet damage and photocarcinogenesis by vitamin D compounds. Adv. Exp. Med. Biol. 2014, 810, 303–328. [Google Scholar] [PubMed]
- Petersen, B.; Wulf, H.C.; Triguero-Mas, M.; Philipsen, P.A.; Thieden, E.; Olsen, P.; Heydenreich, J.; Dadvand, P.; Basagana, X.; Liljendahl, T.S.; et al. Sun and ski holidays improve vitamin D status, but are associated with high levels of DNA damage. J. Investig. Dermatol. 2014, 134, 2806–2813. [Google Scholar] [CrossRef] [PubMed]
- Wimalawansa, S.J. Vitamin D. What clinicians would like to know. Sri Lanka Journal of Diabetes, Endocrinology and Metabolism 2012, 1, 73–88. [Google Scholar] [CrossRef]
- Mark, K.A.; Dumas, K.J.; Bhaumik, D.; Schilling, B.; Davis, S.; Oron, T.R.; Sorensen, D.J.; Lucanic, M.; Brem, R.B.; Melov, S.; et al. Vitamin D Promotes Protein Homeostasis and Longevity via the Stress Response Pathway Genes skn-1, ire-1, and xbp-1. Cell Rep. 2016, 17, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, W.B.; Boucher, B.J.; Bhattoa, H.P.; Lahore, H. Why vitamin D clinical trials should be based on 25-hydroxyvitamin D concentrations. J. Steroid Biochem. Mol. Biol. 2018, 177, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Kroll, M.H.; Bi, C.; Garber, C.C.; Kaufman, H.W.; Liu, D.; Caston-Balderrama, A.; Zhang, K.; Clarke, N.; Xie, M.; Reitz, R.E.; et al. Temporal relationship between vitamin D status and parathyroid hormone in the United States. PLoS ONE 2015, 10, e0118108. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Prosser, D.E.; Kaufmann, M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch. Biochem. Biophys. 2012, 523, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.R.; DeCosta, B.R.; Holick, M.F. Sunlight regulates the cutaneous production of vitamin D3 by causing its photodegradation. J. Clin. Endocrinol. Metab. 1989, 68, 882–887. [Google Scholar] [CrossRef]
- Armbrecht, H.J.; Hodam, T.L.; Boltz, M.A.; Partridge, N.C.; Brown, A.J.; Kumar, V.B. Induction of the vitamin D 24-hydroxylase (CYP24) by 1,25-dihydroxyvitamin D3 is regulated by parathyroid hormone in UMR106 osteoblastic cells. Endocrinology 1998, 139, 3375–3381. [Google Scholar] [CrossRef]
- Miller, W.L. Genetic disorders of Vitamin D biosynthesis and degradation. J. Steroid Biochem. Mol. Biol. 2017, 165, 101–108. [Google Scholar] [CrossRef]
- Mithal, A.; Wahl, D.A.; Bonjour, J.P.; Burckhardt, P.; Dawson-Hughes, B.; Eisman, J.A.; El-Hajj Fuleihan, G.; Josse, R.G.; Lips, P.; Morales-Torres, J. Global vitamin D status and determinants of hypovitaminosis D. Osteoporos. Int. 2009, 20, 1807–1820. [Google Scholar] [CrossRef] [Green Version]
- Wimalawansa, S.J. Vitamin D in the new millennium. Curr. Osteoporos. Rep. 2012, 10, 4–15. [Google Scholar] [CrossRef]
- Grant, W.B.; Wimalawansa, S.J.; Holick, M.F. Vitamin D supplements and reasonable solar UVB should be recommended to prevent escalating incidence of chronic diseases. Br. Med. J. 2015, 350, h321. [Google Scholar]
- Holick, M.F. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin. Proc. 2006, 81, 353–373. [Google Scholar] [CrossRef]
- Calzavara-Pinton, P.; Ortel, B.; Venturini, M. Non-melanoma skin cancer, sun exposure and sun protection. Giornale Italiano di Dermatologia e Venereologia 2015, 150, 369–378. [Google Scholar]
- Cascinelli, N.; Krutmann, J.; MacKie, R.; Pierotti, M.; Prota, G.; Rosso, S.; Young, A. European School of Oncology advisory report. Sun exposure, UVA lamps and risk of skin cancer. Eur. J. Cancer 1994, 30A, 548–560. [Google Scholar] [PubMed]
