Phylogenetic Analyses of Sites in Different Protein Structural Environments Result in Distinct Placements of the Metazoan Root



Abstract
1. Introduction
- H0:
- Conflicting signals are randomly distributed with respect to functionally defined subsets of the data. This hypothesis predicts that separate analyses of those functionally defined subsets of the data matrix will yield trees with the same topology (probably with lower support than the analysis of the complete dataset due to the smaller size of the subsets).
- HA:
- Conflicting signals are associated with functionally defined data subsets. Different subsets of the data matrix defined using functional information are associated with distinct signals (i.e., analyses of those subsets support different topologies when the subsets are analyzed separately).
2. Materials and Methods
2.1. Dataset
2.2. Structural Class Assignment
2.3. Phylogenetic Analyses
2.4. Model Estimation and Model Comparisons
- Two relative solvent accessibility (RSA) based classes (EXPOSED and BURIED).
- Three secondary structure-based classes (HELIX, SHEET, and COIL).
- Six classes, combining RSA and secondary structure (HELIX_EXP, HELIX_BUR, SHEET_EXP, SHEET_BUR, COIL_EXP, and COIL_BUR).
2.5. Analyses Using Site-Heterogeneous CAT-Type Models
2.6. Compositional Heterogeneity and Data Recoding
3. Results
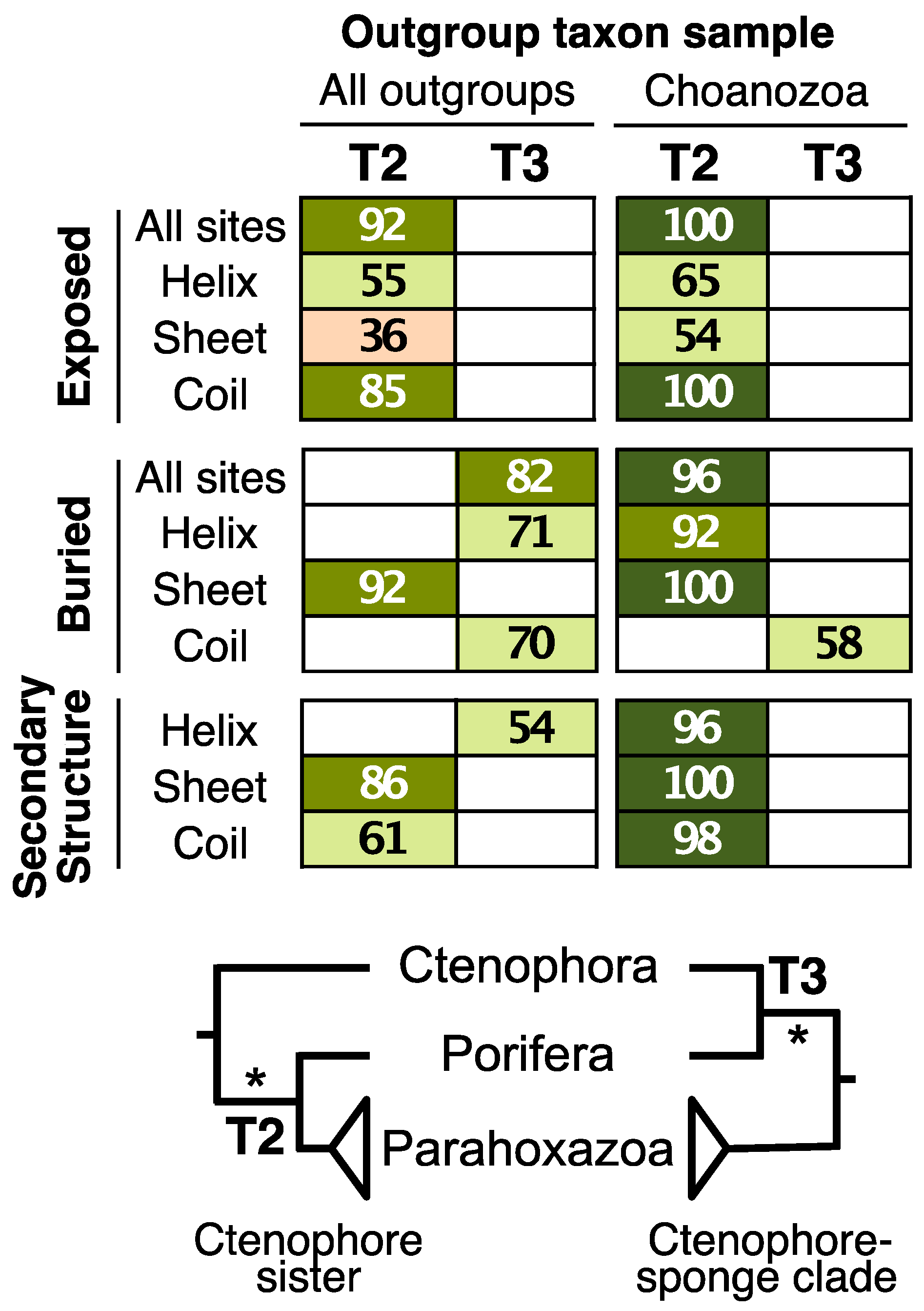
3.1. Sites in Distinct Structural Environments Have Different Signals
3.2. Decisive Sites Reveal Conflicts within Each Structural Class
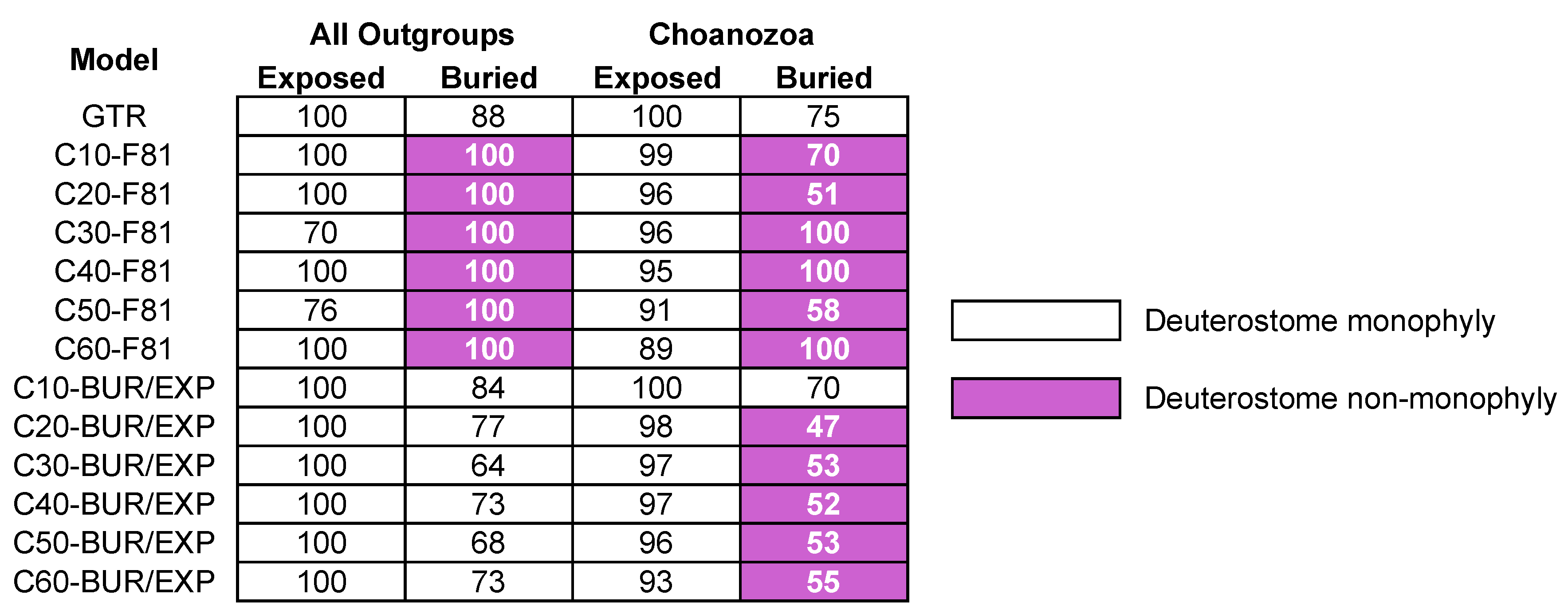
3.3. Reduced Outgroup Sampling also Reduces Some Differences in Signal
3.4. Sites in Different Structural Environments Exhibit Distinct Patterns of Sequence Evolution
3.5. Site-Heterogeneous Profile Mixture Models can Yield Surprising Topological Changes
3.6. Binary Recoding Eliminates the Observed Differences in Signal
4. Discussion
4.1. Different Models for Different Structural Environments
4.2. Site-Heterogeneous CAT-Type Models do not Increase Congruence in Signal for Different Structural Classes
4.3. Amino Acid Recoding Increases Congruence in Signal for Different Structural Classes
4.4. Phylogenetic Implications
4.5. Why Are Different Structural Environments Associated with Distinct Signals?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gee, H. Ending incongruence. Nature 2003, 425, 782. [Google Scholar] [CrossRef]
- Rokas, A.; Williams, B.I.; King, N.; Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef]
- Nishihara, H.; Okada, N.; Hasegawa, M.; Rokas, A.; Williams, B.; King, N.; Carroll, S.; Soltis, D.; Albert, V.; Savolainen, V.; et al. Rooting the eutherian tree: The power and pitfalls of phylogenomics. Genome Biol. 2007, 8, R199. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, S.; Barker, M.S.; Burleigh, J.G.; Gitzendanner, M.A.; et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 2014, 111, E4859–E4868. [Google Scholar] [CrossRef]
- Ryan, J.F.; Pang, K.; Schnitzler, C.E.; Nguyen, A.D.; Moreland, R.T.; Simmons, D.K.; Koch, B.J.; Francis, W.R.; Havlak, P.; Smith, S.A.; et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 2013, 342, 1242592. [Google Scholar] [CrossRef]
- Moroz, L.L.; Kocot, K.M.; Citarella, M.R.; Dosung, S.; Norekian, T.P.; Povolotskaya, I.S.; Grigorenko, A.P.; Dailey, C.; Berezikov, E.; Buckley, K.M.; et al. The ctenophore genome and the evolutionary origins of neural systems. Nature 2014, 510, 109–114. [Google Scholar] [CrossRef]
- Dunn, C.W.; Giribet, G.; Edgecombe, G.D.; Hejnol, A. Animal phylogeny and its evolutionary implications. Annu. Rev. Ecol. Evol. Syst. 2014, 45, 371–395. [Google Scholar] [CrossRef]
- Feuda, R.; Dohrmann, M.; Pett, W.; Philippe, H.; Rota-Stabelli, O.; Lartillot, N.; Wörheide, G.; Pisani, D. Improved modeling of compositional heterogeneity supports sponges as sister to all other animals. Curr. Biol. 2017, 27, 3864–3870. [Google Scholar] [CrossRef]
