Impairments of Synaptic Plasticity Induction Threshold and Network Oscillatory Activity in the Hippocampus Underlie Memory Deficits in a Non-Transgenic Mouse Model of Amyloidosis

 , , ,
, , ,
Abstract
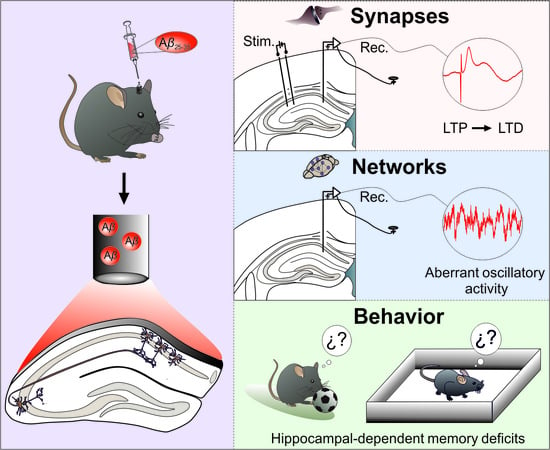
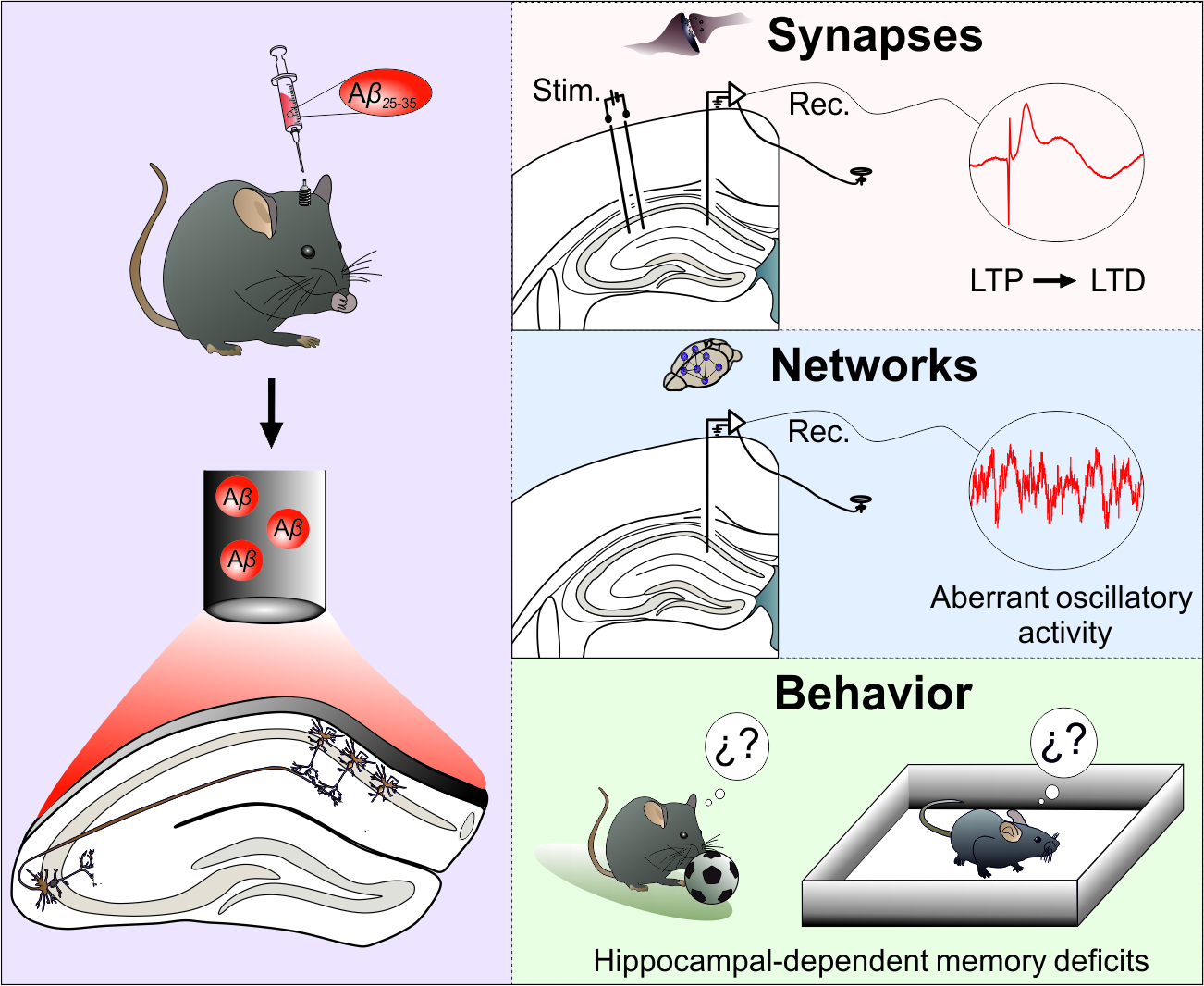{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Surgery for Chronic Recording and Aβ Injection Experiments
2.3. Electrophysiological Recordings in Behaving Animals
2.4. Open Field Habituation Task
2.5. Novel Object Recognition Test
2.6. Rotarod Performance Test
2.7. Locomotion Test
2.8. Drugs
2.9. Histology
2.10. Data Collection and Analysis
2.11. Statistical Analysis
3. Results
3.1. Aβ Transforms LTP of Synaptic Excitation or Inhibition into LTD in the Hippocampal CA3-CA1 Synapse of Behaving Mice
3.2. CA1 Hippocampal Oscillatory Activity Is Synchronized by Aβ in Behaving Mice
3.3. Hippocampal-Dependent Memory Is Disrupted by Aβ
4. Discussion
4.1. HFS-Induced LTP Is Transformed into LTD by Aβ25-35 in Behaving Mice
4.2. Aβ25-35-Induced Network Dysfunction and Memory Impairments
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goedert, M.; Spillantini, M.G. A century of alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed]
- Nava-Mesa, M.O.; Jimenez-Diaz, L.; Yajeya, J.; Navarro-Lopez, J.D. Gabaergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of alzheimer’s disease. Front. Cell. Neurosci. 2014, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.C.; Kuperstein, I.; Wilkinson, H.; Maes, E.; Vanbrabant, M.; Jonckheere, W.; Van, G.P.; Hartmann, D.; D’Hooge, R.; De, S.B.; et al. Lipids revert inert abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008, 27, 224–233. [Google Scholar] [CrossRef]
- Selkoe, D.J. Preventing alzheimer’s disease. Science 2012, 337, 1488–1492. [Google Scholar] [CrossRef]
- Jan, A.; Hartley, D.M.; Lashuel, H.A. Preparation and characterization of toxic abeta aggregates for structural and functional studies in alzheimer’s disease research. Nat. Protoc. 2010, 5, 1186–1209. [Google Scholar] [CrossRef]
- Stephan, A.; Laroche, S.; Davis, S. Generation of aggregated beta-amyloid in the rat hippocampus impairs synaptic transmission and plasticity and causes memory deficits. J. Neurosci. 2001, 21, 5703–5714. [Google Scholar] [CrossRef]
- Gruden, M.A.; Davidova, T.B.; Malisauskas, M.; Sewell, R.D.E.; Voskresenskaya, N.I.; Wilhelm, K.; Elistratova, E.I.; Sherstnev, V.V.; Morozova-Roche, L.A. Differential neuroimmune markers to the onset of Alzheimer’s disease neurodegeneration and dementia: Autoantibodies to Aβ(25–35) oligomers, S100b and neurotransmitters. J. Neuroimmunol. 2007, 186, 181–192. [Google Scholar] [CrossRef]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and biological activities of amyloid beta peptide 25-35. Curr. Protein Pept. Sci. 2010, 11, 54–67. [Google Scholar] [CrossRef]
- Pike, C.J.; Burdick, D.; Walencewicz, A.J.; Glabe, C.G.; Cotman, C.W. Neurodegeneration induced by beta-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 1993, 13, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Nabeshima, T. Animal models of alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacol. Ther. 2000, 88, 93–113. [Google Scholar] [CrossRef]
