The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes

 ,
, 
 and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of Diabetes
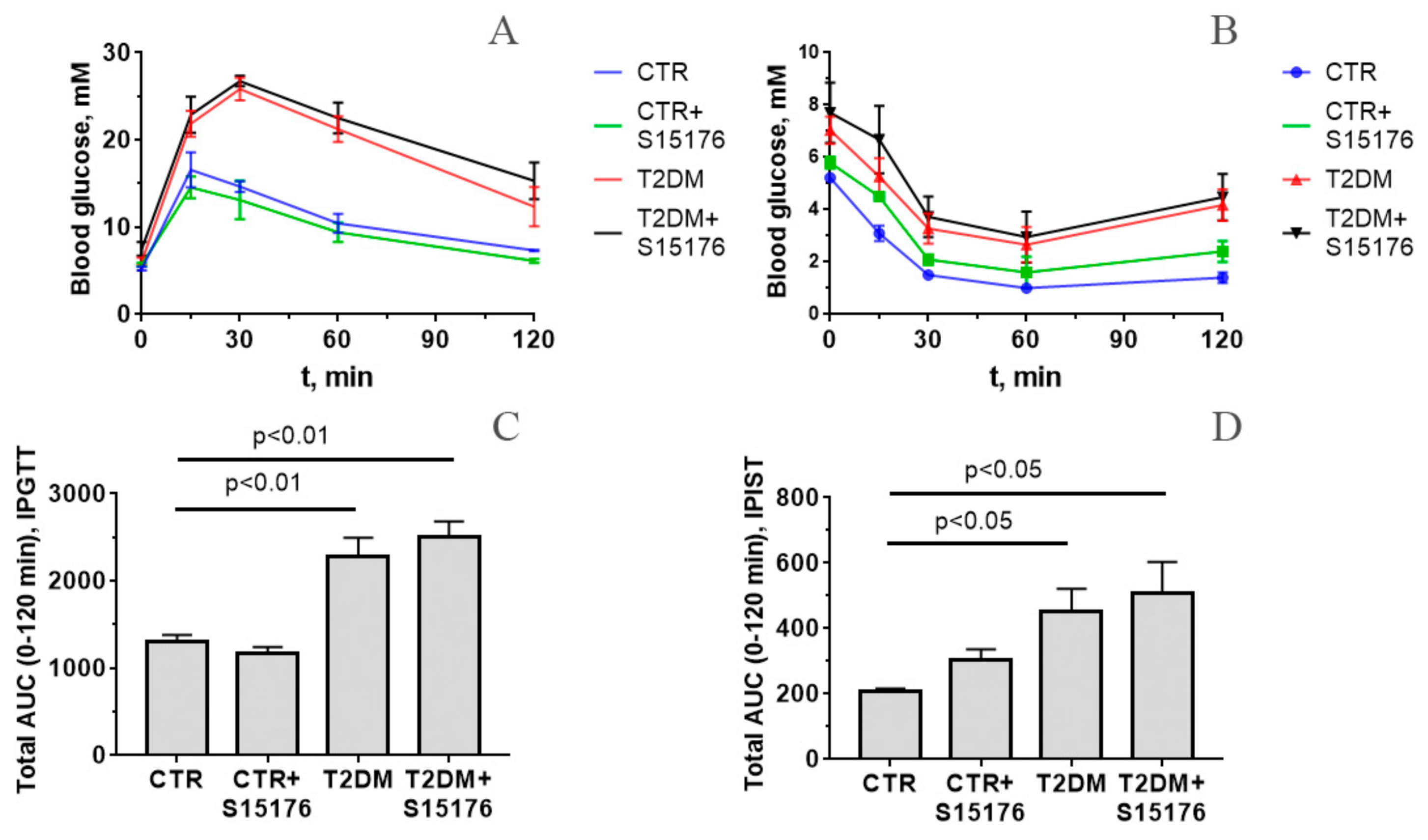
2.3. Intraperitoneal Glucose Tolerance Test and Intraperitoneal Insulin Sensitivity Test
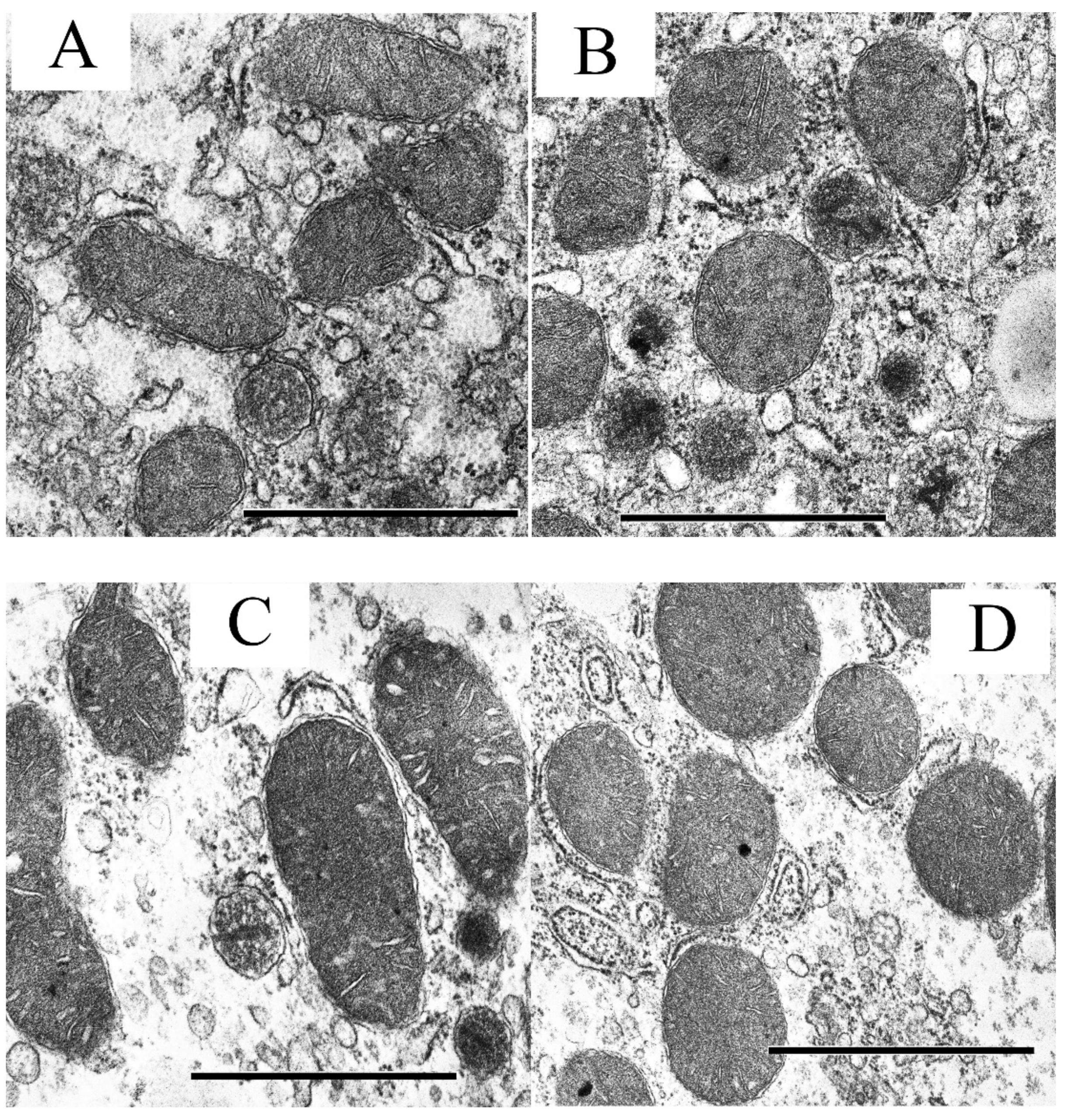
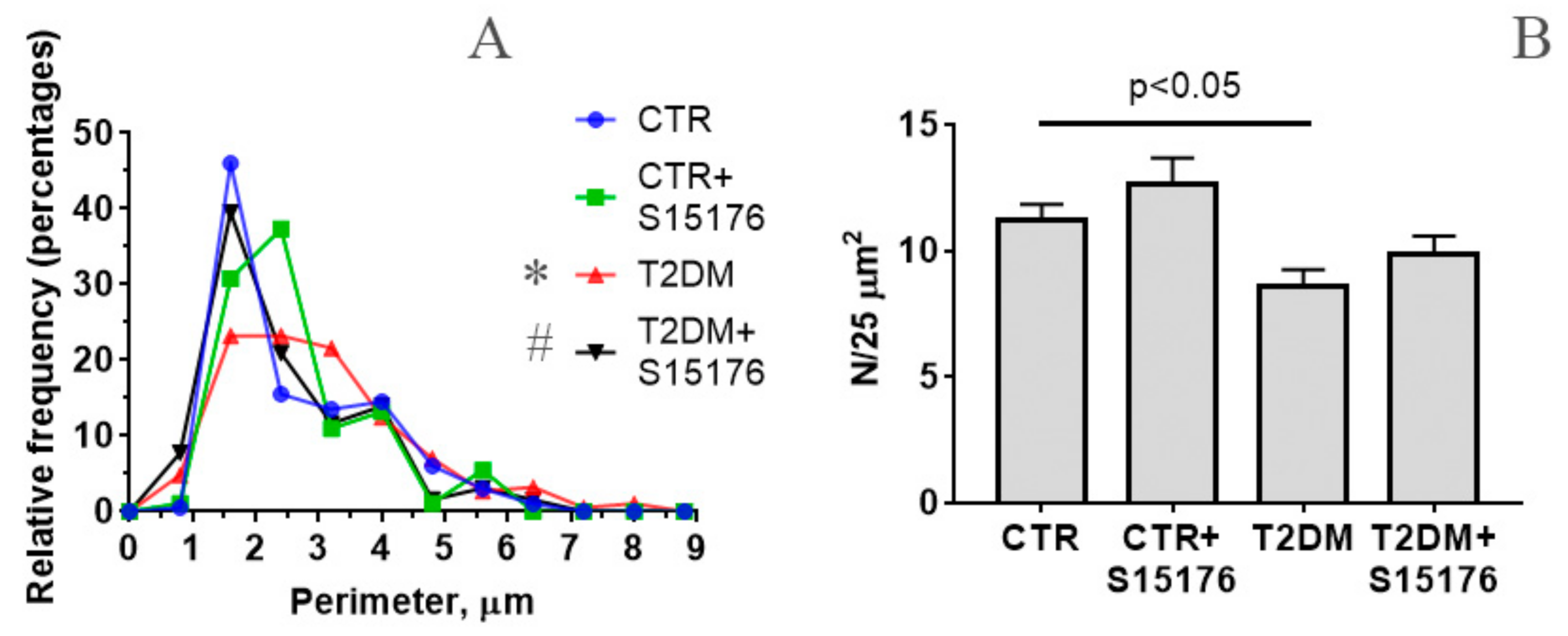
2.4. Electron Microscopy
2.5. RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR
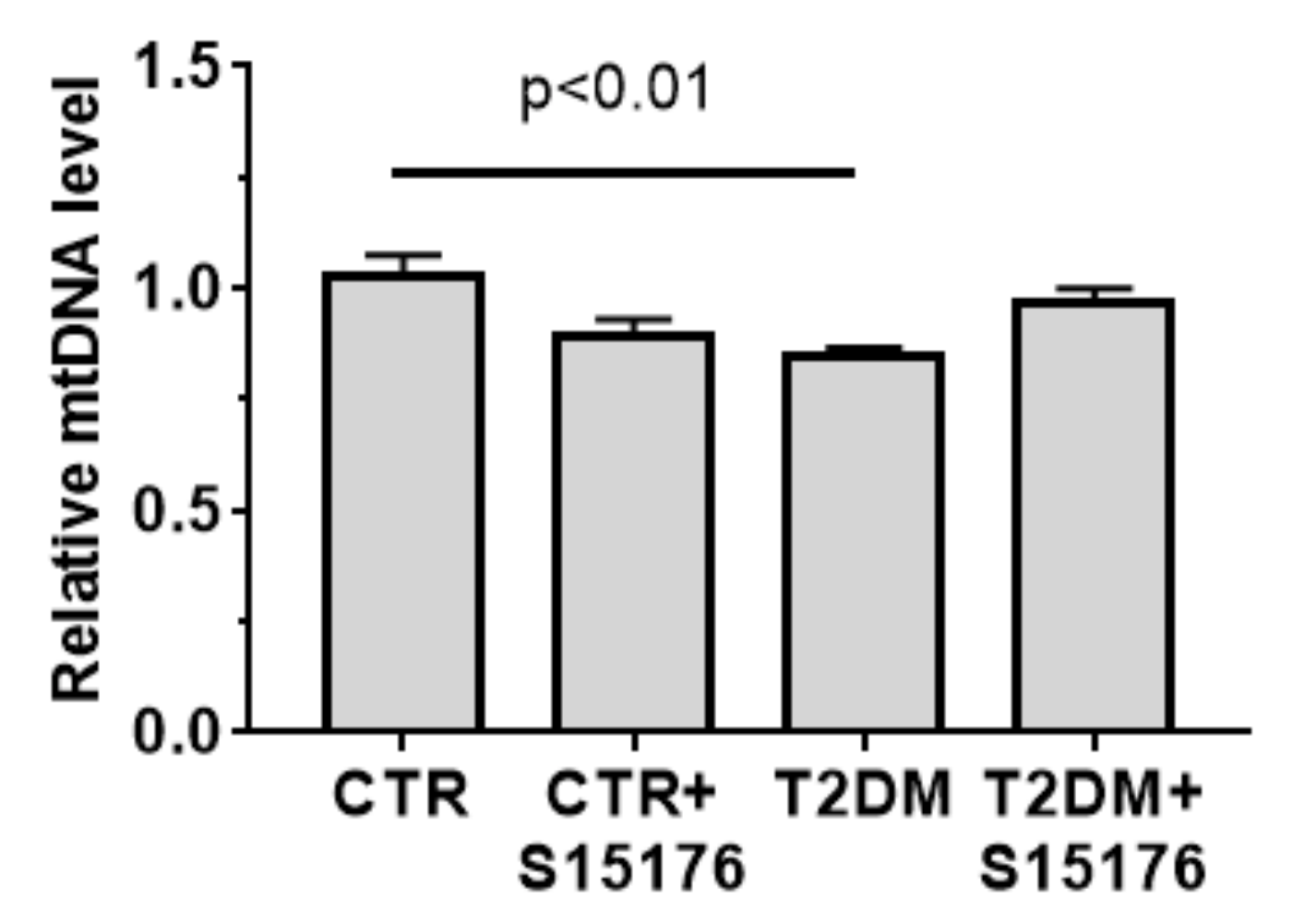
2.6. Quantification of Mitochondrial DNA
2.7. Isolation of Liver Mitochondria
2.8. Mitochondrial Respiration and Oxidative Phosphorylation
2.9. Lipid Peroxidation
2.10. Statistical Analysis
3. Results
3.1. Somatic and Biochemical Characteristics of Mice
3.2. The Effect of S-15176 Difumarate Salt on Ultrastructural Changes in Liver Mitochondria of Mice with Experimental T2DM
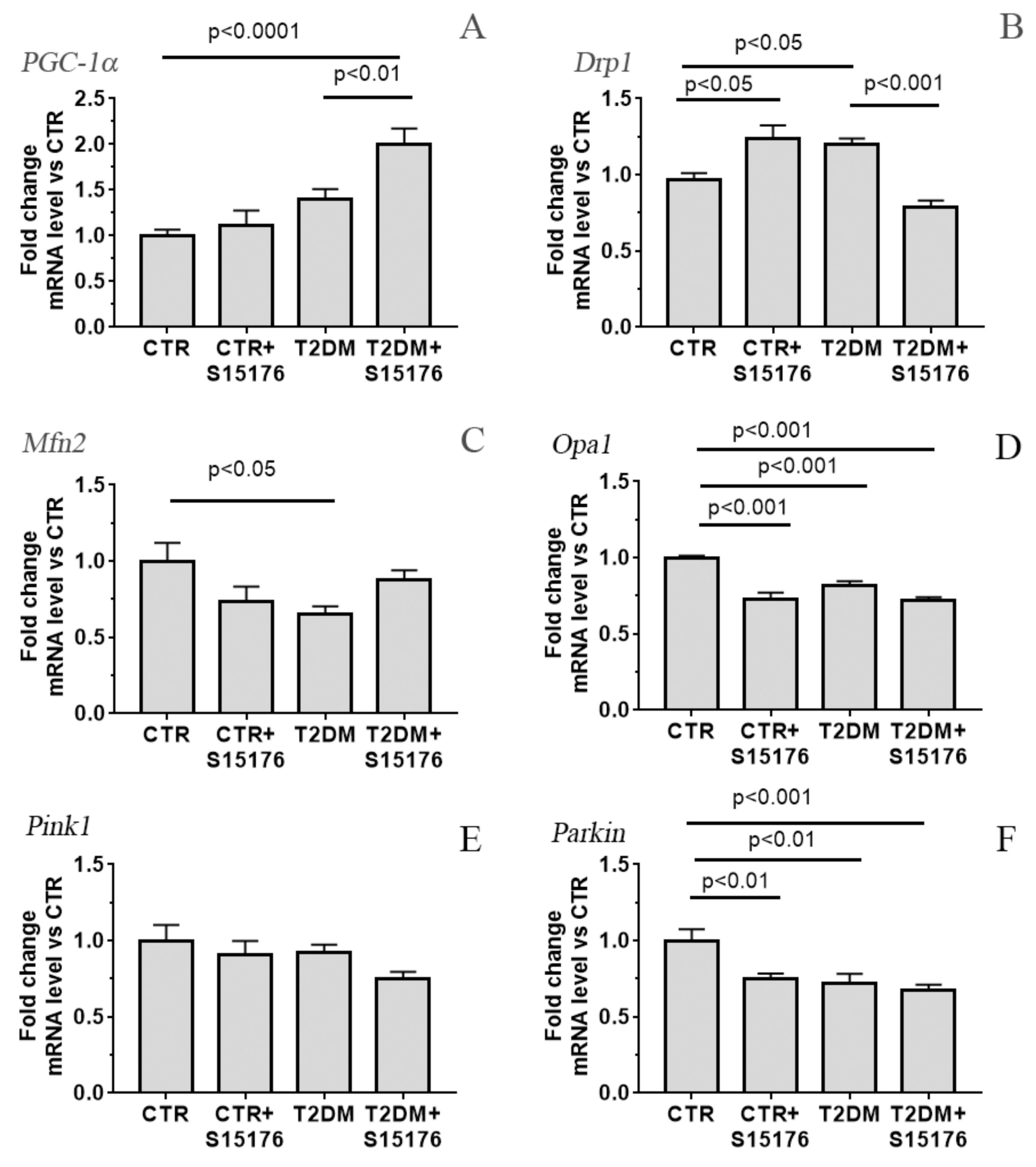
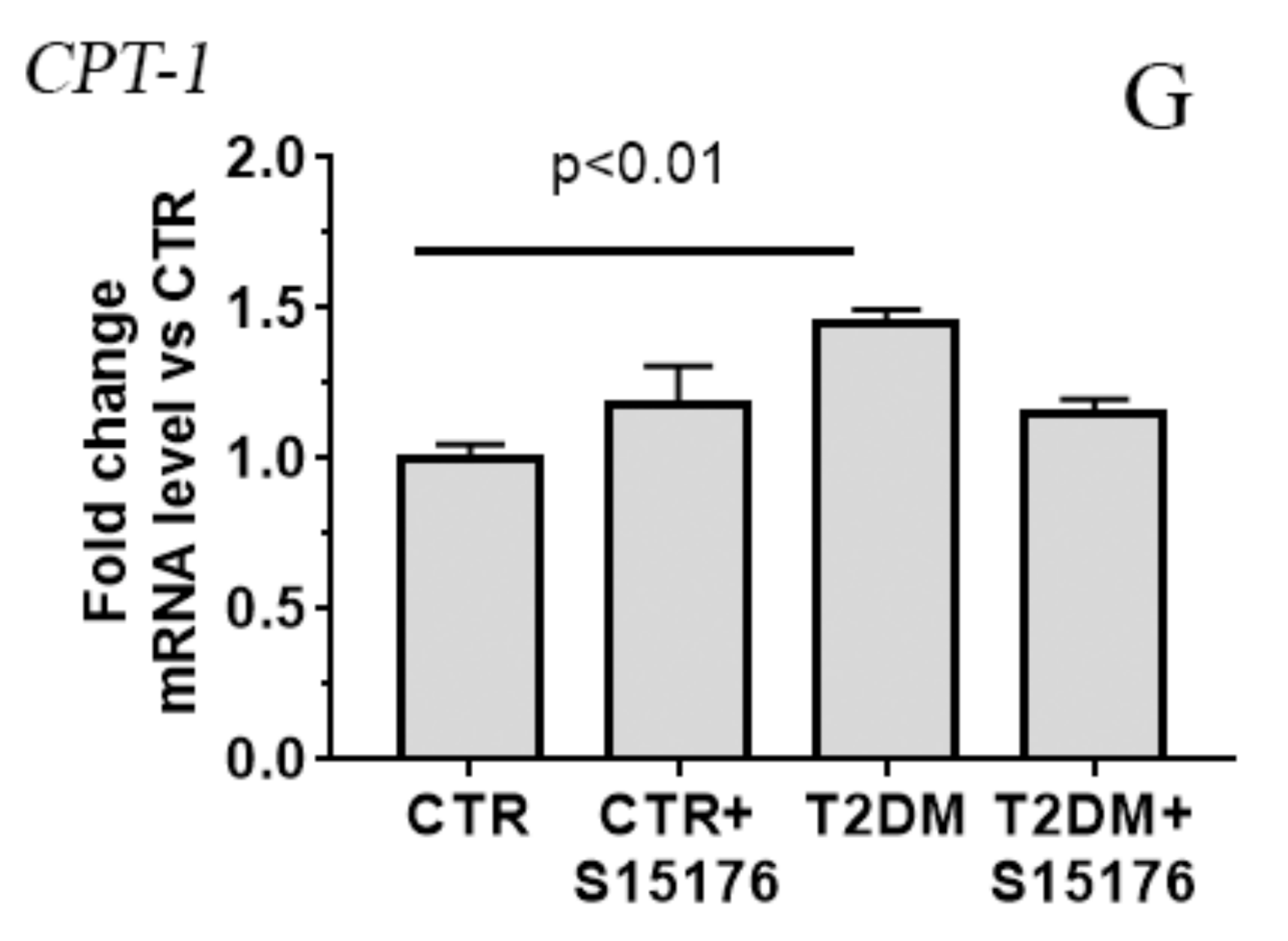
3.3. Effect of S-15176 Difumarate Salt on Diabetes-Induced Changes in the Expression Level of Proteins Responsible for Mitochondrial Biogenesis and Mitochondrial Dynamics
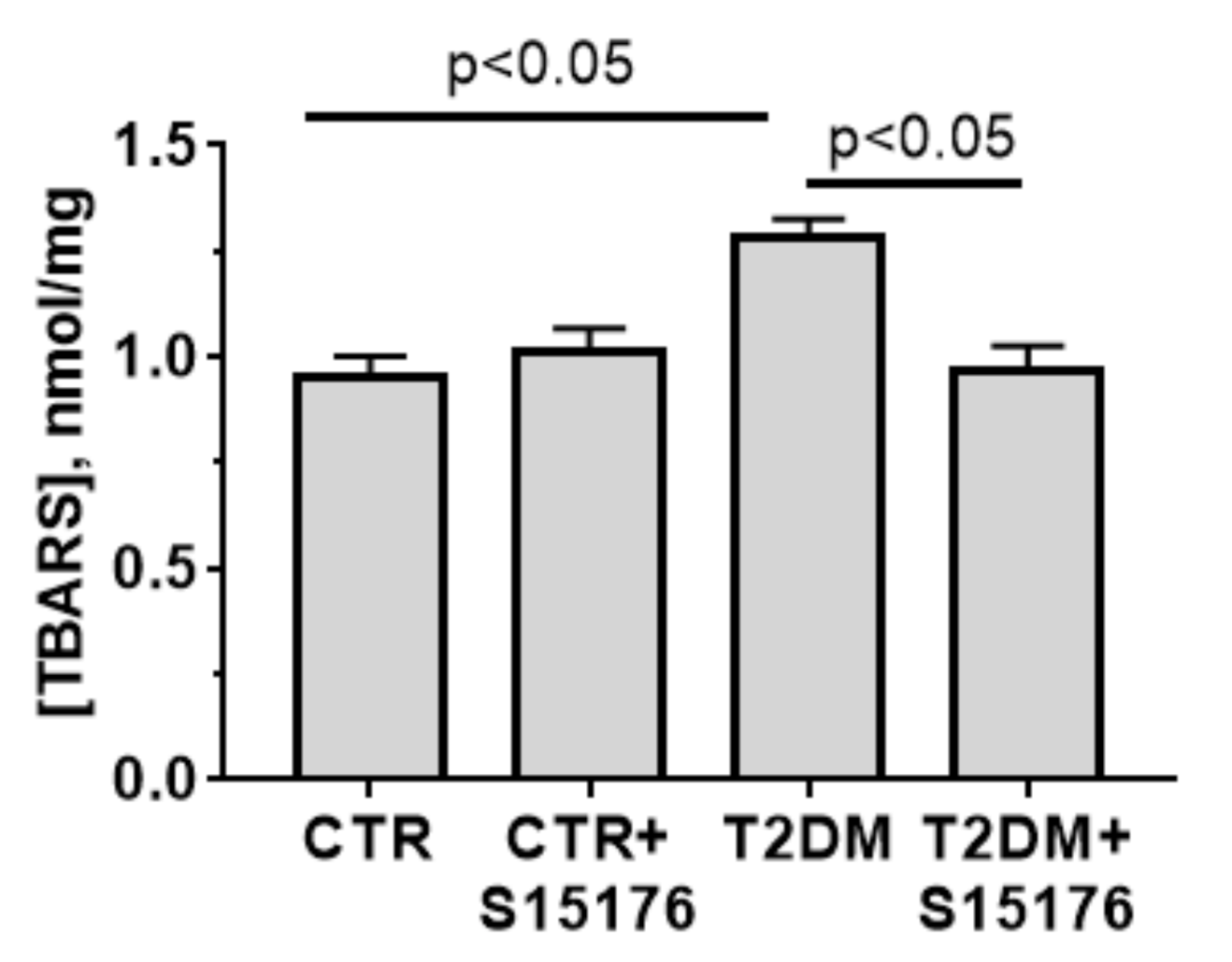
3.4. Effects of T2DM and S-15176 on the Functioning of Mouse Liver Mitochondria
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Winer, N.; Sowers, J.R. Epidemiology of Diabetes. J. Clin. Pharmacol. 2004, 44, 397–405. [Google Scholar] [CrossRef]