- King, N.; Rokas, A. Embracing uncertainty in reconstructing early animal evolution. Curr. Biol. 2017, 27, R1081–R1088. [Google Scholar] [CrossRef]
- Simion, P.; Philippe, H.; Baurain, D.; Jager, M.; Richter, D.J.; Di Franco, A.; Roure, B.; Satoh, N.; Quéinnec, É.; Ereskovsky, A.; et al. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr. Biol. 2017, 27, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Zool. 1978, 27, 401–410. [Google Scholar] [CrossRef]
- Hendy, M.D.; Penny, D. A framework for the quantitative study of evolutionary trees. Syst. Zool. 1989, 38, 297–309. [Google Scholar] [CrossRef]
- Phillips, M.J.; Delsuc, F.; Penny, D. Genome-scale phylogeny and the detection of systematic biases. Mol. Biol. Evol. 2004, 21, 1455–1458. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Goldman, N.; Thorne, J.L.; Jones, D.T. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 1998, 149, 445–458. [Google Scholar]
- Thorne, J.L.; Goldman, N.; Jones, D.T. Combining protein evolution and secondary structure. Mol. Biol. Evol. 1996, 13, 666–673. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. Accounting for solvent accessibility and secondary structure in protein phylogenetics is clearly beneficial. Syst. Biol. 2010, 59, 277–287. [Google Scholar] [CrossRef]
- Le, S.Q.; Lartillot, N.; Gascuel, O. Phylogenetic mixture models for proteins. Philos. Trans. R. Soc. B 2008, 363, 3965–3976. [Google Scholar] [CrossRef]
- Blanquart, S.; Lartillot, N. A Bayesian compound stochastic process for modeling nonstationary and nonhomogeneous sequence evolution. Mol. Biol. Evol. 2006, 23, 2058–2071. [Google Scholar] [CrossRef]
- Blanquart, S.; Lartillot, N. A site- and time-heterogeneous model of amino acid replacement. Mol. Biol. Evol. 2008, 25, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Groussin, M.; Boussau, B.; Gouy, M. A branch-heterogeneous model of protein evolution for efficient inference of ancestral sequences. Syst. Biol. 2013, 62, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Whelan, N.V.; Halanych, K.M. Who let the CAT out of the bag? Accurately dealing with substitutional heterogeneity in phylogenomic analyses. Syst. Biol. 2016, 66, 232–255. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Kimball, R.T.; Braun, E.L. Error in phylogenetic estimation for bushes in the tree of life. J. Phylogenetics Evol. Biol. 2013, 1, 110. [Google Scholar] [CrossRef]
- Maddison, W.P. Gene trees in species trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Slowinski, J.B.; Page, R.D.M. How should species phylogenies be inferred from sequence data ? Syst. Biol. 2007, 48, 814–825. [Google Scholar] [CrossRef]
- Edwards, S.V. Is a new and general theory of molecular systematics emerging? Evolution 2009, 63, 1–19. [Google Scholar] [CrossRef]
- Reddy, S.; Kimball, R.T.; Pandey, A.; Hosner, P.A.; Braun, M.J.; Hackett, S.J.; Han, K.-L.; Harshman, J.; Huddleston, C.J.; Kingston, S.; et al. Why do phylogenomic data sets yield conflicting trees? Data type influences the avian tree of life more than taxon sampling. Syst. Biol. 2017, 51, 588–598. [Google Scholar] [CrossRef]
- Braun, E.L.; Cracraft, J.; Houde, P. Resolving the avian tree of life from top to bottom: The promise and potential boundaries of the phylogenomic era. In Avian Genomics in Ecology and Evolution—From the Lab into the Wild; Kraus, R.H.S., Ed.; Springer: Cham, Switzerland, 2019; pp. 151–210. [Google Scholar]
- Raymann, K.; Brochier-Armanet, C.; Gribaldo, S. The two-domain tree of life is linked to a new root for the Archaea. Proc. Natl. Acad. Sci. USA 2015, 112, 6670–6675. [Google Scholar] [CrossRef]
- He, D.; Fiz-Palacios, O.; Fu, C.J.; Tsai, C.C.; Baldauf, S.L. An alternative root for the eukaryote tree of life. Curr. Biol. 2014, 24, 465–470. [Google Scholar] [CrossRef]
