Transcription Factors in Cartilage Homeostasis and Osteoarthritis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
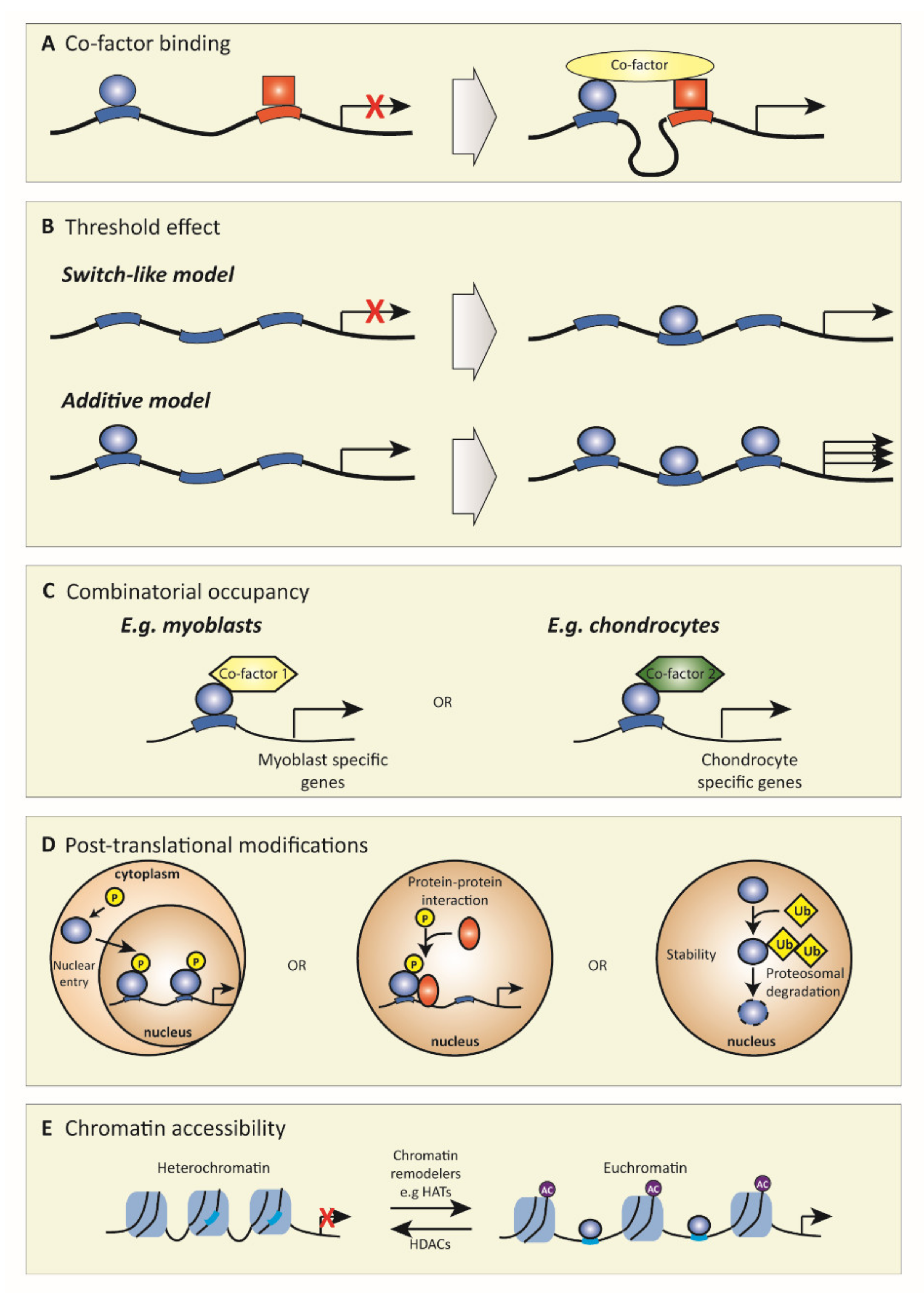
2. Transcription Factors
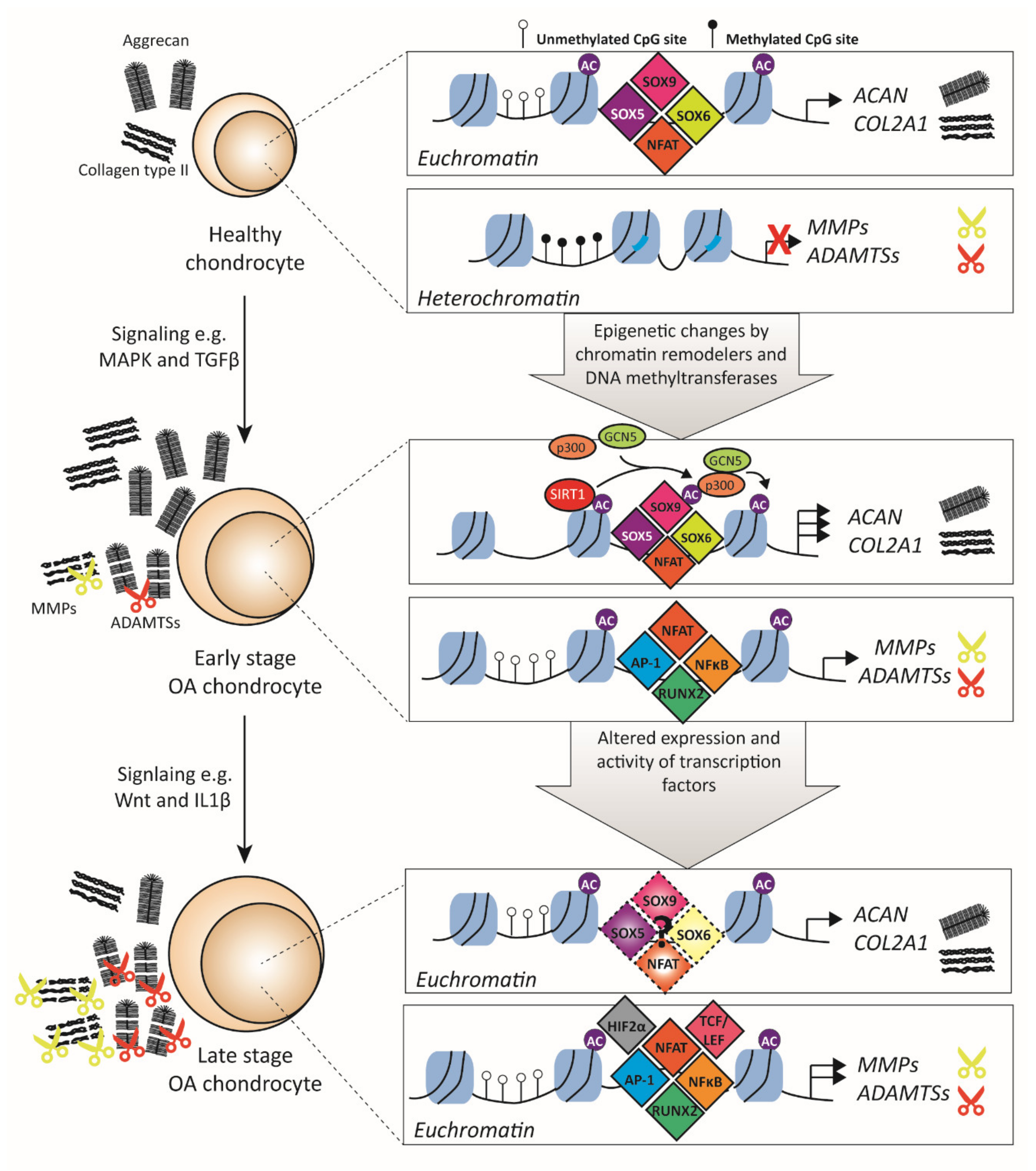
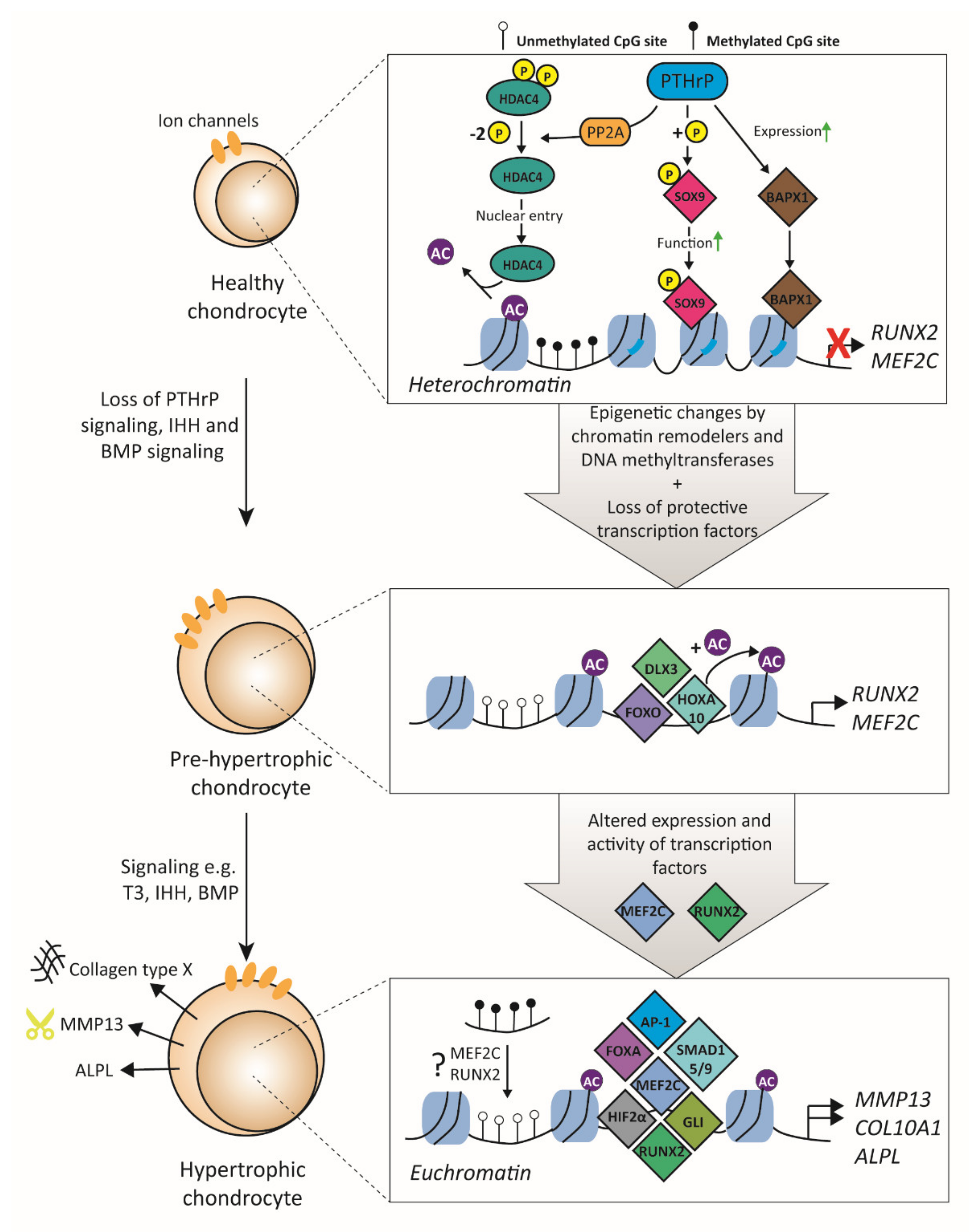
3. Transcription Factors in OA
3.1. Transcription Factors Related to ECM Metabolism
3.2. Transcription Factors Related to Hypertrophy
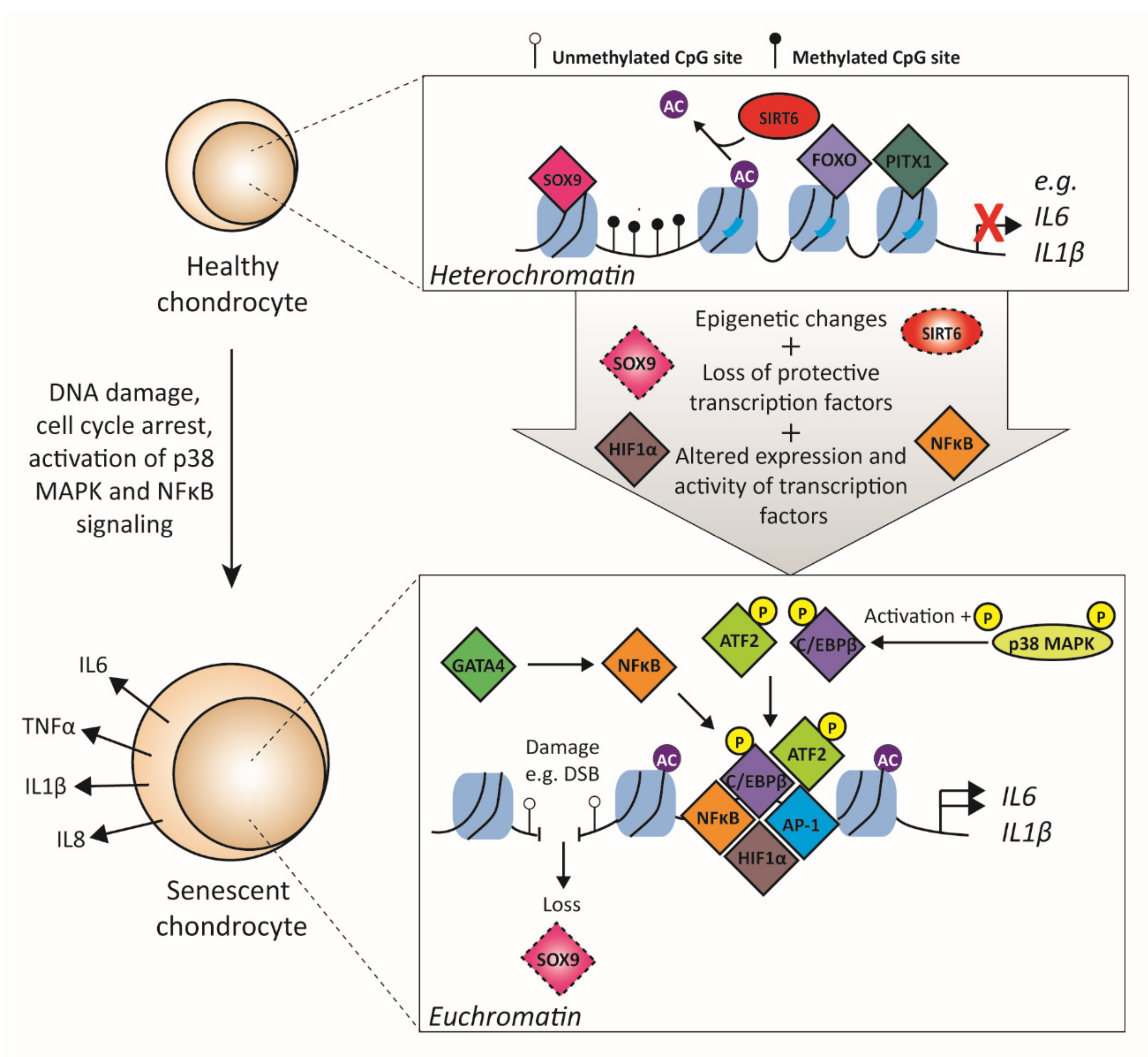
3.3. Transcription Factors Related to Cellular Senescence
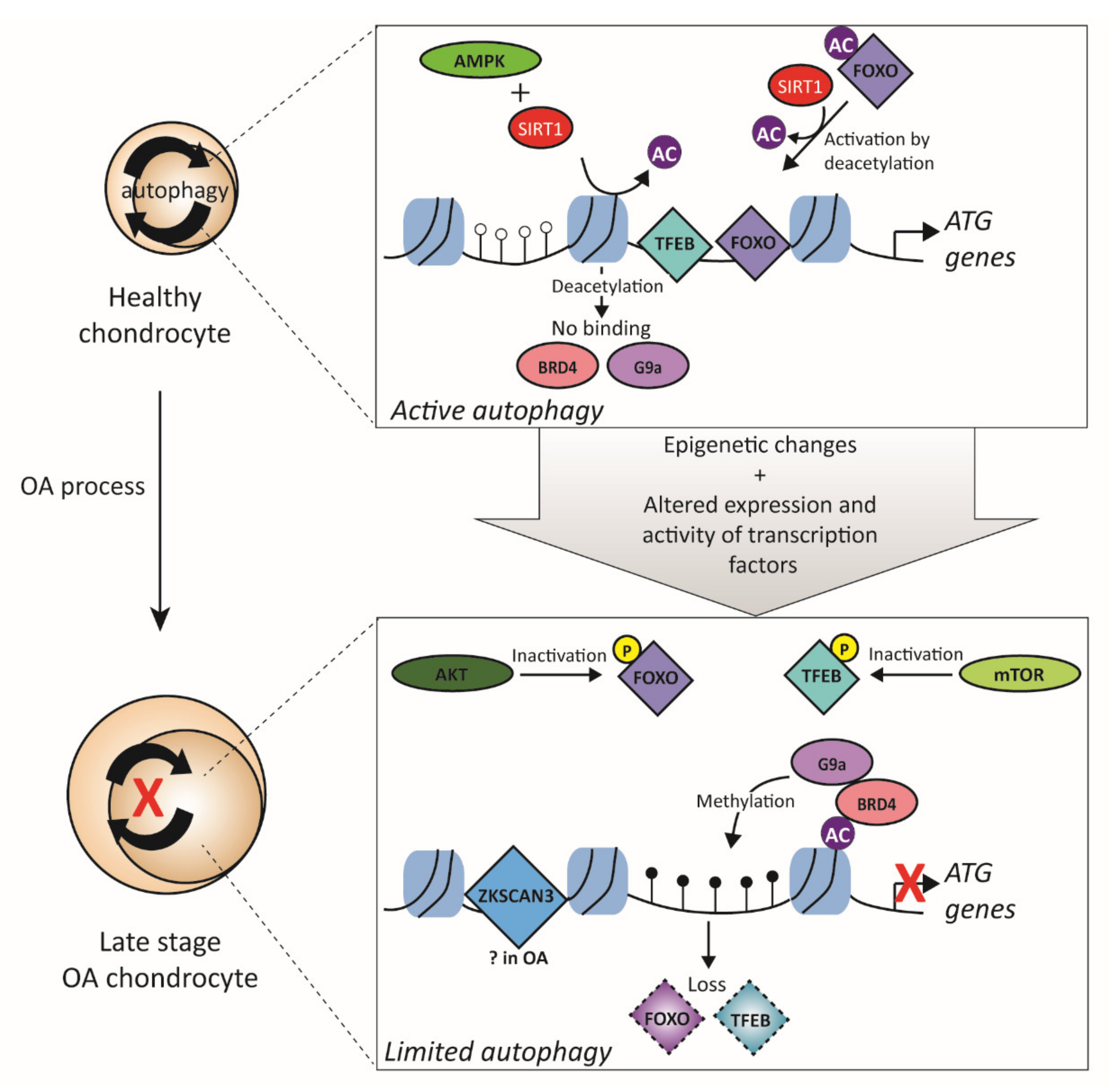
3.4. Transcription Factors Related to Autophagy
4. Transcription Factors as Therapeutic Targets
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACAN | aggrecan |
| ADAMTS | a disintegrin and metalloproteinase with thrombospondin motif |
| ALPL | alkaline phosphatase |
| AP-1 | activator protein 1 |
| ATF2 | activating transcription factor 2 |
| ATG | autophagy-related |
| BAPX-1 | homeobox protein Nkx-3.2 |
| C/EBP | CCAAT-enhancer-binding protein |
| COL10A1 | collagen type X |
| COL2A1 | collagen type II |
| CREB | cAMP response element binding protein |
| DMM | destabilization of the medial meniscus |
| ECM | extracellular matrix |
| ENaC | epithelial sodium channel |
| ETS | erythroblast transformation specific |
| FOXA | forkhead box transcription factor class A |
| FOXO | forkhead box transcription factor class O |
| GATA4 | GATA-binding factor 4 |
| GLI | glioma-associated oncogene |
| HAT | histone acetyltransferase |
| HDAC | histone deacetylase |
| HIF | hypoxia-inducible factor |
| IHH | Indian hedgehog |
| IKK | IκB kinase |
| IL | interleukin |
| KCa | Ca2+ activated potassium channels |
| LC3 | light chain 3 |
| MAPK | mitogen-activated protein kinase |
| MEF2C | myocyte-specific enhancer factor 2C |
| MMP | matrix matalloproteinase |
| MR | master regulator |
| mTOR | mechanistic target of rapamycin kinase |
| MYOD | myoblast determination protein |
| NFAT | nuclear factor of activated T-cell |
| NFκB | nuclear factor kappa B |
| OA | osteoarthritis |
| PAX | paired box |
| PI3K | phosphatidylinositol 3-kinase |
| PITX1 | paired-like homeodomain 1 |
| PKA | cAMP-dependent protein kinase A |
| PTHrP | parathyroid hormone-like protein |
| RUNX2 | runt-related transcription factor 2 |
| SASP | Senescence-associated secretory phenotype |
| siRNA | small interfering RNA |
| SIRT1 | sirtuin 1 |
| SMAD3 | SMAD family member 3 |
| SOX9 | SRY-BOX transcription factor 9 |
| SP1 | specificity protein 1 |
| STAT3 | signal transducer and activator of transcription 3 |
| T3 | active thyroid hormone |
| TASK-2 | Acid-sensing potassium channel |
| tBHP | tert-butyl-hydroperoxide |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TF | transcription factor |
| TFD ODN | transcription factor decoy oligodeoxynucleotides |
| TFEB | transcription factor EB |
| TGFβ | transforming growth factor β |
| TMEM16 | Ca2+ activated chloride channel |
| TNFα | tumor necrosis factor alpha |
| TSS | transcription start site |
| ULK1/2 | unc-51-like autophagy activating kinase 1 and 2 |
| ZKSCAN3 | zinc-finger protein with KRAB and SCAN domain 3 |
References
- Hunter, D.J.; Schofield, D.; Callander, E. The individual and socioeconomic impact of osteoarthritis. Nat. Rev. Rheumatol. 2014, 10, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Oo, W.M.; Yu, S.P.; Daniel, M.S.; Hunter, D.J. Disease-modifying drugs in osteoarthritis: Current understanding and future therapeutics. Expert Opin. Emerg. Drugs 2018, 23, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Carballo, C.B.; Nakagawa, Y.; Sekiya, I.; Rodeo, S.A. Basic Science of Articular Cartilage. Clin. Sports Med. 2017, 36, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.M.; Jackson, D.W. Articular Cartilage: Injury Pathways and Treatment Options. Sports Med. Arthrosc. Rev. 2018, 26, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sports Health 2009, 1, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Cucchiarini, M.; de Girolamo, L.; Filardo, G.; Oliveira, J.M.; Orth, P.; Pape, D.; Reboul, P. Basic science of osteoarthritis. J. Exp. Orthop. 2016, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef] [Green Version]
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Pattabiraman, D.R.; McGirr, C.; Shakhbazov, K.; Barbier, V.; Krishnan, K.; Mukhopadhyay, P.; Hawthorne, P.; Trezise, A.; Ding, J.; Grimmond, S.M.; et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 2014, 123, 2682–2690. [Google Scholar] [CrossRef] [Green Version]
- Boyadjiev, S.A.; Jabs, E.W. Online Mendelian Inheritance in Man (OMIM) as a knowledgebase for human developmental disorders. Clin. Genet. 2000, 57, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Fan, C.; Topol, S.E.; Topol, E.J.; Wang, Q. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science 2003, 302, 1578–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akirav, E.M.; Ruddle, N.H.; Herold, K.C. The role of AIRE in human autoimmune disease. Nat. Rev. Endocrinol. 2011, 7, 25–33. [Google Scholar] [CrossRef]
- Garrett-Sinha, L.A.; Kearly, A.; Satterthwaite, A.B. The Role of the Transcription Factor Ets1 in Lupus and Other Autoimmune Diseases. Crit. Rev. Immunol. 2016, 36, 485–510. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 175, 598–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latchman, D.S. Transcription factors: An overview. Int. J. Exp. Pathol. 1993, 74, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Dynlacht, B.D. Regulation of transcription by proteins that control the cell cycle. Nature 1997, 389, 149–152. [Google Scholar] [CrossRef]