- Ashenafi, S.; Fuente, A.; Criado, J.M.; Riolobos, A.S.; Heredia, M.; Yajeya, J. Beta-amyloid peptide25-35 depresses excitatory synaptic transmission in the rat basolateral amygdala “in vitro”. Neurobiol. Aging 2005, 26, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Lerma, A.I.; Ordaz, B.; Peña-Ortega, F. Amyloid beta peptides differentially affect hippocampal theta rhythms in vitro. Int. J. Pept. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Leao, R.N.; Colom, L.V.; Borgius, L.; Kiehn, O.; Fisahn, A. Medial septal dysfunction by abeta-induced kcnq channel-block in glutamatergic neurons. Neurobiol. Aging 2012, 33, 2046–2061. [Google Scholar] [CrossRef]
- Nava-Mesa, M.O.; Jimenez-Diaz, L.; Yajeya, J.; Navarro-Lopez, J.D. Amyloid-beta induces synaptic dysfunction through g protein-gated inwardly rectifying potassium channels in the fimbria-ca3 hippocampal synapse. Front. Cell. Neurosci. 2013, 7, 117. [Google Scholar] [CrossRef]
- Peña-Ortega, F.; Ordaz, B.; Balleza-Tapia, H.; Bernal-Pedraza, R.; Márquez-Ramos, A.; Carmona-Aparicio, L.; Giordano, M. Beta-amyloid protein (25-35) disrupts hippocampal network activity: Role of FYN-kinase. Hippocampus 2009, 20, 78–96. [Google Scholar] [CrossRef]
- Freir, D.B.; Holscher, C.; Herron, C.E. Blockade of long-term potentiation by beta-amyloid peptides in the CA1 region of the rat hippocampus in vivo. J. Neurophysiol. 2001, 85, 708–713. [Google Scholar] [CrossRef]
- Chumakov, I.; Nabirotchkin, S.; Cholet, N.; Milet, A.; Boucard, A.; Toulorge, D.; Pereira, Y.; Graudens, E.; Traore, S.; Foucquier, J.; et al. Combining two repurposed drugs as a promising approach for alzheimer’s disease therapy. Sci. Rep. 2015, 5, 7608. [Google Scholar] [CrossRef]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide protects against the amyloid-beta25-35-induced learning and memory impairment in mice, an experimental model of alzheimer disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef]
- Chang, Z.; Luo, Y.; Zhang, Y.; Wei, G. Interactions of abeta25-35 beta-barrel-like oligomers with anionic lipid bilayer and resulting membrane leakage: An all-atom molecular dynamics study. J. Phys. Chem. B 2011, 115, 1165–1174. [Google Scholar] [CrossRef]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Azimova, R.; Kagan, B.L.; Nussinov, R.; Lal, R. Truncated -amyloid peptide channels provide an alternative mechanism for Alzheimer’s Disease and Down syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 6538–6543. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rodriguez, I.; Temprano-Carazo, S.; Najera, A.; Djebari, S.; Yajeya, J.; Gruart, A.; Delgado-Garcia, J.M.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. Activation of g-protein-gated inwardly rectifying potassium (kir3/girk) channels rescues hippocampal functions in a mouse model of early amyloid-beta pathology. Sci. Rep. 2017, 7, 14658. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Rodríguez, I.; Djebari, S.; Temprano-Carazo, S.; Vega-Avelaira, D.; Jiménez-Herrera, R.; Iborra-Lázaro, G.; Yajeya, J.; Jiménez-Díaz, L.; López, J.D.D.N. Hippocampal long-term synaptic depression and memory deficits induced in early amyloidopathy are prevented by enhancing G-protein-gated inwardly rectifying potassium channel activity. J. Neurochem. 2020, 153, 362–376. [Google Scholar] [CrossRef]