- Lesk, A.M.; Chothia, C.H. The response of protein structures to amino-acid sequence changes. Philos. Trans. R. Soc. 1986, 317, 345–356. [Google Scholar]
- Illergård, K.; Ardell, D.H.; Elofsson, A. Structure is three to ten times more conserved than sequence—A study of structural response in protein cores. Proteins 2009, 77, 499–508. [Google Scholar]
- Magnan, C.; Baldi, P. SSpro/ACCpro 5: Almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning, and structural similarity. Bioinformatics 2014, 30, 2592–2597. [Google Scholar] [CrossRef]
- Nielsen, C. Early animal evolution: A morphologist’s view. R. Soc. Open Sci. 2019, 6, 190638. [Google Scholar] [CrossRef]
- Pisani, D.; Pett, W.; Dohrmann, M.; Feuda, R.; Rota-Stabelli, O.; Philippe, H.; Lartillot, N.; Wörheide, G. Genomic data do not support comb jellies as the sister group to all other animals. Proc. Natl. Acad. Sci. USA 2015, 112, 15402–15407. [Google Scholar] [CrossRef]
- Philippe, H.; Derelle, R.; Lopez, P.; Pick, K.; Borchiellini, C.; Boury-Esnault, N.; Vacelet, J.; Renard, E.; Houliston, E.; Quéinnec, E.; et al. Phylogenomics revives traditional views on deep animal relationships. Curr. Biol. 2009, 19, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Pick, K.S.; Philippe, H.; Schreiber, F.; Erpenbeck, D.; Jackson, D.J.; Wrede, P.; Wiens, M.; Alié, A.; Morgenstern, B.; Manuel, M.; et al. Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Mol. Biol. Evol. 2010, 27, 1983–1987. [Google Scholar] [CrossRef]
- Nosenko, T.; Schreiber, F.; Adamska, M.; Adamski, M.; Eitel, M.; Hammel, J.; Maldonado, M.; Müller, W.E.G.; Nickel, M.; Schierwater, B.; et al. Deep metazoan phylogeny: When different genes tell different stories. Mol. Phylogenet. Evol. 2013, 67, 223–233. [Google Scholar] [CrossRef]
- Dunn, C.W.; Hejnol, A.; Matus, D.Q.; Pang, K.; Browne, W.E.; Smith, S.A.; Seaver, E.; Rouse, G.W.; Obst, M.; Edgecombe, G.D.; et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 2008, 452, 745–749. [Google Scholar] [CrossRef]
- Hejnol, A.; Obst, M.; Stamatakis, A.; Ott, M.; Rouse, G.W.; Edgecombe, G.D.; Martinez, P.; Baguna, J.; Bailly, X.; Jondelius, U.; et al. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. R. Soc. B 2009, 276, 4261–4270. [Google Scholar] [CrossRef]
- Borowiec, M.L.; Lee, E.K.; Chiu, J.C.; Plachetzki, D.C. Extracting phylogenetic signal and accounting for bias in whole-genome data sets supports the Ctenophora as sister to remaining Metazoa. BMC Genomics 2015, 16, 987. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.F.; Pang, K.; NISC Comparative Sequencing Program; Mullikin, J.C.; Martindale, M.Q.; Baxevanis, A.D. The homeodomain complement of the ctenophore Mnemiopsis leidyi suggests that Ctenophora and Porifera diverged prior to the ParaHoxozoa. Evodevo 2010, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformat. 2003. Available online: https://currentprotocols.onlinelibrary.wiley.com/action/showCitFormats?doi=10.1002%2F0471250953.bi0203s00 (accessed on 9 March 2020).
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, 72–76. [Google Scholar] [CrossRef]
- Pollastri, G.; Przybylski, D.; Rost, B.; Baldi, P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins 2002, 47, 228–235. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G. Position-based sequence weights. J. Mol. Biol. 1994, 243, 574–578. [Google Scholar] [CrossRef]