- Moan, J.; Porojnicu, A.C.; Dahlback, A.; Setlow, R.B. Addressing the health benefits and risks, involving vitamin D or skin cancer, of increased sun exposure. Proc. Natl. Acad. Sci. USA 2008, 105, 668–673. [Google Scholar] [CrossRef] [Green Version]
- Omura, Y. Clinical Significance of Human Papillomavirus Type 16 for Breast Cancer & Adenocarcinomas of Various Internal Organs and Alzheimer’s Brain with Increased beta-amyloid (1-42); Combined Use of Optimal Doses of Vitamin D3 and Taurine 3 times/day Has Significant Beneficial Effects of Anti-Cancer, Anti-Ischemic Heart, and Memory & Other Brain Problems By Significant Urinary Excretion of Viruses, Bacteria, and Toxic Metals & Substances. Acupunct. Electro-Ther. Res. 2016, 41, 127–134. [Google Scholar]
- Gilad, L.A.; Bresler, T.; Gnainsky, J.; Smirnoff, P.; Schwartz, B. Regulation of vitamin D receptor expression via estrogen-induced activation of the ERK 1/2 signaling pathway in colon and breast cancer cells. J. Endocrinol. 2005, 185, 577–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robsahm, T.E.; Tretli, S.; Dahlback, A.; Moan, J. Vitamin D3 from sunlight may improve the prognosis of breast-, colon- and prostate cancer (Norway). Cancer Causes Control 2004, 15, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Shin, E.A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdivielso, J.M. The physiology of vitamin D receptor activation. Contrib. Nephrol. 2009, 163, 206–212. [Google Scholar] [PubMed]
- Farnham, P.J. Thematic minireview series on results from the ENCODE Project: Integrative global analyses of regulatory regions in the human genome. J. Biol. Chem. 2012, 287, 30885–30887. [Google Scholar] [CrossRef]
- Washington, S.D.; Edenfield, S.I.; Lieux, C.; Watson, Z.L.; Taasan, S.M.; Dhummakupt, A.; Bloom, D.C.; Neumann, D.M. Depletion of the insulator protein CTCF results in HSV-1 reactivation in vivo. J. Virol. 2018. [Google Scholar] [CrossRef]
- MacPherson, M.J.; Sadowski, P.D. The CTCF insulator protein forms an unusual DNA structure. BMC Mol. Biol. 2010, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Genome-wide (over)view on the actions of vitamin D. Front. Physiol. 2014, 5, 167. [Google Scholar] [CrossRef]
- Narvaez, C.J.; Matthews, D.; LaPorta, E.; Simmons, K.M.; Beaudin, S.; Welsh, J. The impact of vitamin D in breast cancer: Genomics, pathways, metabolism. Front. Physiol. 2014, 5, 213. [Google Scholar] [CrossRef]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612. [Google Scholar] [CrossRef]
- Pusceddu, I.; Farrell, C.J.; Di Pierro, A.M.; Jani, E.; Herrmann, W.; Herrmann, M. The role of telomeres and vitamin D in cellular aging and age-related diseases. Clin. Chem. Lab. Med. 2015, 53, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hua, F.; Wang, J.; Sayeed, I.; Wang, X.; Chen, Z.; Yousuf, S.; Atif, F.; Stein, D.G. Progesterone and vitamin D: Improvement after traumatic brain injury in middle-aged rats. Horm. Behav. 2013, 64, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; He, Y.; Shen, Y.; Zhang, Q.; Chen, D.; Zuo, C.; Qin, J.; Wang, H.; Wang, J.; Yu, Y. Vitamin D inhibits COX-2 expression and inflammatory response by targeting thioesterase superfamily member 4. J. Biol. Chem. 2014, 289, 11681–11694. [Google Scholar] [CrossRef] [PubMed]
- Myszka, M.; Klinger, M. The immunomodulatory role of Vitamin D. Postepy Hig. Med. Dosw. 2014, 68, 865–878. [Google Scholar] [CrossRef]
- Watanabe, R.; Inoue, D. Current Topics on Vitamin D. Anti-cancer effects of vitamin D. Clin. Calcium 2015, 25, 373–380. [Google Scholar]