- Flores, G.V.; Gruart, A.; Delgado-García, J.M. Involvement of the GABAergic septo-hippocampal pathway in brain stimulation reward. PLoS ONE 2014, 9, e113787. [Google Scholar] [CrossRef]
- Gruart, A.; Muñoz, M.D.; Delgado-García, J.M. Involvement of the CA3–CA1 synapse in the acquisition of associative learning in behaving mice. J. Neurosci. 2006, 26, 1077–1087. [Google Scholar] [CrossRef]
- Sanchez-Rodriguez, I.; Gruart, A.; Delgado-Garcia, J.M.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. Role of girk channels in long-term potentiation of synaptic inhibition in an in vivo mouse model of early amyloid-beta pathology. Int. J. Mol. Sci. 2019, 20, 1168. [Google Scholar] [CrossRef]
- Flores, G.V.; Rubio, S.E.; Jurado-Parras, M.T.; Gómez-Climent, M.Á.; Hampe, C.S.; Manto, M.; Soriano, E.; Pascual, M.; Gruart, A.; Delgado-García, J.M. The GABAergic septohippocampal pathway is directly involved in internal processes related to operant reward learning. Cereb. Cortex 2013, 24, 2093–2107. [Google Scholar] [CrossRef]
- Zussy, C.; Brureau, A.; Keller, E.; Marchal, S.; Blayo, C.; Delair, B.; Ixart, G.; Maurice, T.; Givalois, L. Alzheimer’s disease related markers, cellular toxicity and behavioral deficits induced six weeks after oligomeric amyloid-beta peptide injection in rats. PLoS ONE 2013, 8, e53117. [Google Scholar] [CrossRef]
- Mayordomo-Cava, J.; Yajeya, J.; Navarro-Lopez, J.D.; Jimenez-Diaz, L. Amyloid-beta (25-35) modulates the expression of girk and kcnq channel genes in the hippocampus. PLoS ONE 2015, 10, e0134385. [Google Scholar] [CrossRef]
- Santos-Torres, J.; Fuente, A.; Criado, J.M.; Riolobos, A.; Heredia, M.; Yajeya, J. Glutamatergic synaptic depression by synthetic amyloid β-peptide in the medial septum. J. Neurosci. Res. 2007, 85, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar] [CrossRef]
- Bliss, T.V.; Collingridge, G.L.; Morris, R. Synaptic plasticity in the hippocampus. In The Hippocampus Book; Andersen, P., Morris, R., Amaral, D.G., Bliss, T., O’Keefe, J., Eds.; Oxford University Press: New York, NY, USA, 2007; pp. 343–474. [Google Scholar]
- Leussis, M.; Bolivar, V. Habituation in rodents: A review of behavior, neurobiology, and genetics. Neurosci. Biobehav. Rev. 2006, 30, 1045–1064. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.R.; Cammarota, M.; Gruart, A.; Izquierdo, I.; Delgado-García, J.M. Plastic modifications induced by object recognition memory processing. Proc. Natl. Acad. Sci. USA 2010, 107, 2652–2657. [Google Scholar] [CrossRef] [PubMed]
- Fanselow, M.S.; Dong, H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef]
- Bliss, T.; Collingridge, G.L.; Morris, R.; Reymann, K.G. Long-term potentiation in the hippocampus: Discovery, mechanisms and function. Neuroforum 2018, 24, A103–A120. [Google Scholar] [CrossRef]
- Bischofberger, J.; Engel, D.; Li, L.; Geiger, J.R.; Jonas, P. Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat. Protoc. 2006, 1, 2075–2081. [Google Scholar] [CrossRef]
- Marsden, K.C.; Beattie, J.B.; Friedenthal, J.; Carroll, R.C. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABAA receptors. J. Neurosci. 2007, 27, 14326–14337. [Google Scholar] [CrossRef]