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Fulton, D.L.; Sundararajan, S.; Badis, G.; Hughes, T.R.; Wasserman, W.W.; Roach, J.C.; Sladek, R. TFCat: The curated catalog of mouse and human transcription factors. Genome Biol. 2009, 10, R29. [Google Scholar] [CrossRef] [Green Version]
- Babu, M.M.; Luscombe, N.M.; Aravind, L.; Gerstein, M.; Teichmann, S.A. Structure and evolution of transcriptional regulatory networks. Curr. Opin. Struct. Biol. 2004, 14, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Roeder, R.G. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 1996, 21, 327–335. [Google Scholar] [CrossRef]
- Chan, S.S.; Kyba, M. What is a Master Regulator? J. Stem. Cell Res. Ther. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Iwafuchi-Doi, M.; Zaret, K.S. Cell fate control by pioneer transcription factors. Development 2016, 143, 1833–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassar, A.B.; Paterson, B.M.; Weintraub, H. Transfection of a DNA locus that mediates the conversion of 10T1/2 fibroblasts to myoblasts. Cell 1986, 47, 649–656. [Google Scholar] [CrossRef]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genom. Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Shendure, J. Mechanisms of Interplay between Transcription Factors and the 3D Genome. Mol. Cell 2019, 76, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inukai, S.; Kock, K.H.; Bulyk, M.L. Transcription factor-DNA binding: Beyond binding site motifs. Curr. Opin. Genet. Dev. 2017, 43, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Mulero, M.C.; Shahabi, S.; Ko, M.S.; Schiffer, J.M.; Huang, D.B.; Wang, V.Y.; Amaro, R.E.; Huxford, T.; Ghosh, G. Protein Cofactors Are Essential for High-Affinity DNA Binding by the Nuclear Factor kappaB RelA Subunit. Biochemistry 2018, 57, 2943–2957. [Google Scholar] [CrossRef]
- Fiering, S.; Northrop, J.P.; Nolan, G.P.; Mattila, P.S.; Crabtree, G.R.; Herzenberg, L.A. Single cell assay of a transcription factor reveals a threshold in transcription activated by signals emanating from the T-cell antigen receptor. Genes Dev. 1990, 4, 1823–1834. [Google Scholar] [CrossRef] [Green Version]
- Giorgetti, L.; Siggers, T.; Tiana, G.; Caprara, G.; Notarbartolo, S.; Corona, T.; Pasparakis, M.; Milani, P.; Bulyk, M.L.; Natoli, G. Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol. Cell 2010, 37, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Loven, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; DeKoter, R.P.; Young, R.A. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 2011, 147, 565–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.F.; Williams, S.A.; Mu, Y.; Nakano, H.; Duerr, J.M.; Buckbinder, L.; Greene, W.C. NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol. Cell. Biol. 2005, 25, 7966–7975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, W.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; de Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Dvir-Ginzberg, M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res. 2017, 58, 2–14. [Google Scholar] [CrossRef]
- Ashraf, S.; Cha, B.H.; Kim, J.S.; Ahn, J.; Han, I.; Park, H.; Lee, S.H. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthr. Cartil. 2016, 24, 196–205. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Chaboissier, M.C.; Martin, J.F.; Schedl, A.; de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002, 16, 2813–2828. [Google Scholar] [CrossRef] [Green Version]
- Csukasi, F.; Duran, I.; Zhang, W.; Martin, J.H.; Barad, M.; Bamshad, M.; Weis, M.A.; Eyre, D.; Krakow, D.; Cohn, D.H. Dominant-negative SOX9 mutations in campomelic dysplasia. Hum. Mutat. 2019, 40, 2344–2352. [Google Scholar] [CrossRef]
- Tew, S.R.; Clegg, P.D.; Brew, C.J.; Redmond, C.M.; Hardingham, T.E. SOX9 transduction of a human chondrocytic cell line identifies novel genes regulated in primary human chondrocytes and in osteoarthritis. Arthritis Res. Ther. 2007, 9, R107. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Huang, X.; Karperien, M.; Post, J.N. Correlation between Gene Expression and Osteoarthritis Progression in Human. Int. J. Mol. Sci. 2016, 17, 1126. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chang, J.C.; Hon, C.C.; Fukui, N.; Tanaka, N.; Zhang, Z.; Lee, M.T.M.; Minoda, A. Chromatin accessibility landscape of articular knee cartilage reveals aberrant enhancer regulation in osteoarthritis. Sci. Rep. 2018, 8, 15499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisch, K.M.; Gamini, R.; Alvarez-Garcia, O.; Akagi, R.; Saito, M.; Muramatsu, Y.; Sasho, T.; Koziol, J.A.; Su, A.I.; Lotz, M.K. Identification of transcription factors responsible for dysregulated networks in human osteoarthritis cartilage by global gene expression analysis. Osteoarthr. Cartil. 2018, 26, 1531–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Sun, Y.; Li, X. Identification of key gene modules and transcription factors for human osteoarthritis by weighted gene co-expression network analysis. Exp. Ther. Med. 2019, 18, 2479–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thielen, N.G.M.; van der Kraan, P.M.; van Caam, A.P.M. TGFbeta/BMP Signaling Pathway in Cartilage Homeostasis. Cells 2019, 8, 969. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Davis, F.J.; Sharrocks, A.D.; Im, H.J. Emerging roles of SUMO modification in arthritis. Gene 2010, 466, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasadam, I.; Mao, X.; Wang, Y.; Shi, W.; Crawford, R.; Xiao, Y. Inhibition of p38 pathway leads to OA-like changes in a rat animal model. Rheumatology (Oxford) 2012, 51, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.Y.; Hu, K.Z.; Yin, Z.S. Inhibition of the p38-MAPK signaling pathway suppresses the apoptosis and expression of proinflammatory cytokines in human osteoarthritis chondrocytes. Cytokine 2017, 90, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Soder, S.; Oehler, S.; Fundel, K.; Aigner, T. Activation of interleukin-1 signaling cascades in normal and osteoarthritic articular cartilage. Am. J. Pathol. 