- Groop, L.C.; Eriksson, J.G. The etiology and pathogenesis of non-insulin-dependent diabetes. Ann. Med. 1992, 24, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimaki, S.; Kuwabara, T. Diabetes-induced dysfunction of mitochondria and stem cells in skeletal muscle and the nervous system. Int. J. Mol. Sci. 2017, 18, 2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Garikipati, V.N.S.; Kishore, R. Mitochondrial dysfunction and its impact on diabetic heart. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.F.; Natali, A.J.; Da Silva, E.; Gomes, G.J.; Teodoro, B.G.; Cunha, D.N.; Drummond, L.R.; Drummond, F.R.; Moura, A.G.; Belfort, F.G.; et al. Attenuation of Ca2+ homeostasis, oxidative stress, and mitochondrial dysfunctions in diabetic rat heart: Insulin therapy or aerobic exercise? J. Appl. Physiol. 2015, 119, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.C.; Chau, Y.Y.; Ng, H.Y.; Chen, C.H.; Wang, P.W.; Liou, C.W.; Lin, T.K.; Chen, J.B. Empagliflozin protects HK-2 cells from high glucose-mediated injuries via a mitochondrial mechanism. Cells 2019, 8, 1085. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Mitochondrial dysfunction in diabetes and the regulatory roles of antidiabetic agents on the mitochondrial function. J. Cell Physiol. 2019, 234, 8402–8410. [Google Scholar] [CrossRef]