- Pennacchietti, F.; Vascon, S.; Nieus, T.; Rosillo, C.; Das, S.; Tyagarajan, S.K.; Diaspro, A.; Del Bue, A.; Petrini, E.M.; Barberis, A.; et al. Nanoscale molecular reorganization of the inhibitory postsynaptic density is a determinant of GABAergic synaptic potentiation. J. Neurosci. 2017, 37, 1747–1756. [Google Scholar] [CrossRef]
- Dembitskaya, Y.; Wu, Y.W.; Semyanov, A. Tonic GABAA conductance favors spike-timing-dependent over theta-burst-induced long-term potentiation in the hippocampus. J. Neurosci. 2020, 40, 4266–4276. [Google Scholar] [CrossRef]
- Freir, D.B.; Costello, D.A.; Herron, C.E. A beta 25-35-induced depression of long-term potentiation in area ca1 in vivo and in vitro is attenuated by verapamil. J. Neurophysiol. 2003, 89, 3061–3069. [Google Scholar] [CrossRef]
- Moreno-Castilla, P.; Rodriguez-Duran, L.F.; Guzman-Ramos, K.; Barcenas-Femat, A.; Escobar, M.L.; Bermúdez-Rattoni, F. Dopaminergic neurotransmission dysfunction induced by amyloid-β transforms cortical long-term potentiation into long-term depression and produces memory impairment. Neurobiol. Aging 2016, 41, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.N.; Bear, M.F. The BCM theory of synapse modification at 30: Interaction of theory with experiment. Nat. Rev. Neurosci. 2012, 13, 798–810. [Google Scholar] [CrossRef]
- Abraham, W.C. Metaplasticity: Tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 2008, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Park, E.; Zhong, L.R.; Chen, L. Homeostatic synaptic plasticity as a metaplasticity mechanism–a molecular and cellular perspective. Curr. Opin. Neurobiol. 2018, 54, 44–53. [Google Scholar] [CrossRef]
- Tamagnini, F.; Scullion, S.; Brown, J.T.; Randall, A.D. Intrinsic excitability changes induced by acute treatment of hippocampal CA1 pyramidal neurons with exogenous amyloid β peptide. Hippocampus 2015, 25, 786–797. [Google Scholar] [CrossRef]
- Huang, C.S.; Shi, S.H.; Ule, J.; Ruggiu, M.; Barker, L.A.; Darnell, R.B.; Jan, Y.N.; Jan, L.Y. Common molecular pathways mediate long-term potentiation of synaptic excitation and slow synaptic inhibition. Cell 2005, 123, 105–118. [Google Scholar] [CrossRef]
- Malenka, R.C.; Bear, M.F. Ltp and ltd: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Malinow, R.; Malenka, R.C. AMPA Receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mglur5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. Ampar removal underlies abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, C.; Malenka, R.C. NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef]
- Braz, B.Y.; Belforte, J.E.; Murer, M.G.; Galiñanes, G. Properties of the corticostriatal long term depression induced by medial prefrontal cortex high frequency stimulation in vivo. Neuropharmacology 2017, 121, 278–286. [Google Scholar] [CrossRef]
- Abraham, W.C.; Mason-Parker, S.E.; Bear, M.F.; Webb, S.; Tate, W.P. Heterosynaptic metaplasticity in the hippocampus in vivo: A BCM-like modifiable threshold for LTP. Proc. Natl. Acad. Sci. USA 2001, 98, 10924–10929. [Google Scholar] [CrossRef]
- Liu, Y.; Yoo, M.J.; Savonenko, A.; Stirling, W.; Price, D.L.; Borchelt, D.R.; Mamounas, L.; Lyons, W.E.; Blue, M.E.; Lee, M.K. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of alzheimer’s disease. J. Neurosci. 2008, 28, 13805–13814. [Google Scholar] [CrossRef]