2007, 171, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Martel-Pelletier, J.; Mineau, F.; Jovanovic, D.; Di Battista, J.A.; Pelletier, J.P. Mitogen-activated protein kinase and nuclear factor kappaB together regulate interleukin-17-induced nitric oxide production in human osteoarthritic chondrocytes: Possible role of transactivating factor mitogen-activated protein kinase-activated proten kinase (MAPKAPK). Arthritis Rheum. 1999, 42, 2399–2409. [Google Scholar] [CrossRef]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [Green Version]
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyas, J.R.; Adams, M.E.; Huang, D.; Sandell, L.J. Discoordinate gene expression of aggrecan and type II collagen in experimental osteoarthritis. Arthritis Rheum. 1995, 38, 420–425. [Google Scholar] [CrossRef]
- Sandy, J.D.; Adams, M.E.; Billingham, M.E.; Plaas, A.; Muir, H. In vivo and in vitro stimulation of chondrocyte biosynthetic activity in early experimental osteoarthritis. Arthritis Rheum. 1984, 27, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Roach, H.I.; Yamada, N.; Cheung, K.S.; Tilley, S.; Clarke, N.M.; Oreffo, R.O.; Kokubun, S.; Bronner, F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005, 52, 3110–3124. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression--belts, braces, and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef] [Green Version]
- Bui, C.; Barter, M.J.; Scott, J.L.; Xu, Y.; Galler, M.; Reynard, L.N.; Rowan, A.D.; Young, D.A. cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J. 2012, 26, 3000–3011. [Google Scholar] [CrossRef]
- Cheung, K.S.; Hashimoto, K.; Yamada, N.; Roach, H.I. Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatol. Int. 2009, 29, 525–534. [Google Scholar] [CrossRef]
- Richardson, B. Impact of aging on DNA methylation. Ageing Res. Rev. 2003, 2, 245–261. [Google Scholar] [CrossRef]
- Hashimoto, K.; Oreffo, R.O.; Gibson, M.B.; Goldring, M.B.; Roach, H.I. DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis Rheum. 2009, 60, 3303–3313. [Google Scholar] [CrossRef] [Green Version]
- Roach, H.I.; Aigner, T. DNA methylation in osteoarthritic chondrocytes: A new molecular target. Osteoarthr. Cartil. 2007, 15, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.Y.; Lin, H.J.; Chen, H.S.; Cheng, S.Y.; Hsu, H.C.; Tang, C.H. Thrombin promotes matrix metalloproteinase-13 expression through the PKCdelta c-Src/EGFR/PI3K/Akt/AP-1 signaling pathway in human chondrocytes. Mediat. Inflamm. 2013, 2013, 326041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell. Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Yun, K.; Im, S.H. Transcriptional regulation of MMP13 by Lef1 in chondrocytes. Biochem. Biophys. Res. Commun. 2007, 364, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Hirata, M.; Saito, T.; Itoh, S.; Chung, U.I.; Kawaguchi, H. Transcriptional induction of ADAMTS5 protein by nuclear factor-kappaB (NF-kappaB) family member RelA/p65 in chondrocytes during osteoarthritis development. J. Biol. Chem. 2013, 288, 28620–28629. [Google Scholar] [CrossRef] [Green Version]
- Takahata, Y.; Nakamura, E.; Hata, K.; Wakabayashi, M.; Murakami, T.; Wakamori, K.; Yoshikawa, H.; Matsuda, A.; Fukui, N.; Nishimura, R. Sox4 is involved in osteoarthritic cartilage deterioration through induction of ADAMTS4 and ADAMTS5. FASEB J. 2019, 33, 619–630. [Google Scholar] [CrossRef]
- Aikawa, Y.; Morimoto, K.; Yamamoto, T.; Chaki, H.; Hashiramoto, A.; Narita, H.; Hirono, S.; Shiozawa, S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol. 2008, 26, 817–823. [Google Scholar] [CrossRef]
- Motomura, H.; Seki, S.; Shiozawa, S.; Aikawa, Y.; Nogami, M.; Kimura, T. A selective c-Fos/AP-1 inhibitor prevents cartilage destruction and subsequent osteophyte formation. Biochem. Biophys. Res. Commun. 2018, 497, 756–761. [Google Scholar] [CrossRef]
- Haag, J.; Gebhard, P.M.; Aigner, T. SOX gene expression in human osteoarthritic cartilage. Pathobiology 2008, 75, 195–199. [Google Scholar] [CrossRef]
- Choi, M.C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-kappaB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.X.; Lin, L.; Wang, H.J.; Wei, X.L.; Fu, X.; Zhang, J.Y.; Yu, C.L. Suppression of early experimental osteoarthritis by in vivo delivery of the adenoviral vector-mediated NF-kappaBp65-specific siRNA. Osteoarthr. Cartil. 2008, 16, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Murahashi, Y.; Yano, F.; Kobayashi, H.; Makii, Y.; Iba, K.; Yamashita, T.; Tanaka, S.; Saito, T. Intra-articular administration of IkappaBalpha kinase inhibitor suppresses mouse knee osteoarthritis via downregulation of the NF-kappaB/HIF-2alpha axis. Sci. Rep. 2018, 8, 16475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Zhong, L.; van Blitterswijk, C.A.; Post, J.N.; Karperien, M. T cell factor 4 is a pro-catabolic and apoptotic factor in human articular chondrocytes by potentiating nuclear factor kappaB signaling. J. Biol. Chem. 2013, 288, 17552–17558. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, V.; Hu, H.; Barroga, C.; Bossard, C.; Kc, S.; Dellamary, L.; Stewart, J.; Chiu, K.; Ibanez, M.; Pedraza, M.; et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil. 2018, 26, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Yazici, Y.; McAlindon, T.E.; Fleischmann, R.; Gibofsky, A.; Lane, N.E.; Kivitz, A.J.; Skrepnik, N.; Armas, E.; Swearingen, C.J.; DiFrancesco, A.; et al. A novel Wnt pathway inhibitor, SM04690, for the treatment of moderate to severe osteoarthritis of the knee: Results of a 24-week, randomized, controlled, phase 1 study. Osteoarthr. Cartil. 