- Segawa, M.; Sekine, S.; Sato, T.; Ito, K. Increased susceptibility to troglitazone-induced mitochondrial permeability transition in type 2 diabetes mellitus model rat. J. Toxicol. Sci. 2018, 43, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Chacko, B.K.; Reily, C.; Srivastava, A.; Johnson, M.S.; Ye, Y.; Ulasova, E.; Agarwal, A.; Zinn, K.R.; Murphy, M.P.; Kalyanaraman, B.; et al. Prevention of diabetic nephropathy in Ins2(+/)⁻(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010, 432, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Kalinovich, A.V.; Mattsson, C.L.; Youssef, M.R.; Petrovic, N.; Ost, M.; Skulachev, V.P.; Shabalina, I.G. Mitochondria-targeted dodecyltriphenylphosphonium (C12TPP) combats high-fat-diet-induced obesity in mice. Int. J. Obes. 2016, 40, 1864–1874. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Chen, F.; Zhang, X. Cyclosporine A attenuates retinal inflammation by inhibiting HMGB-1 formation in rats with type 2 diabetes mellitus. BMC Pharmacol. Toxicol. 2020, 21, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groen, A.K.; van Roermund, C.W.; Vervoorn, R.C.; Tager, J.M. Control of gluconeogenesis in rat liver cells. Flux control coefficients of the enzymes in the gluconeogenic pathway in the absence and presence of glucagon. Biochem. J. 1986, 237, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Grantham, B.D.; Zammit, V.A. Role of carnitine palmitoyltransferase I in the regulation of hepatic ketogenesis during the onset and reversal of chronic diabetes. Biochem. J. 1988, 249, 409–414. [Google Scholar] [CrossRef]