- Booze, R.M.; Mactutus, C.F.; Gutman, C.R.; Davis, J.N. Frequency analysis of catecholamine axonal morphology in human brain. J. Neurol. Sci. 1993, 119, 99–109. [Google Scholar] [CrossRef]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fulop, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkanen, A.; et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef]
- Keck, T.; Hübener, M.; Bonhoeffer, T. Interactions between synaptic homeostatic mechanisms: An attempt to reconcile BCM theory, synaptic scaling, and changing excitation/inhibition balance. Curr. Opin. Neurobiol. 2017, 43, 87–93. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What happens with the circuit in alzheimer’s disease in mice and humans? Annu. Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Goutagny, R.; Gu, N.; Cavanagh, C.; Jackson, J.; Chabot, J.G.; Quirion, R.; Krantic, S.; Williams, S. Alterations in hippocampal network oscillations and theta-gamma coupling arise before abeta overproduction in a mouse model of alzheimer’s disease. Eur. J. Neurosci. 2013, 37, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Villette, V.; Dutar, P. GABAergic microcircuits in Alzheimer’s disease models. Curr. Alzheimer Res. 2016, 13, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Musaeus, C.S.; Engedal, K.; Høgh, P.; Jelic, V.; Mørup, M.; Naik, M.; Oeksengaard, A.R.; Snaedal, J.; Wahlund, L.O.; Waldemar, G.; et al. EEG theta power is an early marker of cognitive decline in dementia due to Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Gaubert, S.; Raimondo, F.; Houot, M.; Corsi, M.C.; Naccache, L.; Sitt, J.D.; Hermann, B.; Oudiette, D.; Gagliardi, G.; Habert, M.O.; et al. EEG evidence of compensatory mechanisms in preclinical Alzheimer’s disease. Brain 2019, 142, 2096–2112. [Google Scholar] [CrossRef]
- Zhang, X.; Zhong, W.; Brankačk, J.; Weyer, S.W.; Müller, U.C.; Tort, A.B.; Draguhn, A. Impaired theta-gamma coupling in APP-deficient mice. Sci. Rep. 2016, 6, 21948. [Google Scholar] [CrossRef]
- Chavant, F.; Deguil, J.; Pain, S.; Ingrand, I.; Milin, S.; Fauconneau, B.; Perault-Pochat, M.C.; Lafay-Chebassier, C. Imipramine, in part through tumor necrosis factor alpha inhibition, prevents cognitive decline and beta-amyloid accumulation in a mouse model of alzheimer’s disease. J. Pharmacol. Exp. Ther. 2010, 332, 505–514. [Google Scholar] [CrossRef]
- Hölscher, C.; Gengler, S.; Gault, V.; Harriott, P.; Mallot, H.A. Soluble beta-amyloid [25–35] reversibly impairs hippocampal synaptic plasticity and spatial learning. Eur. J. Pharmacol. 2007, 561, 85–90. [Google Scholar] [CrossRef]
- Meunier, J.; Ieni, J.; Maurice, T. The anti-amnesic and neuroprotective effects of donepezil against amyloid beta25-35 peptide-induced toxicity in mice involve an interaction with the sigma1 receptor. Br. J. Pharmacol. 2006, 149, 998–1012. [Google Scholar] [CrossRef]