- Shi, Y.; Au, J.S.; Thongprasert, S.; Srinivasan, S.; Tsai, C.M.; Khoa, M.T.; Heeroma, K.; Itoh, Y.; Cornelio, G.; Yang, P.C. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef]
- O’Malley, M.; King, A.N.; Conte, M.; Ellingrod, V.L.; Ramnath, N. Effects of cigarette smoking on metabolism and effectiveness of systemic therapy for lung cancer. J. Thorac. Oncol. 2014, 9, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Iswariya, G.T.; Paital, B.; Padma, P.R.; Nirmaladevi, R. MicroRNAs: Epigenetic players in cancer and aging. Front. Biosci. (Schol Ed) 2019, 11, 29–55. [Google Scholar]
- Ramnath, N.; Nadal, E.; Jeon, C.K.; Sandoval, J.; Colacino, J.; Rozek, L.S.; Christensen, P.J.; Esteller, M.; Beer, D.G.; Kim, S.H. Epigenetic regulation of vitamin D metabolism in human lung adenocarcinoma. J. Thorac. Oncol. 2014, 9, 473–482. [Google Scholar] [CrossRef]
- Karlic, H.; Varga, F. Impact of vitamin D metabolism on clinical epigenetics. Clin. Epigenet. 2011, 2, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codoner-Franch, P.; Tavarez-Alonso, S.; Simo-Jorda, R.; Laporta-Martin, P.; Carratala-Calvo, A.; Alonso-Iglesias, E. Vitamin D status is linked to biomarkers of oxidative stress, inflammation, and endothelial activation in obese children. J. Pediatr. 2012, 161, 848–854. [Google Scholar] [CrossRef]
- Zeitelhofer, M.; Adzemovic, M.Z.; Gomez-Cabrero, D.; Bergman, P.; Hochmeister, S.; N’Diaye, M.; Paulson, A.; Ruhrmann, S.; Almgren, M.; Tegner, J.N.; et al. Functional genomics analysis of vitamin D effects on CD4+ T cells in vivo in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2017, 114, E1678–E1687. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhao, S. Metabolic changes in cancer: Beyond the Warburg effect. Acta Biochim. Biophys. Sin. 2013, 45, 18–26. [Google Scholar] [CrossRef]
- Garcia-Quiroz, J.; Garcia-Becerra, R.; Barrera, D.; Santos, N.; Avila, E.; Ordaz-Rosado, D.; Rivas-Suarez, M.; Halhali, A.; Rodriguez, P.; Gamboa-Dominguez, A.; et al. Astemizole synergizes calcitriol antiproliferative activity by inhibiting CYP24A1 and upregulating VDR: A novel approach for breast cancer therapy. PLoS ONE 2012, 7, e45063. [Google Scholar] [CrossRef]
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. [Google Scholar] [CrossRef]
- Holmes, S.; Abbassi, B.; Su, C.; Singh, M.; Cunningham, R.L. Oxidative stress defines the neuroprotective or neurotoxic properties of androgens in immortalized female rat dopaminergic neuronal cells. Endocrinology 2013, 154, 4281–4292. [Google Scholar] [CrossRef]
- Petersen, K.S.; Smith, C. Ageing-Associated Oxidative Stress and Inflammation Are Alleviated by Products from Grapes. Oxid. Med. Cell. Longev. 2016, 2016, 6236309. [Google Scholar] [CrossRef]
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D activates the Nrf2-Keap1 antioxidant pathway and ameliorates nephropathy in diabetic rats. Am. J. Hypertens. 2014, 27, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.N.; Mele, J.; Hayes, J.D.; Buffenstein, R. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr. Comp. Biol. 2010, 50, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Tullet, J.M.A.; Green, J.W.; Au, C.; Benedetto, A.; Thompson, M.A.; Clark, E.; Gilliat, A.F.; Young, A.; Schmeisser, K.; Gems, D. The SKN-1/Nrf2 transcription factor can protect against oxidative stress and increase lifespan in C. elegans by distinct mechanisms. Aging Cell 2017, 16, 1191–1194. [Google Scholar] [CrossRef] [PubMed]
- Forster, R.E.; Jurutka, P.W.; Hsieh, J.C.; Haussler, C.A.; Lowmiller, C.L.; Kaneko, I.; Haussler, M.R.; Kerr Whitfield, G. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochem. Biophys. Res. Commun. 2011, 414, 557–562. [Google Scholar] [CrossRef]