- Bressler, R.; Johnson, D. New pharmacological approaches to therapy of NIDDM. Diabetes Care 1992, 15, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Deems, R.O.; Anderson, R.C.; Foley, J.E. Hypoglycemic effects of a novel fatty acid oxidation inhibitor in rats and monkeys. Am. J. Physiol. 1998, 274, R524–R528. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Lindblom, R.S.J.; Higgins, G.C.; Nguyen, T.V.; Arnstein, M.; Henstridge, D.C.; Granata, C.; Snelson, M.; Thallas-Bonke, V.; Cooper, M.E.; Forbes, J.M.; et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin. Sci. 2020, 134, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Hamdan, M.; Urien, S.; Le Louet, H.; Tillement, J.P.; Morin, D. Inhibition of mitochondrial carnitine palmitoyltransferase-1 by a trimetazidine derivative, S-15176. Pharmacol. Res. 2001, 44, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Erdoğan, H.; Tunçdemir, M.; Kelten, B.; Akdemir, O.; Karaoglan, A.; Taşdemiroglu, E. The effects of difumarate salt S-15176 after spinal cord injury in rats. J. Korean Neurosurg. Soc. 2015, 57, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Elimadi, A.; Jullien, V.; Tillement, J.P.; Morin, D.S. S-15176 inhibits mitochondrial permeability transition via a mechanism independent of its antioxidant properties. Eur. J. Pharmacol. 2003, 468, 93–101. [Google Scholar] [CrossRef]