- Maddison, D.R.; Swofford, D.L.; Maddison, W.P. NEXUS: An extensible file format for systematic information. Syst. Biol. 2006, 46, 590–621. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Vingron, M. Modeling amino acid replacement. J. Comput. Biol. 2002, 7, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Kosiol, C.; Goldman, N. Different versions of the Dayhoff rate matrix. Mol. Biol. Evol. 2005, 22, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Dimmic, M.W.; Rest, J.S.; Mindell, D.P.; Goldstein, R.A. rtREV: An amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J. Mol. Evol. 2002, 55, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Pattengale, N.D.; Alipour, M.; Bininda-Emonds, O.R.P.; Moret, B.M.E.; Stamatakis, A. How many bootstrap replicates are necessary? J. Comput. Biol. 2010, 17, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Kimball, R.T.; Wang, N.; Heimer-McGinn, V.; Ferguson, C.; Braun, E.L. Identifying localized biases in large datasets: A case study using the avian tree of life. Mol. Phylogenet. Evol. 2013, 69, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-X.; Hittinger, C.T.; Rokas, A. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat. Ecol. Evol. 2017, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Worth, C.L.; Gong, S.; Blundell, T.L. Structural and functional constraints in the evolution of protein families. Nat. Rev. Mol. Cell Biol. 2009, 10, 709–720. [Google Scholar] [CrossRef]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Toda, H.; Ikeguchi, M. Dependence of α-helical and β-sheet amino acid propensities on the overall protein fold type. BMC Struct. Biol. 2012, 12, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.Y.; Fasman, G.D. Prediction of protein conformation. Biochemistry 1974, 13, 222–245. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O.; Lartillot, N. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics 2008, 24, 2317–2323. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Warnow, T. Computational Phylogenetics: An Introduction to Designing Methods for Phylogeny Estimation, 1st ed.; Cambridge University Press: New York, NY, USA, 2017; ISBN 1107184711. [Google Scholar]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Melamed, D.; Young, D.L.; Gamble, C.E.; Miller, C.R.; Fields, S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly (A)-binding protein. RNA 2013, 19, 1537–1551. [Google Scholar] [CrossRef]
- Pollock, D.D.; Thiltgen, G.; Goldstein, R.A. Amino acid coevolution induces an evolutionary stokes shift. Proc. Natl. Acad. Sci. USA 2012, 109, E1352–E1359. [Google Scholar] [CrossRef]
- Singer, G.A.C.; Hickey, D.A. Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol. Biol. Evol. 2000, 17, 1581–1588. [Google Scholar] [CrossRef]
- Embley, T.M.; Van Der Giezen, M.; Horner, D.S.; Dyal, P.L.; Foster, P.; Tielens, A.G.M.; Martin, W.; Tovar, J.; Douglas, A.E.; Cavalier-Smith, T.; et al. Mitochondria and hydrogenosomes are two forms of the same fundamental organelle. Philos. Trans. R. Soc. B 2003, 358, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Hrdy, I.; Hirt, R.P.; Dolezal, P.; Bardonová, L.; Foster, P.G.; Tachezy, J.; Embley, T.M. Trichomonas hydrogenosomes contain the NADH dehydrogenase module of mitochondrial complex I. Nature 2004, 432, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Achenbach, L.; Rouviere, P.; Mandelco, L. Archaeal Phylogeny: Reexamination of the phylogenetic position of Archaeoglobus fulgidus in light of certain composition-induced artifacts. Syst. Appl. Microbiol. 1991, 14, 364–371. [Google Scholar] [CrossRef]
- Budd, G.E.; Jensen, S. The origin of the animals and a ‘Savannah’ hypothesis for early bilaterian evolution. Biol. Rev. 2017, 92, 446–473. [Google Scholar] [CrossRef] [PubMed]
- Salichos, L.; Rokas, A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 2013, 497, 327–331. [Google Scholar] [CrossRef]
- Brown, J.M.; Thomson, R.C. Bayes factors unmask highly variable information content, bias, and extreme influence in phylogenomic analyses. Syst. Biol. 2016, 66, 517–530. [Google Scholar] [CrossRef]
- Evans, N.M.; Holder, M.T.; Barbeitos, M.S.; Okamura, B.; Cartwright, P. The phylogenetic position of Myxozoa: Exploring conflicting signals in phylogenomic and ribosomal data sets. Mol. Biol. Evol. 2010, 27, 2733–2746. [Google Scholar] [CrossRef]