- Berridge, M.J. Vitamin D: A custodian of cell signalling stability in health and disease. Biochem. Soc. Trans. 2015, 43, 349–358. [Google Scholar] [CrossRef]
- Razzaque, M.S. FGF23, klotho and vitamin D interactions: What have we learned from in vivo mouse genetics studies? Adv. Exp. Med. Biol. 2012, 728, 84–91. [Google Scholar]
- Kuro-o, M. Klotho and aging. Biochim. Biophys. Acta 2009, 1790, 1049–1058. [Google Scholar] [CrossRef]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Wang, L.; Lewis, T.; Zhang, Y.L.; Khodier, C.; Magesh, S.; Chen, L.; Inoyama, D.; Chen, Y.; Zhen, J.; Hu, L.; et al. The identification and characterization of non-reactive inhibitor of Keap1-Nrf2 interaction through HTS using a fluorescence polarization assay. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Berridge, M.J. Vitamin D deficiency: Infertility and neurodevelopmental diseases (attention deficit hyperactivity disorder, autism, and schizophrenia). Am. J. Physiol. Cell Physiol. 2018, 314, C135–C151. [Google Scholar] [CrossRef]
- Ryan, Z.C.; Craig, T.A.; Folmes, C.D.; Wang, X.; Lanza, I.R.; Schaible, N.S.; Salisbury, J.L.; Nair, K.S.; Terzic, A.; Sieck, G.C.; et al. 1alpha,25-Dihydroxyvitamin D3 Regulates Mitochondrial Oxygen Consumption and Dynamics in Human Skeletal Muscle Cells. J. Biol. Chem. 2016, 291, 1514–1528. [Google Scholar] [CrossRef]
- Bouillon, R.; Verstuyf, A. Vitamin D, mitochondria, and muscle. J. Clin. Endocrinol. Metab. 2013, 98, 961–963. [Google Scholar] [CrossRef]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Berridge, M.J. Vitamin D cell signalling in health and disease. Biochem. Biophys. Res. Commun. 2015, 460, 53–71. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Rocha, K.K.; Lopes, G.S.; Bincoletto, C.; Smaili, S.S. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox Signal. 2014, 21, 123–137. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Fu, B.; Zhang, J.; Zhao, J.; Yuan, M.; Peng, W.; Zhang, Y.; Wu, H. Sodium fluoride induces nephrotoxicity via oxidative stress-regulated mitochondrial SIRT3 signaling pathway. Sci. Rep. 2017, 7, 672. [Google Scholar] [CrossRef]
- Wei, R.; Christakos, S. Mechanisms Underlying the Regulation of Innate and Adaptive Immunity by Vitamin D. Nutrients 2015, 7, 8251–8260. [Google Scholar] [CrossRef] [Green Version]
- George, N.; Kumar, T.P.; Antony, S.; Jayanarayanan, S.; Paulose, C.S. Effect of vitamin D3 in reducing metabolic and oxidative stress in the liver of streptozotocin-induced diabetic rats. Br. J. Nutr. 2012, 108, 1410–1418. [Google Scholar] [CrossRef] [Green Version]
- Shelton, R.C.; Claiborne, J.; Sidoryk-Wegrzynowicz, M.; Reddy, R.; Aschner, M.; Lewis, D.A.; Mirnics, K. Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol. Psychiatry 2011, 16, 751–762. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.H.; Verkhratsky, A. Calcium and ATP control multiple vital functions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150418. [Google Scholar] [CrossRef] [Green Version]
- Petrosillo, G.; Di Venosa, N.; Moro, N.; Colantuono, G.; Paradies, V.; Tiravanti, E.; Federici, A.; Ruggiero, F.M.; Paradies, G. In vivo hyperoxic preconditioning protects against rat-heart ischemia/reperfusion injury by inhibiting mitochondrial permeability transition pore opening and cytochrome c release. Free Radic. Biol. Med. 2011, 50, 477–483. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Finch, C.E.; Pike, M.C. Maximum life span predictions from the Gompertz mortality model. J. Gerontol. A Biol. Sci. Med. Sci. 1996, 51, B183–B194. [Google Scholar] [CrossRef]