- Morin, D.; Zini, R.; Berdeaux, A.; Tillement, J.P. Effect of the mitochondrial transition pore inhibitor, S-15176, on rat liver mitochondria: ATP synthase modulation and mitochondrial uncoupling induction. Biochem. Pharmacol. 2006, 72, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Settaf, A.; Zahidy, M.; Elimadi, A.; Sapena, R.; Alsamad, I.A.; Tillement, J.; Morin, D. S-15176 reduces the hepatic injury in rats subjected to experimental ischemia and reperfusion. Eur. J. Pharmacol. 2000, 406, 281–292. [Google Scholar] [CrossRef]
- Nath, S.; Ghosh, S.K.; Choudhury, Y. A murine model of type 2 diabetes mellitus developed using a combination of high fat diet and multiple low doses of streptozotocin treatment mimics the metabolic characteristics of type 2 diabetes mellitus in humans. J. Pharmacol. Toxicol. Methods. 2017, 84, 20–30. [Google Scholar] [CrossRef]
- Kokura, S.; Adachi, S.; Manabe, E.; Mizushima, K.; Hattori, T.; Okuda, T.; Nakabe, N.; Handa, O.; Takagi, T.; Naito, Y.; et al. Whole body hyperthermia improves obesity-induced insulin resistance in diabetic mice. Int. J. Hyperth. 2007, 23, 259–265. [Google Scholar] [CrossRef]
- Allison, D.B.; Paultre, F.; Maggio, C.; Mezzitis, N.; Pi-Sunyer, F.X. The use of areas under curves in diabetes research. Diabetes Care 1995, 18, 245–250. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, I. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA ratio in mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Agafonov, A.V.; Pavlik, L.L.; Dubinin, M.V. Effect of bedaquiline on the functions of rat liver mitochondria. Biochim. Biophys. Acta Biomembr. 2019, 1861, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, A.L.; Pérez, S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxidative Med. Cell Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makrecka-Kuka, M.; Liepinsh, E.; Murray, A.J.; Lemieux, H.; Dambrova, M.; Tepp, K.; Puurand, M.; Kaambre, T.; Han, W.H.; de Goede, P.; et al. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. 2020, 228, e13430. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Luan, Z.; Fu, X.; Xu, X. Overexpression of uncoupling protein 2 inhibits the high glucose-induced apoptosis of human umbilical vein endothelial cells. Int. J. Mol. Med. 2016, 37, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Brady, L.J.; Brady, P.S. Regulation of carnitine palmitoyltransferase synthesis in spontaneously diabetic BB Wistar rats. Diabetes 1989, 38, 65–69. [Google Scholar] [CrossRef]
- Lavrentyev, E.N.; Matta, S.G.; Cook, G.A. Expression of three carnitine palmitoyltransferase-I isoforms in 10 regions of the rat brain during feeding, fasting, and diabetes. Biochem. Biophys. Res. Commun. 2004, 315, 174–178. [Google Scholar] [CrossRef]