2017, 25, 1598–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martel-Pelletier, J.; Boileau, C.; Pelletier, J.P.; Roughley, P.J. Cartilage in normal and osteoarthritis conditions. Best Pract. Res. Clin. Rheumatol. 2008, 22, 351–384. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Codina, M.; Fisher, S. Multiple enhancers associated with ACAN suggest highly redundant transcriptional regulation in cartilage. Matrix Biol. 2012, 31, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ji, Q.; Wang, X.; Kang, L.; Fu, Y.; Yin, Y.; Li, Z.; Liu, Y.; Xu, X.; Wang, Y. SOX9 is a regulator of ADAMTSs-induced cartilage degeneration at the early stage of human osteoarthritis. Osteoarthr. Cartil. 2015, 23, 2259–2268. [Google Scholar] [CrossRef] [Green Version]
- Dunn, S.L.; Soul, J.; Anand, S.; Schwartz, J.M.; Boot-Handford, R.P.; Hardingham, T.E. Gene expression changes in damaged osteoarthritic cartilage identify a signature of non-chondrogenic and mechanical responses. Osteoarthr. Cartil. 2016, 24, 1431–1440. [Google Scholar] [CrossRef] [Green Version]
- Zwickl, H.; Niculescu-Morzsa, E.; Halbwirth, F.; Bauer, C.; Jeyakumar, V.; Reutterer, A.; Berger, M.; Nehrer, S. Correlation Analysis of SOX9, -5, and -6 as well as COL2A1 and Aggrecan Gene Expression of Collagen I Implant-Derived and Osteoarthritic Chondrocytes. Cartilage 2016, 7, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Zhou, X.; Lefebvre, V.; de Crombrugghe, B. Phosphorylation of SOX9 by cyclic AMP-dependent protein kinase A enhances SOX9’s ability to transactivate a Col2a1 chondrocyte-specific enhancer. Mol. Cell. Biol. 2000, 20, 4149–4158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvir-Ginzberg, M.; Gagarina, V.; Lee, E.J.; Hall, D.J. Regulation of cartilage-specific gene expression in human chondrocytes by SirT1 and nicotinamide phosphoribosyltransferase. J. Biol. Chem. 2008, 283, 36300–36310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Yang, H.; Hu, B.; Zhang, M. Sirt1 regulates apoptosis and extracellular matrix degradation in resveratrol-treated osteoarthritis chondrocytes via the Wnt/beta-catenin signaling pathways. Exp. Ther. Med. 2017, 14, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Sasaki, H.; Takayama, K.; Ishida, K.; Matsumoto, T.; Kubo, S.; Matsuzaki, T.; Nishida, K.; Kurosaka, M.; Kuroda, R. The overexpression of SIRT1 inhibited osteoarthritic gene expression changes induced by interleukin-1beta in human chondrocytes. J. Orthop. Res. 2013, 31, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Takayama, K.; Kurosaka, M.; Kuroda, R. Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J. Orthop. Res. 2011, 29, 511–515. [Google Scholar] [CrossRef]
- Hattori, T.; Kishino, T.; Stephen, S.; Eberspaecher, H.; Maki, S.; Takigawa, M.; de Crombrugghe, B.; Yasuda, H. E6-AP/UBE3A protein acts as a ubiquitin ligase toward SOX9 protein. J. Biol. Chem. 2013, 288, 35138–35148. [Google Scholar] [CrossRef] [Green Version]
- Furumatsu, T.; Tsuda, M.; Taniguchi, N.; Tajima, Y.; Asahara, H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 2005, 280, 8343–8350. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Lu, Q.; Budden, T.; Wang, J. NFAT1 protects articular cartilage against osteoarthritic degradation by directly regulating transcription of specific anabolic and catabolic genes. Bone Joint Res. 2019, 8, 90–100. [Google Scholar] [CrossRef]
- Ghayor, C.; Chadjichristos, C.; Herrouin, J.F.; Ala-Kokko, L.; Suske, G.; Pujol, J.P.; Galera, P. Sp3 represses the Sp1-mediated transactivation of the human COL2A1 gene in primary and de-differentiated chondrocytes. J. Biol. Chem. 2001, 276, 36881–36895. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gardner, B.M.; Lu, Q.; Rodova, M.; Woodbury, B.G.; Yost, J.G.; Roby, K.F.; Pinson, D.M.; Tawfik, O.; Anderson, H.C. Transcription factor Nfat1 deficiency causes osteoarthritis through dysfunction of adult articular chondrocytes. J. Pathol. 2009, 219, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Ghayor, C.; Herrouin, J.F.; Chadjichristos, C.; Ala-Kokko, L.; Takigawa, M.; Pujol, J.P.; Galera, P. Regulation of human COL2A1 gene expression in chondrocytes. Identification of C-Krox-responsive elements and modulation by phenotype alteration. J. Biol. Chem. 2000, 275, 27421–27438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadjichristos, C.; Ghayor, C.; Kypriotou, M.; Martin, G.; Renard, E.; Ala-Kokko, L.; Suske, G.; de Crombrugghe, B.; Pujol, J.P.; Galera, P. Sp1 and Sp3 transcription factors mediate interleukin-1 beta down-regulation of human type II collagen gene expression in articular chondrocytes. J. Biol. Chem. 2003, 278, 39762–39772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnson, K.W.; Parker, W.L.; ten Dijke, P.; Thorikay, M.; Philip, A. ALK1 opposes ALK5/Smad3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Miner. Res. 2008, 23, 896–906. [Google Scholar] [CrossRef] [PubMed]
- de Kroon, L.M.; Narcisi, R.; van den Akker, G.G.; Vitters, E.L.; Blaney Davidson, E.N.; van Osch, G.J.; van der Kraan, P.M. SMAD3 and SMAD4 have a more dominant role than SMAD2 in TGFbeta-induced chondrogenic differentiation of bone marrow-derived mesenchymal stem cells. Sci. Rep. 2017, 7, 43164. [Google Scholar] [CrossRef] [Green Version]