- Schalchian-Tabrizi, K.; Minge, M.A.; Espelund, M.; Orr, R.; Ruden, T.; Jakobsen, K.S.; Cavalier-Smith, T. Multigene phylogeny of Choanozoa and the origin of animals. PLoS ONE 2008, 3, e2098. [Google Scholar] [CrossRef]
- Carr, M.; Leadbeater, B.S.C.; Hassan, R.; Nelson, M.; Baldauf, S.L. Molecular phylogeny of choanoflagellates, the sister group to Metazoa. Proc. Natl. Acad. Sci. USA 2008, 105, 16641–16646. [Google Scholar] [CrossRef]
- Wilke, C.O. Bringing molecules back into molecular evolution. PLoS Comput. Biol. 2012, 8, e1002572. [Google Scholar] [CrossRef]
- Crooks, G.E.; Brenner, S.E. An alternative model of amino acid replacement. Bioinformatics 2005, 21, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.; Sonnhammer, E.L.; Chothia, C. Volume changes in protein evolution. J. Mol. Biol. 1994, 236, 1067–1078. [Google Scholar] [CrossRef]
- Philippe, H.; Brinkmann, H.; Copley, R.R.; Moroz, L.L.; Nakano, H.; Poustka, A.J.; Wallberg, A.; Peterson, K.J.; Telford, M.J. Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature 2011, 470, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Egger, B.; Lapraz, F.; Tomiczek, B.; Müller, S.; Dessimoz, C.; Girstmair, J.; Škunca, N.; Rawlinson, K.A.; Cameron, C.B.; Beli, E.; et al. A transcriptomic-phylogenomic analysis of the evolutionary relationships of flatworms. Curr. Biol. 2015, 25, 1347–1353. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L.; Kubatko, L.; Pearl, D.K.; Edwards, S.V. Coalescent methods for estimating phylogenetic trees. Mol. Phylogenet. Evol. 2009, 53, 320–328. [Google Scholar] [CrossRef]
- Tsagkogeorga, G.; Turon, X.; Hopcroft, R.R.; Tilak, M.K.; Feldstein, T.; Shenkar, N.; Loya, Y.; Huchon, D.; Douzery, E.J.; Delsuc, F. An updated 18S rRNA phylogeny of tunicates based on mixture and secondary structure models. BMC Evol. Biol. 2009, 9, 187. [Google Scholar] [CrossRef]
- Finet, C.; Timme, R.E.; Delwiche, C.F.; Marlétaz, F. Multigene phylogeny of the green lineage reveals the origin and diversification of land plants. Curr. Biol. 2010, 20, 2217–2222. [Google Scholar] [CrossRef]
- Philippe, H.; Brinkmann, H.; Lavrov, D.V.; Littlewood, D.T.J.; Manuel, M.; Wörheide, G.; Baurain, D. Resolving difficult phylogenetic questions: Why more sequences are not enough. PLoS Biol. 2011, 9, e1000602. [Google Scholar] [CrossRef]
- Lartillot, N.; Brinkmann, H.; Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 2007, 7 (Suppl. S1), S4. [Google Scholar] [CrossRef]
- Brinkmann, H.; Philippe, H. Animal phylogeny and large-scale sequencing: Progress and pitfalls. J. Syst. Evol. 2008, 46, 274–286. [Google Scholar]
- Roure, B.; Baurain, D.; Philippe, H. Impact of missing data on phylogenies inferred from empirical phylogenomic data sets. Mol. Biol. Evol. 2013, 30, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Roscoe, B.P.; Bolon, D.N.A. Systematic exploration of ubiquitin sequence, E1 activation efficiency, and experimental fitness in yeast. J. Mol. Biol. 2014, 426, 2854–2870. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Young, D.L.; Islam, M.; Kitzman, J.O.; Gullingsrud, J.; Hause, R.J.; Fowler, D.M.; Parvin, J.D.; Shendure, J.; Fields, S. Massively parallel functional analysis of BRCA1 RING domain variants. Genetics 2015, 200, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Mighell, T.L.; Evans-Dutson, S.; O’Roak, B.J. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am. J. Hum. Genet. 2018, 102, 943–955. [Google Scholar] [CrossRef]
- Roure, B.; Philippe, H. Site-specific time heterogeneity of the substitution process and its impact on phylogenetic inference. BMC Evol. Biol. 2011, 11, 17. [Google Scholar] [CrossRef]
- Telford, M.J.; Budd, G.E.; Philippe, H. Phylogenomic insights into animal evolution. Curr. Biol. 2015, 25, R876–R887. [Google Scholar] [CrossRef]
- Halanych, K.M.; Whelan, N.V.; Kocot, K.M.; Kohn, A.B.; Moroz, L.L. Miscues misplace sponges. Proc. Natl. Acad. Sci. USA 2016, 113, E946–E947. [Google Scholar] [CrossRef]