- Molinari, F.; Pin, F.; Gorini, S.; Chiandotto, S.; Pontecorvo, L.; Penna, F.; Rizzuto, E.; Pisu, S.; Musarò, A.; Costelli, P.; et al. The mitochondrial metabolic reprogramming agent trimetazidine as an ‘exercise mimetic’ in cachectic C26-bearing mice. J. Cachex Sarcopenia Muscle 2017, 8, 954–973. [Google Scholar] [CrossRef]
- Wu, S.; Chang, G.; Gao, L.; Jiang, D.; Wang, L.; Li, G.; Luo, X.; Qin, S.; Guo, X.; Zhang, D. Trimetazidine protects against myocardial ischemia/reperfusion injury by inhibiting excessive autophagy. J. Mol. Med. 2018, 96, 791–806. [Google Scholar] [CrossRef]
- Stacchiotti, A.; Favero, G.; Lavazza, A.; Golic, I.; Aleksic, M.; Korac, A.; Rodella, L.F.; Rezzani, R. Hepatic macrosteatosis is partially converted to microsteatosis by melatonin supplementation in ob/ob mice non-alcoholic fatty liver disease. PLoS ONE. 2016, 11, e0148115. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Nd4 | ATTATTATTACCCGATGAGGGAACC | ATTAAGATGAGGGCAATTAGCAGT |
| Gadph | GTGAGGGAGATGCYCAGTGT | CTGGCATTGCTCTCAATGAC |
| Drp1 | TTACAGCACACAGGAATTGT | TTGTCACGGGCAACCTTTTA |
| Mfn2 | CACGCTGATGCAGACGGAGAA | ATCCCAGCGGTTGTTCAGG |
| Ppargc1a | CTGCCATTGTTAAGACCGAG | GTGTGAGGAGGGTCATCGTT |
| Cpt1a | GGCATAAACGCAGAGCATTCCTG | CAGTGTCCATCCTCTGAGTAGC |
| Pink1 | TTGCCCCACACCCTAACATC | GCAGGGTACAGGGGTAGTTCT |
| Prkn | AGCCAGAGGTCCAGCAGTTA | GAGGGTTGCTTGTTTGCAGG |
| Opa1 | GGACCCAAGAGCAGTGTGTT | CGAGACTCCAGGTTCTTCCG |
| Rplp2 | CGGCTCAACAAGGTCATCAGTGA | AGCAGAAACAGCCACAGCCCCAC |
| CTR | CTR + S15176 | T2DM | T2DM + S15176 | |
|---|---|---|---|---|