- Blaney Davidson, E.N.; Remst, D.F.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retting, K.N.; Song, B.; Yoon, B.S.; Lyons, K.M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.M.; Beier, F. Chondrocyte hypertrophy in skeletal development, growth, and disease. Birth Defects Res. C Embryo Today 2014, 102, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Ripmeester, E.G.J.; Timur, U.T.; Caron, M.M.J.; Welting, T.J.M. Recent Insights into the Contribution of the Changing Hypertrophic Chondrocyte Phenotype in the Development and Progression of Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Hall, A.C. The Role of Chondrocyte Morphology and Volume in Controlling Phenotype-Implications for Osteoarthritis, Cartilage Repair, and Cartilage Engineering. Curr. Rheumatol. Rep. 2019, 21, 38. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.; Feetham, C.H.; Barrett-Jolley, R. Cell volume regulation in chondrocytes. Cell. Physiol. Biochem. 2011, 28, 1111–1122. [Google Scholar] [CrossRef]
- Mobasheri, A.; Lewis, R.; Ferreira-Mendes, A.; Rufino, A.; Dart, C.; Barrett-Jolley, R. Potassium channels in articular chondrocytes. Channels (Austin) 2012, 6, 416–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soul, J.; Dunn, S.L.; Anand, S.; Serracino-Inglott, F.; Schwartz, J.M.; Boot-Handford, R.P.; Hardingham, T.E. Stratification of knee osteoarthritis: Two major patient subgroups identified by genome-wide expression analysis of articular cartilage. Ann. Rheum. Dis. 2018, 77, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, C.; Dehne, T.; Lindahl, A.; Brittberg, M.; Pruss, A.; Sittinger, M.; Ringe, J. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthr. Cartil. 2010, 18, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakibaei, M.; John, T.; De Souza, P.; Rahmanzadeh, R.; Merker, H.J. Signal transduction by beta1 integrin receptors in human chondrocytes in vitro: Collaboration with the insulin-like growth factor-I receptor. Biochem. J. 1999, 342 Pt 3, 615–623. [Google Scholar] [CrossRef]
- Sitara, D.; Aliprantis, A.O. Transcriptional regulation of bone and joint remodeling by NFAT. Immunol. Rev. 2010, 233, 286–300. [Google Scholar] [CrossRef]
- Dolmetsch, R.E.; Lewis, R.S.; Goodnow, C.C.; Healy, J.I. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 1997, 386, 855–858. [Google Scholar] [CrossRef]
- Nishimori, S.; Lai, F.; Shiraishi, M.; Kobayashi, T.; Kozhemyakina, E.; Yao, T.P.; Lassar, A.B.; Kronenberg, H.M. PTHrP targets HDAC4 and HDAC5 to repress chondrocyte hypertrophy. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Takeda, S.; Bonnamy, J.P.; Owen, M.J.; Ducy, P.; Karsenty, G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001, 15, 467–481. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.A.; Kim, Y.; Czubryt, M.P.; Phan, D.; McAnally, J.; Qi, X.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell 2007, 12, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Cao, K.; Wei, L.; Zhang, Z.; Guo, L.; Zhang, C.; Li, Y.; Sun, C.; Sun, X.; Wang, S.; Li, P.; et al. Decreased histone deacetylase 4 is associated with human osteoarthritis cartilage degeneration by releasing histone deacetylase 4 inhibition of runt-related transcription factor-2 and increasing osteoarthritis-related genes: A novel mechanism of human osteoarthritis cartilage degeneration. Arthritis Res. Ther. 2014, 16, 491. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.Q.; Tare, R.; Lee, S.H.; Mandeville, M.; Weiner, B.; Montecino, M.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. HOXA10 controls osteoblastogenesis by directly activating bone regulatory and phenotypic genes. Mol. Cell. Biol. 2007, 27, 3337–3352. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.Q.; Tare, R.S.; Lee, S.H.; Mandeville, M.; Morasso, M.I.; Javed, A.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. BMP2 commitment to the osteogenic lineage involves activation of Runx2 by DLX3 and a homeodomain transcriptional network. J. Biol. Chem. 2006, 281, 40515–40526. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.M.; Lee, E.H. Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng. Part B Rev. 2013, 19, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushton, M.D.; Reynard, L.N.; Barter, M.J.; Refaie, R.; Rankin, K.S.; Young, D.A.; Loughlin, J. Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol. 2014, 66, 2450–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, S.J.; Aubourg, G.; Sorial, A.K.; Almarza, D.; Tselepi, M.; Deehan, D.J.; Reynard, L.N.; Loughlin, J. Identification of a novel, methylation-dependent, RUNX2 regulatory region associated with osteoarthritis risk. Hum. Mol. Genet. 2018, 27, 3464–3474. [Google Scholar] [CrossRef]
- Bi, W.; Huang, W.; Whitworth, D.J.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; de Crombrugghe, B. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl. Acad. Sci. USA 2001, 98, 6698–6703. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Chung, U.I.; Kronenberg, H.M.; de Crombrugghe, B. The chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-related peptide in the growth plate of endochondral bones. Proc. Natl. Acad. Sci. USA 2001, 98, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.M.; Emans, P.J.; Surtel, D.A.; van der Kraan, P.M.; van Rhijn, L.W.; Welting, T.J. BAPX-1/NKX-3.2 acts as a chondrocyte hypertrophy molecular switch in osteoarthritis. Arthritis Rheumatol. 2015, 67, 2944–2956. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Andoh, M.; Ueno-Kudoh, H.; Sato, T.; Miyaki, S.; Asahara, H. Sox9 directly promotes Bapx1 gene expression to repress Runx2 in chondrocytes. Exp. Cell Res. 2009, 315, 2231–2240. [Google Scholar] [CrossRef] [Green Version]