- Macedo, J.C.; Vaz, S.; Logarinho, E. Mitotic Dysfunction Associated with Aging Hallmarks. Adv. Exp. Med. Biol. 2017, 1002, 153–188. [Google Scholar]
- Jallali, N.; Ridha, H.; Thrasivoulou, C.; Underwood, C.; Butler, P.E.; Cowen, T. Vulnerability to ROS-induced cell death in ageing articular cartilage: The role of antioxidant enzyme activity. Osteoarthr. Cartil. 2005, 13, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Pilz, S.; Marz, W.; Wellnitz, B.; Seelhorst, U.; Fahrleitner-Pammer, A.; Dimai, H.P.; Boehm, B.O.; Dobnig, H. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J. Clin. Endocrinol. Metab. 2008, 93, 3927–3935. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.C.; Offiah, A.; Sprigg, A.; Al-Adnani, M. Vitamin D deficiency and sudden unexpected death in infancy and childhood: A cohort study. Pediatr. Dev. Pathol. 2013, 16, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B. Vitamin D Deficiency May Explain Comorbidity as an Independent Risk Factor for Death Associated with Cancer in Taiwan. Asia Pac. J. Public Health 2015, 27, 572–573. [Google Scholar] [CrossRef] [PubMed]
- Scorza, F.A.; Albuquerque, M.; Arida, R.M.; Terra, V.C.; Machado, H.R.; Cavalheiro, E.A. Benefits of sunlight: Vitamin D deficiency might increase the risk of sudden unexpected death in epilepsy. Med. Hypotheses 2010, 74, 158–161. [Google Scholar] [CrossRef]
- Autier, P.; Mullie, P.; Macacu, A.; Dragomir, M.; Boniol, M.; Coppens, K.; Pizot, C.; Boniol, M. Effect of vitamin D supplementation on non-skeletal disorders: A systematic review of meta-analyses and randomised trials. Lancet Diabetes Endocrinol. 2017, 5, 986–1004. [Google Scholar] [CrossRef]
- Brenner, H.; Jansen, L.; Saum, K.U.; Holleczek, B.; Schottker, B. Vitamin D Supplementation Trials Aimed at Reducing Mortality Have Much Higher Power When Focusing on People with Low Serum 25-Hydroxyvitamin D Concentrations. J. Nutr. 2017, 147, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Autier, P.; Boniol, M.; Pizot, C.; Mullie, P. Vitamin D status and ill health: A systematic review. Lancet Diabetes Endocrinol. 2014, 2, 76–89. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Gluud, L.L.; Nikolova, D.; Whitfield, K.; Krstic, G.; Wetterslev, J.; Gluud, C. Vitamin D supplementation for prevention of cancer in adults. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Pilz, S.; Tomaschitz, A.; Marz, W.; Drechsler, C.; Ritz, E.; Zittermann, A.; Cavalier, E.; Pieber, T.R.; Lappe, J.M.; Grant, W.B.; et al. Vitamin D, cardiovascular disease and mortality. Clin. Endocrinol. 2011, 75, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Van der Reest, J.; Gottlieb, E. Anti-cancer effects of vitamin C revisited. Cell Res. 2016, 26, 269–270. [Google Scholar] [CrossRef] [Green Version]
- Da Luz Dias, R.; Basso, B.; Donadio, M.V.F.; Pujol, F.V.; Bartrons, R.; Haute, G.V.; Gassen, R.B.; Bregolin, H.D.; Krause, G.; Viau, C.; et al. Leucine reduces the proliferation of MC3T3-E1 cells through DNA damage and cell senescence. Toxicol. In Vitro 2018, 48, 1–10. [Google Scholar] [CrossRef]
- Cevenini, E.; Caruso, C.; Candore, G.; Capri, M.; Nuzzo, D.; Duro, G.; Rizzo, C.; Colonna-Romano, G.; Lio, D.; Di Carlo, D.; et al. Age-related inflammation: The contribution of different organs, tissues and systems. How to face it for therapeutic approaches. Curr. Pharm. Des. 2010, 16, 609–618. [Google Scholar] [CrossRef]