- Suzuki, Y.; Glazko, G.V.; Nei, M. Overcredibility of molecular phylogenies obtained by Bayesian phylogenetics. Proc. Natl. Acad. Sci. USA 2002, 99, 16138–16143. [Google Scholar] [CrossRef]
- Simmons, M.P.; Pickett, K.M.; Miya, M. How meaningful are Bayesian support values? Mol. Biol. Evol. 2004, 21, 188–199. [Google Scholar] [CrossRef]
- Liò, P.; Goldman, N. Models of molecular evolution and phylogeny. Genome Res. 1998, 8, 1233–1244. [Google Scholar] [CrossRef]
- Braun, E.L. An evolutionary model motivated by physicochemical properties of amino acids reveals variation among proteins. Bioinformatics 2018, 34, i350–i356. [Google Scholar] [CrossRef] [PubMed]
- Foster, P.G.; Hickey, D.A. Compositional bias may affect both DNA-based and protein-based phylogenetic reconstructions. J. Mol. Evol. 1999, 48, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Katsu, Y.; Braun, E.L.; Guillette, L.J.; Iguchi, T. From reptilian phylogenomics to reptilian genomes: Analyses of c-Jun and DJ-1 proto-oncogenes. Cytogenet. Genome Res. 2010, 127, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Singer, G.A.C.; Hickey, D.A. Mutational bias affects protein evolution in flowering plants. Mol. Biol. Evol. 2004, 21, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Savard, J.; Tautz, D.; Richards, S.; Weinstock, G.M.; Gibbs, R.A.; Werren, J.H.; Tettelin, H.; Lercher, M.J. Phylogenomic analysis reveals bees and wasps (Hymenoptera) at the base of the radiation of Holometabolous insects. Genome Res. 2006, 16, 1334–1338. [Google Scholar] [CrossRef]
- Boussau, B.; Blanquart, S.; Necsulea, A.; Lartillot, N.; Gouy, M. Parallel adaptations to high temperatures in the Archaean eon. Nature 2008, 456, 942–945. [Google Scholar] [CrossRef]
- Hernandez, A.M.; Ryan, J.F. Six-state amino acid recoding is not an effective strategy to offset the effects of compositional heterogeneity and saturation in phylogenetic analyses. BioRxiv 2019, 729103. [Google Scholar] [CrossRef]
- Simmons, M.P. Relative benefits of amino-acid, codon, degeneracy, DNA, and purine-pyrimidine character coding for phylogenetic analyses of exons. J. Syst. Evol. 2017, 55, 85–109. [Google Scholar] [CrossRef]
- Mallet, J.; Besansky, N.; Hahn, M.W. How reticulated are species? BioEssays 2016, 38, 140–149. [Google Scholar] [CrossRef]
- Rivera, M.C.; Lake, J.A. The ring of life provides evidence for a genome fusion origin of eukaryotes. Nature 2004, 431, 152–155. [Google Scholar] [CrossRef]
- Gatesy, J.; Springer, M.S. Phylogenetic analysis at deep timescales: Unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol. Phylogenet. Evol. 2014, 80, 231–266. [Google Scholar] [CrossRef] [PubMed]
- Laumer, C.E.; Gruber-Vodicka, H.; Hadfield, M.G.; Pearse, V.B.; Riesgo, A.; Marioni, J.C.; Giribet, G. Support for a clade of placozoa and cnidaria in genes with minimal compositional bias. eLife 2018, 7, e36278. [Google Scholar] [CrossRef]
- Laumer, C.E.; Fernandez, R.; Lemer, S.; Andrade, C.S.; Combosch, D.; Kocot, K.M.; Riesgo, A.; Sterrer, W.; Sørensen, M.V.; Giribet, G. Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc. R. Soc. B 2019, 286, 20190831. [Google Scholar] [CrossRef] [PubMed]
- Torruella, G.; Derelle, R.; Paps, J.; Lang, B.F.; Roger, A.J.; Shalchian-Tabrizi, K.; Ruiz-Trillo, I. Phylogenetic relationships within the Opisthokonta based on phylogenomic analyses of conserved single-copy protein domains. Mol. Biol. Evol. 2012, 29, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Torruella, G.; De Mendoza, A.; Grau-Bové, X.; Antó, M.; Chaplin, M.A.; Del Campo, J.; Eme, L.; Pérez-Cordón, G.; Whipps, C.M.; Nichols, K.M.; et al. Phylogenomics reveals convergent evolution of lifestyles in close relatives of animals and fungi. Curr. Biol. 2015, 25, 2404–2410. [Google Scholar] [CrossRef] [PubMed]