- Provot, S.; Kempf, H.; Murtaugh, L.C.; Chung, U.I.; Kim, D.W.; Chyung, J.; Kronenberg, H.M.; Lassar, A.B. Nkx3.2/Bapx1 acts as a negative regulator of chondrocyte maturation. Development 2006, 133, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Magee, C.; Nurminskaya, M.; Faverman, L.; Galera, P.; Linsenmayer, T.F. SP3/SP1 transcription activity regulates specific expression of collagen type X in hypertrophic chondrocytes. J. Biol. Chem. 2005, 280, 25331–25338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T.; et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Lu, Y.; Li, F.; Qiao, L.; Wang, Q.; Li, N.; Borgia, J.A.; Deng, Y.; Lei, G.; Zheng, Q. Identification and characterization of the novel Col10a1 regulatory mechanism during chondrocyte hypertrophic differentiation. Cell Death Dis. 2014, 5, e1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, K.; Densmore, M.; Nishimura, R.; Lanske, B. Indian hedgehog signaling regulates transcription and expression of collagen type X via Runx2/Smads interactions. J. Biol. Chem. 2014, 289, 24898–24910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellingman, C.A.; Davidson, E.N.; Koevoet, W.; Vitters, E.L.; van den Berg, W.B.; van Osch, G.J.; van der Kraan, P.M. Smad signaling determines chondrogenic differentiation of bone-marrow-derived mesenchymal stem cells: Inhibition of Smad1/5/8P prevents terminal differentiation and calcification. Tissue Eng. Part A 2011, 17, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Hong, S.H.; Bae, S.C. Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-beta and bone morphogenetic protein. Oncogene 2002, 21, 7156–7163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Ohba, S.; Hojo, H.; McMahon, A.P. AP-1 family members act with Sox9 to promote chondrocyte hypertrophy. Development 2016, 143, 3012–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Kim, J.; Ryu, J.H.; Oh, H.; Chun, C.H.; Kim, B.J.; Min, B.H.; Chun, J.S. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef]
- Nagase, H.; Nagasawa, Y.; Tachida, Y.; Sakakibara, S.; Okutsu, J.; Suematsu, N.; Arita, S.; Shimada, K. Deiodinase 2 upregulation demonstrated in osteoarthritis patients cartilage causes cartilage destruction in tissue-specific transgenic rats. Osteoarthr. Cartil. 2013, 21, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomer, N.; den Hollander, W.; Ramos, Y.F.; Bos, S.D.; van der Breggen, R.; Lakenberg, N.; Pepers, B.A.; van Eeden, A.E.; Darvishan, A.; Tobi, E.W.; et al. Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionescu, A.; Kozhemyakina, E.; Nicolae, C.; Kaestner, K.H.; Olsen, B.R.; Lassar, A.B. FoxA family members are crucial regulators of the hypertrophic chondrocyte differentiation program. Dev. Cell 2012, 22, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, K.; Perez, R.F.; Kim, R.; Xu, L.; Teguh, D.A.; Gamer, L.; Kozhemyakina, E.; Lassar, A.; Li, Y.; Kaestner, K.; et al. The Role of Foxa2 Transcription Factor as Potential Regulator of Articular Cartilage Hypertrophy and Oa Progression. Osteoarthr. Cartil. 2019, 27, S157–S158. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef]
- Xu, M.; Bradley, E.W.; Weivoda, M.M.; Hwang, S.M.; Pirtskhalava, T.; Decklever, T.; Curran, G.L.; Ogrodnik, M.; Jurk, D.; Johnson, K.O.; et al. Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 780–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Rose, J.; Soder, S.; Skhirtladze, C.; Schmitz, N.; Gebhard, P.M.; Sesselmann, S.; Aigner, T. DNA damage, discoordinated gene expression and cellular senescence in osteoarthritic chondrocytes. Osteoarthr. Cartil. 2012, 20, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Mitchell, D.L. Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 1999, 27, 3213–3218. [Google Scholar] [CrossRef] [Green Version]
- Aida, Y.; Maeno, M.; Suzuki, N.; Namba, A.; Motohashi, M.; Matsumoto, M.; Makimura, M.; Matsumura, H. The effect of IL-1beta on the expression of inflammatory cytokines and their receptors in human chondrocytes. Life Sci. 2006, 79, 764–771. [Google Scholar] [CrossRef]
- Nagai, K.; Matsushita, T.; Matsuzaki, T.; Takayama, K.; Matsumoto, T.; Kuroda, R.; Kurosaka, M. Depletion of SIRT6 causes cellular senescence, DNA damage, and telomere dysfunction in human chondrocytes. Osteoarthr. Cartil. 2015, 23, 1412–1420. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, L.; Wang, Y.; Li, W.; Lin, Y.; Yu, D.; Zhang, L.; Li, F.; Pan, Z. Overexpression of Sirtuin 6 suppresses cellular senescence and NF-kappaB mediated inflammatory responses in osteoarthritis development. Sci. Rep. 2015, 5, 17602. [Google Scholar] [CrossRef]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y.; et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhang, S.; Previn, R.; Chen, D.; Jin, Y.; Zhou, G. Role of Forkhead Box O Transcription Factors in Oxidative Stress-Induced Chondrocyte Dysfunction: Possible Therapeutic Target for Osteoarthritis? Int. J. Mol. Sci. 2018, 19, 3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akasaki, Y.; Hasegawa, A.; Saito, M.; Asahara, H.; Iwamoto, Y.; Lotz, M.K. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthr. Cartil. 2014, 22, 162–170. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Huang, P.; Li, G.; Feng, Y.; Zhendong, L.; Zhou, C.; Hu, G.; Xu, Q. Overexpression of Pitx1 attenuates the senescence of chondrocytes from osteoarthritis degeneration cartilage-A self-controlled model for studying the etiology and treatment of osteoarthritis. Bone 2020, 131, 115177. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.; Shin, J.; Cho, Y.; Kim, H.S.; Gu, Y.R.; Kim, H.; You, K.T.; Chang, M.J.; Chang, C.B.; Kang, S.B.; et al. Stress-activated miR-204 governs senescent phenotypes of chondrocytes to promote osteoarthritis development. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Shirakawa, F.; Saito, K.; Bonagura, C.A.; Galson, D.L.; Fenton, M.J.; Webb, A.C.; Auron, P.E. The human prointerleukin 1 beta gene requires DNA sequences both proximal and distal to the transcription start site for tissue-specific induction. Mol. Cell. Biol. 1993, 13, 1332–1344. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Otero, M.; Imagawa, K.; de Andres, M.C.; Coico, J.M.; Roach, H.I.; Oreffo, R.O.; Marcu, K.B.; Goldring, M.B. Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1beta (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J. Biol. Chem. 2013, 288, 10061–10072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coimbra, I.B.; Jimenez, S.A.; Hawkins, D.F.; Piera-Velazquez, S.; Stokes, D.G. Hypoxia inducible factor-1 alpha expression in human normal and osteoarthritic chondrocytes. Osteoarthr. Cartil. 2004, 12, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Salotti, J.; Johnson, P.F. Regulation of senescence and the SASP by the transcription factor C/EBPbeta. Exp. Gerontol. 2019, 128, 110752. [Google Scholar] [CrossRef]
- Shimada, H.; Sakakima, H.; Tsuchimochi, K.; Matsuda, F.; Komiya, S.; Goldring, M.B.; Ijiri, K. Senescence of chondrocytes in aging articular cartilage: GADD45beta mediates p21 expression in association with C/EBPbeta in senescence-accelerated mice. Pathol. Res. Pract. 2011, 207, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Jung, M.; Kim, C.W.; Shin, D.Y. Inactivation of p38 kinase delays the onset of senescence in rabbit articular chondrocytes. Mech. Ageing Dev. 2005, 126, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Zhang, F.J.; Zeng, C.; Luo, W.; Xiao, W.F.; Gao, S.G.; Lei, G.H. Autophagy in osteoarthritis. Joint Bone Spine 2016, 83, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Izzo, V.; Niso-Santano, M.; Vacchelli, E.; Galluzzi, L.; Maiuri, M.C.; Kroemer, G. Regulation of autophagy by stress-responsive transcription factors. Semin. Cancer Biol. 2013, 23, 310–322. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin. Exp. Rheumatol. 2016, 34, 12–16. [Google Scholar]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, H.; Wei, J.; Lin, S.; Zong, Z.; Gong, F.; Huang, X.; Sun, J.; Li, P.; Lin, H.; et al. Inhibition of Nrf2/HO-1 signaling leads to increased activation of the NLRP3 inflammasome in osteoarthritis. Arthritis Res. Ther. 2019, 21, 300. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. (Lausanne) 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Takayama, K.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Fujita, N.; Oka, S.; Kurosaka, M.; Kuroda, R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012, 64, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Kawakami, Y.; Kobayashi, M.; Greco, N.; Cummins, J.H.; Matsushita, T.; Kuroda, R.; Kurosaka, M.; Fu, F.H.; Huard, J. Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res. Ther. 2014, 16, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.T.; Meng, H.; Ma, L.F.; Zhang, L.; Yu, H.M.; Wang, Z.Z.; Guo, A. Role of autophagy in the progression of osteoarthritis: The autophagy inhibitor, 3-methyladenine, aggravates the severity of experimental osteoarthritis. Int. J. Mol. Med. 2017, 39, 1224–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Sakamaki, J.I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517–532. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhu, L.; Zhang, T.; Lu, H.; Wang, C.; Xue, B.; Xu, X.; Liu, Y.; Cai, Z.; Sang, W.; et al. BRD4 has dual effects on the HMGB1 and NF-kappaB signalling pathways and is a potential therapeutic target for osteoarthritis. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 3001–3015. [Google Scholar] [CrossRef]
- Hong, J.; Li, S.; Markova, D.Z.; Liang, A.; Kepler, C.K.; Huang, Y.; Zhou, J.; Yan, J.; Chen, W.; Huang, D.; et al. Bromodomain-containing protein 4 inhibition alleviates matrix degradation by enhancing autophagy and suppressing NLRP3 inflammasome activity in NP cells. J. Cell. Physiol. 2020, 235, 5736–5749. [Google Scholar] [CrossRef]
- Liao, F.X.; Huang, F.; Ma, W.G.; Qin, K.P.; Xu, P.F.; Wu, Y.F.; Wang, H.; Chang, J.; Yin, Z.S. The New Role of Sirtuin1 in Human Osteoarthritis Chondrocytes by Regulating Autophagy. Cartilage 2019, 1947603519847736. [Google Scholar] [CrossRef]
- Sacitharan, P.K.; Bou-Gharios, G.; Edwards, J.R. SIRT1 directly activates autophagy in human chondrocytes. Cell Death Discov. 2020, 6, 41. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, S.; Goodwin, J.G.; Chauhan, S.; Manyam, G.; Wang, J.; Kamat, A.M.; Boyd, D.D. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol. Cell 2013, 50, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Zhan, Y.; Li, X.; Pan, Z.; Zheng, F.; Zhang, Z.; Zhou, Y.; Wu, Y.; Wang, X.; Gao, W.; et al. TFEB, a potential therapeutic target for osteoarthritis via autophagy regulation. Cell Death Dis. 2018, 9, 858. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Vasheghani, F.; Li, Y.H.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; Zeng, C.; Yan, B.; Ouyang, J.; Liu, X.; Sun, Q.; Zhao, C.; Fang, H.; Pan, J.; et al. mTORC1 activation downregulates FGFR3 and PTH/PTHrP receptor in articular chondrocytes to initiate osteoarthritis. Osteoarthr. Cartil. 2017, 25, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Akasaki, Y.; Alvarez-Garcia, O.; Saito, M.; Carames, B.; Iwamoto, Y.; Lotz, M.K. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol. 2014, 66, 3349–3358. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, T.; Alvarez-Garcia, O.; Mokuda, S.; Nagira, K.; Olmer, M.; Gamini, R.; Miyata, K.; Akasaki, Y.; Su, A.I.; Asahara, H.; et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Lin, J.; Wei, M.; Teng, Y.; He, Q.; Yang, G.; Yang, X. Sustained Akt signaling in articular chondrocytes causes osteoarthritis via oxidative stress-induced senescence in mice. Bone Res. 2019, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Van Spil, W.E.; Kubassova, O.; Boesen, M.; Bay-Jensen, A.C.; Mobasheri, A. Osteoarthritis phenotypes and novel therapeutic targets. Biochem. Pharmacol. 2019, 165, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Launoit, Y.; Baert, J.L.; Chotteau-Lelievre, A.; Monte, D.; Coutte, L.; Mauen, S.; Firlej, V.; Degerny, C.; Verreman, K. The Ets transcription factors of the PEA3 group: Transcriptional regulators in metastasis. Biochim. Biophys. Acta 2006, 1766, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Rahim, S.; Beauchamp, E.M.; Kong, Y.; Brown, M.L.; Toretsky, J.A.; Uren, A. YK-4-279 inhibits ERG and ETV1 mediated prostate cancer cell invasion. PLoS ONE 2011, 6, e19343. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Punchihewa, C.; Mistry, P.; Ooi, A.T.; Yang, D. Novel DNA bis-intercalation by MLN944, a potent clinical bisphenazine anticancer drug. J. Biol. Chem. 2004, 279, 46096–46103. [Google Scholar] [CrossRef] [Green Version]
- Hecker, M.; Wagner, A.H. Transcription factor decoy technology: A therapeutic update. Biochem. Pharmacol. 2017, 144, 29–34. [Google Scholar] [CrossRef]
- Miyake, T.; Miyake, T.; Sakaguchi, M.; Nankai, H.; Nakazawa, T.; Morishita, R. Prevention of Asthma Exacerbation in a Mouse Model by Simultaneous Inhibition of NF-kappaB and STAT6 Activation Using a Chimeric Decoy Strategy. Mol. Ther. Nucleic Acids 2018, 10, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, O.; Deschenes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, S.H.; Na, H.S.; Kwon, J.Y.; Kim, G.Y.; Jung, K.; Cho, K.H.; Kim, S.A.; Go, E.J.; Park, M.J.; et al. The Therapeutic Effect of STAT3 Signaling-Suppressed MSC on Pain and Articular Cartilage Damage in a Rat Model of Monosodium Iodoacetate-Induced Osteoarthritis. Front. Immunol. 2018, 9, 2881. [Google Scholar] [CrossRef]
- Ouyang, Y.; Wang, W.; Tu, B.; Zhu, Y.; Fan, C.; Li, Y. Overexpression of SOX9 alleviates the progression of human osteoarthritis in vitro and in vivo. Drug Des. Dev. Ther. 2019, 13, 2833–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aini, H.; Itaka, K.; Fujisawa, A.; Uchida, H.; Uchida, S.; Fukushima, S.; Kataoka, K.; Saito, T.; Chung, U.I.; Ohba, S. Messenger RNA delivery of a cartilage-anabolic transcription factor as a disease-modifying strategy for osteoarthritis treatment. Sci. Rep. 2016, 6, 18743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iachettini, S.; Trisciuoglio, D.; Rotili, D.; Lucidi, A.; Salvati, E.; Zizza, P.; Di Leo, L.; Del Bufalo, D.; Ciriolo, M.R.; Leonetti, C.; et al. Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis. 2018, 9, 996. [Google Scholar] [CrossRef] [PubMed]
- Zhi, L.Q.; Yao, S.X.; Liu, H.L.; Li, M.; Duan, N.; Ma, J.B. Hydroxytyrosol inhibits the inflammatory response of osteoarthritis chondrocytes via SIRT6-mediated autophagy. Mol. Med. Rep. 2018, 17, 4035–4042. [Google Scholar] [CrossRef] [PubMed]
- Cusanovich, D.A.; Pavlovic, B.; Pritchard, J.K.; Gilad, Y. The functional consequences of variation in transcription factor binding. PLoS Genet. 2014, 10, e1004226. [Google Scholar] [CrossRef]
- Wei, N.; Li, J.; Fang, C.; Chang, J.; Xirou, V.; Syrigos, N.K.; Marks, B.J.; Chu, E.; Schmitz, J.C. Targeting colon cancer with the novel STAT3 inhibitor bruceantinol. Oncogene 2019, 38, 1676–1687. [Google Scholar] [CrossRef]
- Shang, Y.; Brown, M. Molecular determinants for the tissue specificity of SERMs. Science 2002, 295, 2465–2468. [Google Scholar] [CrossRef]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Medeiros Alencar, V.H.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Kavanaugh, T.E.; Werfel, T.A.; Cho, H.; Hasty, K.A.; Duvall, C.L. Particle-based technologies for osteoarthritis detection and therapy. Drug Deliv. Transl. Res. 2016, 6, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Eymard, F.; Chevalier, X. Pharmacological treatments of knee osteoarthritis. Rev. Prat. 2019, 69, 515–519. [Google Scholar] [PubMed]
- Bottini, M.; Bhattacharya, K.; Fadeel, B.; Magrini, A.; Bottini, N.; Rosato, N. Nanodrugs to target articular cartilage: An emerging platform for osteoarthritis therapy. Nanomedicine 2016, 12, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.C.; Wang, S.; Padera, R.F., Jr.; Grodzinsky, A.J.; Hammond, P.T. Cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment of osteoarthritis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neefjes, M.; van Caam, A.P.M.; van der Kraan, P.M. Transcription Factors in Cartilage Homeostasis and Osteoarthritis. Biology 2020, 9, 290. https://doi.org/10.3390/biology9090290
Neefjes M, van Caam APM, van der Kraan PM. Transcription Factors in Cartilage Homeostasis and Osteoarthritis. Biology. 2020; 9(9):290. https://doi.org/10.3390/biology9090290
Chicago/Turabian StyleNeefjes, Margot, Arjan P. M. van Caam, and Peter M. van der Kraan. 2020. "Transcription Factors in Cartilage Homeostasis and Osteoarthritis" Biology 9, no. 9: 290. https://doi.org/10.3390/biology9090290
APA StyleNeefjes, M., van Caam, A. P. M., & van der Kraan, P. M. (2020). Transcription Factors in Cartilage Homeostasis and Osteoarthritis. Biology, 9(9), 290. https://doi.org/10.3390/biology9090290