- Talmor, Y.; Bernheim, J.; Klein, O.; Green, J.; Rashid, G. Calcitriol blunts pro-atherosclerotic parameters through NFkappaB and p38 in vitro. Eur. J. Clin. Investig. 2008, 38, 548–554. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab. Brain Dis. 2014, 29, 19–36. [Google Scholar] [CrossRef]
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013, 11, 200. [Google Scholar] [CrossRef]
- Beilfuss, J.; Berg, V.; Sneve, M.; Jorde, R.; Kamycheva, E. Effects of a 1-year supplementation with cholecalciferol on interleukin-6, tumor necrosis factor-alpha and insulin resistance in overweight and obese subjects. Cytokine 2012, 60, 870–874. [Google Scholar] [CrossRef]
- Calton, E.K.; Keane, K.N.; Soares, M.J.; Rowlands, J.; Newsholme, P. Prevailing vitamin D status influences mitochondrial and glycolytic bioenergetics in peripheral blood mononuclear cells obtained from adults. Redox Biol. 2016, 10, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hyde, A.S.; Simpson, M.A.; Barycki, J.J. Emerging regulatory paradigms in glutathione metabolism. Adv. Cancer Res. 2014, 122, 69–101. [Google Scholar] [Green Version]
- Weipoltshammer, K.; Schofer, C.; Almeder, M.; Philimonenko, V.V.; Frei, K.; Wachtler, F.; Hozak, P. Intranuclear anchoring of repetitive DNA sequences: Centromeres, telomeres, and ribosomal DNA. J. Cell Biol. 1999, 147, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Consiglio, M.; Viano, M.; Casarin, S.; Castagnoli, C.; Pescarmona, G.; Silvagno, F. Mitochondrial and lipogenic effects of vitamin D on differentiating and proliferating human keratinocytes. Exp. Dermatol. 2015, 24, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Wyckelsma, V.L.; Levinger, I.; McKenna, M.J.; Formosa, L.E.; Ryan, M.T.; Petersen, A.C.; Anderson, M.J.; Murphy, R.M. Preservation of skeletal muscle mitochondrial content in older adults: Relationship between mitochondria, fibre type and high-intensity exercise training. J. Physiol. 2017, 595, 3345–3359. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, A.A.; Abbas, M.; Kassem, M.; Gleizes, C.; Kreutter, G.; Schini-Kerth, V.; Mitrea, I.L.; Toti, F.; Kessler, L. Differential influence of tacrolimus and sirolimus on mitochondrial-dependent signaling for apoptosis in pancreatic cells. Mol. Cell. Biochem. 2016, 418, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Matera, M.; Casanova, G.; Ruggiero, F.M.; Paradies, G. Mitochondrial dysfunction in rat brain with aging Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 2008, 53, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Matera, M.; Moro, N.; Ruggiero, F.M.; Paradies, G. Mitochondrial complex I dysfunction in rat heart with aging: Critical role of reactive oxygen species and cardiolipin. Free Radic. Biol. Med. 2009, 46, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, S.; Kim, A.; Yoshihara, T.; Tobita, S.; Takeuchi, T.; Higuchi, M. Mitochondrial respiratory function induces endogenous hypoxia. PLoS ONE 2014, 9, e88911. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Rezin, G.T.; Carvalho, A.F.; Streck, E.L.; Berk, M.; Quevedo, J. Mitochondrial dysfunction in bipolar disorder: Evidence, pathophysiology and translational implications. Neurosci. Biobehav. Rev. 2016, 68, 694–713. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.S.; Xia, L.; Mills, G.B.; Lowell, C.A.; Touw, I.P.; Corey, S.J. G-CSF induced reactive oxygen species involves Lyn-PI3-kinase-Akt and contributes to myeloid cell growth. Blood 2006, 107, 1847–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Berg, A.H.; Iyengar, P.; Lam, T.K.; Giacca, A.; Combs, T.P.; Rajala, M.W.; Du, X.; Rollman, B.; Li, W.; et al. The hyperglycemia-induced inflammatory response in adipocytes: The role of reactive oxygen species. J. Biol. Chem. 2005, 280, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Agalakova, A.A.; Gusev, G.P. Molecular mechanisms of cytotoxicity and apoptosis induced by inorganic fluoride. ISRN Cell Biol. 2012, 2012, 403835. [Google Scholar] [CrossRef]