- Hehenberger, E.; Tikhonenkov, D.V.; Kolisko, M.; del Campo, J.; Esaulov, A.S.; Mylnikov, A.P.; Keeling, P.J. Novel predators reshape holozoan phylogeny and reveal the presence of a two-component signaling system in the ancestor of animals. Curr. Biol. 2017, 27, 2043–2050. [Google Scholar] [CrossRef]
- Brown, M.W.; Heiss, A.A.; Kamikawa, R.; Inagaki, Y.; Yabuki, A.; Tice, A.K.; Shiratori, T.; Ishida, K.I.; Hashimoto, T.; Simpson, A.G.B.; et al. Phylogenomics places orphan protistan lineages in a novel eukaryotic super-group. Genome Biol. Evol. 2018, 10, 427–433. [Google Scholar] [CrossRef]
- Bull, J.J.; Huelsenbeck, J.P.; Cunningham, C.W.; Swofford, D.L.; Waddell, P.J. Partitioning and combining data in phylogenetic analysis. Syst. Biol. 2010, 42, 384–397. [Google Scholar] [CrossRef]
- Steel, M.; Penny, D. Parsimony, likelihood, and the role of models in molecular phylogenetics. Mol. Biol. Evol. 2000, 17, 839–850. [Google Scholar] [CrossRef]
- Farris, J.S. The retention index and the rescaled consistency index. Cladistics 1989, 5, 417–419. [Google Scholar] [CrossRef]
- Fisher, R.A. The correlation between relatives on the supposition of mendelian inheritance. Trans. R. Soc. Edinb. 1919, 52, 399–433. [Google Scholar] [CrossRef]
- Starr, T.N.; Thornton, J.W. Epistasis in protein evolution. Protein Sci. 2016, 25, 1204–1218. [Google Scholar] [CrossRef]
- Podgornaia, A.I.; Laub, M.T. Pervasive degeneracy and epistasis in a protein-protein interface. Science 2015, 347, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Gatesy, J. A tenth crucial question regarding model use in phylogenetics. Trends Ecol. Evol. 2007, 22, 509–510. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.J.; Kim, J. Parametric phylogenetics? Syst. Biol. 2000, 49, 817–829. [Google Scholar] [CrossRef]
- Steel, M. Should phylogenetic models be trying to “fit an elephant”? Trends Genet. 2005, 21, 307–309. [Google Scholar] [CrossRef]
- Dyson, F. A meeting with Enrico Fermi. Nature 2004, 427, 297. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Subset | Model | T2 b | T3 b | Cni+Bil c | Cni+Pla c | lnL | AICc |
|---|---|---|---|---|---|---|---|
| Exposed | GTR | 92 | - | - | 98 | −1,212,232.985 | 2,424,956.474 |
| LG | 100 | - | - | 100 | −1,222,489.017 | 2,445,088.162 | |
| WAG | 87 | - | - | 100 | −1,225,898.553 | 2,451,907.235 | |
| VT | 96 | - | - | 98 | −1,225,672.633 | 2,451,455.395 | |
| rtREV | 90 | - | - | 98 | −1,222,733.929 | 2,445,577.986 | |
| JTTDCMUT | 94 | - | - | 98 | −1,229,028.451 | 2,458,167.031 | |
| Buried | GTR | - | 82 | 64 | - | −1,045,694.924 | 2,091,880.072 |
| LG | - | 87 | 62 | - | −1,050,022.577 | 2,100,155.267 | |
| WAG | - | 85 | 58 | - | −1,054,829.256 | 2,109,768.626 | |
| VT | - | 81 | 61 | - | −1,059,148.671 | 2,118,407.455 | |
| rtREV | - | 89 | 52 | - | −1,054,432.123 | 2,108,974.359 | |
| JTTDCMUT | - | 81 | - | 54 | −1,061,103.295 | 2,122,316.704 |
| Site Class | Ctenophore Sister (T2) | Ctenophore + Porifera (T3) |
|---|---|---|
| Exposed | 172 | 150 |
| Buried | 167 | 205 |
| LGL-F81 Model | Outgroup | Dataset | T2 | T3 | Cni+Bil | Cni+Pla | Pori + Pla | lnL | AICc |
|---|---|---|---|---|---|---|---|---|---|
| C10 | All outgroups (RG) | Exposed | 100 | 100 | −1,225,103.471 | 2,450,339.126 | |||
| C20 | Exposed | 63 | 100 | −1,216,444.36 | 2,433,040.964 | ||||
| C30 | Exposed | 100 | 72 | −1,214,319.593 | 2,428,811.499 | ||||
| C40 | Exposed | 100 | 96 | −1,212,974.829 | 2,426,142.047 | ||||
| C50 | Exposed | 100 | 73 | −1,212,360.091 | 2,424,932.657 | ||||
| C60 | Exposed | 100 | 93 | −1,211,470.44 | 2,423,173.448 | ||||
| C10 | Buried | 83 | 78 | −1,053,873.212 | 2,107,878.588 | ||||
| C20 | Buried | 97 | 80 | −1,048,007.721 | 2,096,167.66 | ||||
| C30 | Buried | 94 | 73 | −1,046,099.005 | 2,092,370.288 | ||||
| C40 | Buried | 92 | 83 | −1,044,155.469 | 2,088,503.283 | ||||