- Kemp, A.; Manahan-Vaughan, D. Hippocampal long-term depression: Master or minion in declarative memory processes? Trends Neurosci. 2007, 30, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.; Peineau, S.; Howland, J.G.; Wang, Y.T. Long-term depression in the CNS. Nat. Rev. Neurosci. 2010, 11, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Malleret, G.; Alarcon, J.M.; Martel, G.; Takizawa, S.; Vronskaya, S.; Yin, D.; Chen, I.Z.; Kandel, E.R.; Shumyatsky, G.P. Bidirectional regulation of hippocampal long-term synaptic plasticity and its influence on opposing forms of memory. J. Neurosci. 2010, 30, 3813–3825. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic nr2b-containing nmda receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef] [PubMed]
- Grady, C.L.; Furey, M.L.; Pietrini, P.; Horwitz, B.; Rapoport, S.I. Altered brain functional connectivity and impaired short-term memory in Alzheimer’s disease. Brain 2001, 124, 739–756. [Google Scholar] [CrossRef]
- Ameen-Ali, K.E.; Simpson, J.E.; Wharton, S.B.; Heath, P.R.; Sharp, P.S.; Brezzo, G.; Berwick, J. The time course of recognition memory impairment and glial pathology in the hAPP-J20 mouse model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 68, 609–624. [Google Scholar] [CrossRef]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.I.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef]
- Lisman, J.E.; Jensen, O. The theta-gamma neural code. Neuron 2013, 77, 1002–1016. [Google Scholar] [CrossRef]
- Li, Q.; Navakkode, S.; Rothkegel, M.; Soong, T.W.; Sajikumar, S.; Korte, M. Metaplasticity mechanisms restore plasticity and associativity in an animal model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 5527–5532. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayordomo-Cava, J.; Iborra-Lázaro, G.; Djebari, S.; Temprano-Carazo, S.; Sánchez-Rodríguez, I.; Jeremic, D.; Gruart, A.; Delgado-García, J.M.; Jiménez-Díaz, L.; Navarro-López, J.D. Impairments of Synaptic Plasticity Induction Threshold and Network Oscillatory Activity in the Hippocampus Underlie Memory Deficits in a Non-Transgenic Mouse Model of Amyloidosis. Biology 2020, 9, 175. https://doi.org/10.3390/biology9070175
Mayordomo-Cava J, Iborra-Lázaro G, Djebari S, Temprano-Carazo S, Sánchez-Rodríguez I, Jeremic D, Gruart A, Delgado-García JM, Jiménez-Díaz L, Navarro-López JD. Impairments of Synaptic Plasticity Induction Threshold and Network Oscillatory Activity in the Hippocampus Underlie Memory Deficits in a Non-Transgenic Mouse Model of Amyloidosis. Biology. 2020; 9(7):175. https://doi.org/10.3390/biology9070175
Chicago/Turabian StyleMayordomo-Cava, Jennifer, Guillermo Iborra-Lázaro, Souhail Djebari, Sara Temprano-Carazo, Irene Sánchez-Rodríguez, Danko Jeremic, Agnès Gruart, José María Delgado-García, Lydia Jiménez-Díaz, and Juan D. Navarro-López. 2020. "Impairments of Synaptic Plasticity Induction Threshold and Network Oscillatory Activity in the Hippocampus Underlie Memory Deficits in a Non-Transgenic Mouse Model of Amyloidosis" Biology 9, no. 7: 175. https://doi.org/10.3390/biology9070175
APA StyleMayordomo-Cava, J., Iborra-Lázaro, G., Djebari, S., Temprano-Carazo, S., Sánchez-Rodríguez, I., Jeremic, D., Gruart, A., Delgado-García, J. M., Jiménez-Díaz, L., & Navarro-López, J. D. (2020). Impairments of Synaptic Plasticity Induction Threshold and Network Oscillatory Activity in the Hippocampus Underlie Memory Deficits in a Non-Transgenic Mouse Model of Amyloidosis. Biology, 9(7), 175. https://doi.org/10.3390/biology9070175