- Agalakova, N.I.; Gusev, G.P. Fluoride induces oxidative stress and ATP depletion in the rat erythrocytes in vitro. Environ. Toxicol. Pharmacol. 2012, 34, 334–337. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Liu, Z.; Cadenas, E. Mitochondrial function in ageing: Coordination with signalling and transcriptional pathways. J. Physiol. 2016, 594, 2025–2042. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015, 13, 68. [Google Scholar] [CrossRef]
- Ames, B.N. Optimal micronutrients delay mitochondrial decay and age-associated diseases. Mech. Ageing Dev. 2010, 131, 473–479. [Google Scholar] [CrossRef]
- Manna, P.; Achari, A.E.; Jain, S.K. Vitamin D supplementation inhibits oxidative stress and upregulate SIRT1/AMPK/GLUT4 cascade in high glucose-treated 3T3L1 adipocytes and in adipose tissue of high fat diet-fed diabetic mice. Arch. Biochem. Biophys. 2017, 615, 22–34. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.L.; Festuccia, C.; Popov, V.M.; Colapietro, E.A.; Sanita, P.; Musio, D.; De Felice, F.; Lenzi, A.; Jannini, E.A.; et al. Vitamin D protects endothelial cells from irradiation-induced senescence and apoptosis by modulating MAPK/SirT1 axis. J. Endocrinol. Investig. 2016, 39, 411–422. [Google Scholar] [CrossRef]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Guarente, L. Ten years of NAD-dependent SIR2 family deacetylases: Implications for metabolic diseases. Trends Pharmacol. Sci. 2010, 31, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Binkley, N.; Ramamurthy, R.; Krueger, D. Low vitamin D status: Definition, prevalence, consequences, and correction. Rheum. Dis. Clin. N. Am. 2012, 38, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Hollis, B.W.; Wagner, C.L. Clinical review: The role of the parent compound vitamin D with respect to metabolism and function: Why clinical dose intervals can affect clinical outcomes. J. Clin. Endocrinol. Metab. 2013, 98, 4619–4628. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M.; Endocrine, S. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [PubMed]
- Wimalawansa, S.J. Vitamin D: Everything You Need to Know; Karunaratne & Sons: Homagama, Sri Lanka, 2012; ISBN 978-955-9098-94-2. [Google Scholar]
- Heyden, E.L.; Wimalawansa, S.J. Vitamin D: Effects on human reproduction, pregnancy, and fetal well-being. J. Steroid Biochem. Mol. Biol. 2018, 180, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hewison, M.; Wagner, C.L.; Hollis, B.W. Vitamin D Supplementation in Pregnancy and Lactation and Infant Growth. N. Engl. J. Med. 2018, 379, 1880–1881. [Google Scholar]


© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wimalawansa, S.J. Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology 2019, 8, 30. https://doi.org/10.3390/biology8020030
Wimalawansa SJ. Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology. 2019; 8(2):30. https://doi.org/10.3390/biology8020030
Chicago/Turabian StyleWimalawansa, Sunil J. 2019. "Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging" Biology 8, no. 2: 30. https://doi.org/10.3390/biology8020030
APA StyleWimalawansa, S. J. (2019). Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology, 8(2), 30. https://doi.org/10.3390/biology8020030