| C50 | Buried | 93 | 86 | −1,043,172.44 | 2,086,557.3 | ||||
| C60 | Buried | 93 | 89 | −1,042,207.261 | 2,084,647.025 | ||||
| C10 | Choanozoa only | Exposed | 100 | 100 | −964,491.9951 | 1,929,100.133 | |||
| C20 | Exposed | 98 | 74 | −957,706.2984 | 1,915,548.793 | ||||
| C30 | Exposed | 96 | 69 | −956,161.8764 | 1,912,480.01 | ||||
| C40 | Exposed | 95 | 71 | −955,297.444 | 1,910,771.215 | ||||
| C50 | Exposed | 92 | 51 | −953,519.0992 | 1,907,234.604 | ||||
| C60 | Exposed | 90 | 56 | −952,582.9203 | 1,905,382.332 | ||||
| C10 | Buried | 50 | 46 | −837,871.4589 | 1,675,859.045 | ||||
| C20 | Buried | 37 | 34 | −833,463.787 | 1,667,063.748 | ||||
| C30 | Buried | 75 | 63 | −832,000.2662 | 1,664,156.761 | ||||
| C40 | Buried | 66 | 63 | −830,714.1456 | 1,661,604.582 | ||||
| C50 | Buried | 45 | 45 | −829,970.749 | 1,660,137.858 | ||||
| C60 | Buried | 74 | 79 | −829,121.6382 | 1,658,459.713 |
| LGL Model | Outgroup | Dataset | T2 | T3 | Cni+Pla | FO Weight | lnL | AICc |
|---|---|---|---|---|---|---|---|---|
| EXP-C10 | All outgroups (RG) | Exposed | 100 | 100 | 0.2774 | −1,209,875.9003 | 2,419,883.9851 | |
| EXP-C20 | Exposed | 100 | 99 | 0.1808 | −1,206,472.3633 | 2,413,096.9708 | ||
| EXP-C30 | Exposed | 100 | 99 | 0.126 | −1,204,598.8783 | 2,409,370.0689 | ||
| EXP-C40 | Exposed | 100 | 99 | 0.105 | −1,203,943.3823 | 2,408,079.1535 | ||
| EXP-C50 | Exposed | 100 | 99 | 0.1139 | −1,204,270.9338 | 2,408,754.3413 | ||
| EXP-C60 | Exposed | 63 | 100 | 0.1146 | −1,203,752.4217 | 2,407,737.4104 | ||
| BUR-C10 | Buried | 93 | 73 | 0.3637 | −1,043,644.2586 | 2,087,420.6812 | ||
| BUR-C20 | Buried | 95 | 81 | 0.1584 | −1,037,855.6842 | 2,075,863.5853 | ||
| BUR-C30 | Buried | 95 | 84 | 0.0741 | −1,037,721.2711 | 2,075,614.8197 | ||
| BUR-C40 | Buried | 91 | 86 | 0.0797 | −1,036,144.9852 | 2,072,482.3158 | ||
| BUR-C50 | Buried | 95 | 79 | 0.0806 | −1,035,169.3326 | 2,070,551.0859 | ||
| BUR-C60 | Buried | 95 | 84 | 0.0892 | −1,034,522.1735 | 2,069,276.8507 | ||
| EXP-C10 | Choanozoa only | Exposed | 92 | 97 | 0.2551 | −952,094.7392 | 1,904,305.6212 | |
| EXP-C20 | Exposed | 87 | 97 | 0.176 | −949,973.8488 | 1,900,083.8935 | ||
| EXP-C30 | Exposed | 77 | 91 | 0.0604 | −948,352.7546 | 1,896,861.7664 | ||
| EXP-C40 | Exposed | 77 | 98 | 0.0629 | −947,967.3623 | 1,896,111.0517 | ||
| EXP-C50 | Exposed | 76 | 95 | 0.0711 | −948,154.1412 | 1,896,504.6876 | ||
| EXP-C60 | Exposed | 72 | 96 | 0.0566 | −947,765.6493 | 1,895,747.7905 | ||
| BUR-C10 | Buried | 67 | 96 | 0.2376 | −826,008.6321 | 1,652,133.391 | ||
| BUR-C20 | Buried | 48 a | 42 a | 93 | 0.1838 | −822,299.4844 | 1,644,735.1427 | |
| BUR-C30 | Buried | 74 | 97 | 0.0981 | −821,989.872 | 1,644,135.9726 | ||
| BUR-C40 | Buried | 38 a | 37 a | 96 | 0.1211 | −820,830.2779 | 1,641,836.8464 | |
| BUR-C50 | Buried | 74 | 96 | 0.0950 | −820,251.2702 | 1,640,698.9004 | ||
| BUR-C60 | Buried | 78 | 91 | 0.1125 | −819,722.1126 | 1,639,660.6619 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, A.; Braun, E.L. Phylogenetic Analyses of Sites in Different Protein Structural Environments Result in Distinct Placements of the Metazoan Root. Biology 2020, 9, 64. https://doi.org/10.3390/biology9040064
Pandey A, Braun EL. Phylogenetic Analyses of Sites in Different Protein Structural Environments Result in Distinct Placements of the Metazoan Root. Biology. 2020; 9(4):64. https://doi.org/10.3390/biology9040064
Chicago/Turabian StylePandey, Akanksha, and Edward L. Braun. 2020. "Phylogenetic Analyses of Sites in Different Protein Structural Environments Result in Distinct Placements of the Metazoan Root" Biology 9, no. 4: 64. https://doi.org/10.3390/biology9040064
APA StylePandey, A., & Braun, E. L. (2020). Phylogenetic Analyses of Sites in Different Protein Structural Environments Result in Distinct Placements of the Metazoan Root. Biology, 9(4), 64. https://doi.org/10.3390/biology9040064