| Initial BW, g | 16.4 ± 1.0 | 15.6 ± 1.1 | 14.5 ± 0.8 | 16.1 ± 0.9 |
| Final BW, g (after two months) | 25.2 ± 0.8 (↑ 153.7%) | 25.3 ± 1.0 (↑ 161.9%) | 24.5 ± 0.6 * (↑ 172.4%) * | 25.5 ± 0.6 (↑ 158.4%) |
| BG (fed state) | 9.7 ± 0.6 | 9.3 ± 0.6 | 20.0 ± 2.2 * | 20.6 ± 1.4 * |
| Group | V Respiration, nmol O2 × min−1 × mg−1 Protein | RCR | ||
|---|---|---|---|---|
| State 2 | State 3 | State 4 | ||
| CTR | 22.0 ± 1.7 | 82.6 ± 1.9 | 19.9 ± 0.9 | 4.2 ± 0.2 |
| CTR+ S-15176 | 19.5 ± 2.2 | 85.2 ± 4.1 | 24.2 ± 2.1 | 3.5 ± 0.3 |
| T2DM | 22.5 ± 1.3 | 85.4 ± 8.3 | 26.1 ± 1.5 * | 3.4 ± 0.2 * |
| T2DM+S-15176 | 19.4 ± 0.9 | 110.1 ± 4.4 *# | 28.4 ± 1.1 * | 3.9 ± 0.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosludtseva, N.V.; Starinets, V.S.; Pavlik, L.L.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtsev, K.N. The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes. Biology 2020, 9, 309. https://doi.org/10.3390/biology9100309
Belosludtseva NV, Starinets VS, Pavlik LL, Mikheeva IB, Dubinin MV, Belosludtsev KN. The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes. Biology. 2020; 9(10):309. https://doi.org/10.3390/biology9100309
Chicago/Turabian StyleBelosludtseva, Natalia V., Vlada S. Starinets, Lyubov L. Pavlik, Irina B. Mikheeva, Mikhail V. Dubinin, and Konstantin N. Belosludtsev. 2020. "The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes" Biology 9, no. 10: 309. https://doi.org/10.3390/biology9100309