CRISPRLearner: A Deep Learning-Based System to Predict CRISPR/Cas9 sgRNA On-Target Cleavage Efficiency

 ,
, 
 ,
,  ,
,  and
and
Abstract
:1. Introduction
1.1. Background
1.2. Related Works
2. Materials and Methods
2.1. Datasets
- Chari dataset [7], consisting in 1234 guides targeting Human 293T cells
- A new version of Doench et al. dataset [6], consisting in 2333 guides targeting CCDC101, MED12, TADA2B, TADA1, HPRT, CUL3, NF1, and NF2 genes from Human A375 cells
- Hart dataset [22], consisting in 4239 guides targeting 829 genes in Human Hct116 cells
- Moreno-Mateos dataset [10], consisting in 1020 guides targeting 128 genes in Zebrafish genome
- Gandhi dataset, consisting in 72 guides targeting different genes in Ciona genome
- Farboud dataset [23], consisting in 50 guides targeting different genes in Caenorhabditis elegans genome
- Varshney dataset [24], consisting in 102 guides targeting different genes in Zebrafish genome
- Gagnon dataset [25], consisting in 111 guides targeting different genes in Zebrafish genome
2.2. Software
3. System Description
3.1. Dataset Creation
3.1.1. Data extraction
- dataset column, containing the name of the dataset where the sequence was extracted from
- seq column, containing the actual sgRNA sequence
- modFreq column, containing the efficiency value
- longSeq100Bp column, the extended 100-bp sequence
3.1.2. Standardizing sgRNA Sequences
3.1.3. Standardizing Efficiency Measurement
3.1.4. Data augmentation Technique
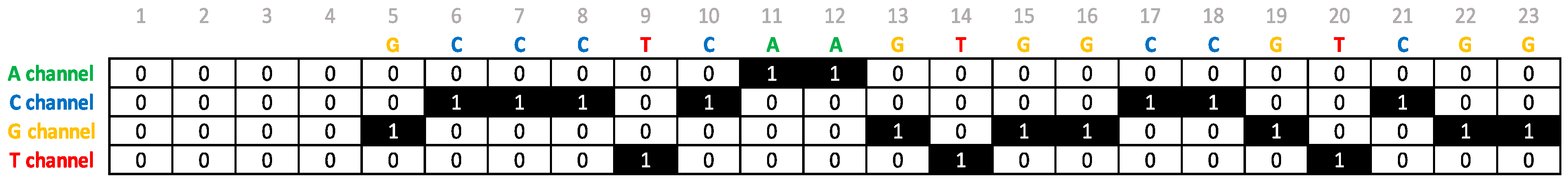
3.2. Data Representation for Training
3.3. Description of the Convolutional Neural Network Architecture
4. Results
5. Conclusion and Future Work
Author Contributions
Funding
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Zahra, H.; Ali, M.; Hui, W.; Dawei, L.; Yasin, O.; Honghua, R.; Qiang, Z. Strategies to Increase On-Target and Reduce Off-Target Effects of the CRISPR/Cas9 System in Plants. Int. J. Mol. Sci. 2019, 20, 3718. [Google Scholar] [CrossRef] [Green Version]
- Aach, J.; Mali, P.; Church, G.M. CasFinder: Flexible algorithm for identifying specific Cas9 targets in genomes. bioRxiv 2014, 005074. [Google Scholar] [CrossRef] [Green Version]
- Labun, K.; Montague, T.G.; Gagnon, J.A.; Thyme, S.B.; Valen, E. CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016, 44, W272–W276. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Chari, R.; Mali, P.; Moosburner, M.; Church, G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods 2015, 12, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Chari, R.; Yeo, N.C.; Chavez, A.; Church, G.M. sgRNA Scorer 2.0: A Species-Independent Model To Predict CRISPR/Cas9 Activity. ACS Synth. Biol. 2017, 6, 902–904. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Xiao, T.; Chen, C.-H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015, 25, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.-D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.W.; Wang, Y.; Zhang, X.X.; Wang, J.Q. Prediction of the RBP binding sites on lncRNAs using the high-order nucleotide encoding convolutional neural network. Anal. Biochem. 2019, 583, 113364. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Mao, W.; Shao, D.; Zhang, W.; Gong, H. DeepConPred2: An Improved Method for the Prediction of Protein Residue Contacts. Comput. Struct. Biotechnol. J. 2018, 16, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Le, N.Q.K.; Ho, Q.T.; Ou, Y.Y. Incorporating deep learning with convolutional neural networks and position specific scoring matrices for identifying electron transport proteins. Comput. Chem. 2017, 38. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wong, K.-C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Oxf. Acad. Bioinform. 2018, 34, i656–i663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 80. [Google Scholar] [CrossRef]
- Xue, L.; Tang, B.; Chen, W.; Luo, J. Prediction of CRISPR sgRNA activity using a deep convolutional neural network. J. Chem. Inf. Modeling 2019, 59, 615–624. [Google Scholar] [CrossRef]
- Dimauro, G. A new image quality metric based on human visual system. In Proceedings of the 2012 IEEE International Conf. on Virtual Environments Human-Computer Interfaces and Measurement Systems, Tianjin, China, 2–4 July 2012; pp. 69–73. [Google Scholar] [CrossRef]
- Casalino, G.; Castellano, G.; Consiglio, A.; Liguori, M.; Nuzziello, N.; Primiceri, e.D. Analysis of microRNA expressions for pediatric multiple sclerosis detection. In Modeling Decisions for Artificial Intelligence. MDAI2019; Lecture Notes in Computer Science. LNAI 11676; Springer: Cham, Switzerland, 2019; pp. 177–188. [Google Scholar] [CrossRef]
- Haeussler, M.; Schonig, K.; Eckert, H.; Eschstruth, A.; Mianne, J.; Renaud, J.-B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off- target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [Green Version]
- Farboud, B.; Meyer, B.J. Dramatic Enhancement of Genome Editing by CRISPR/Cas9 Through Improved Guide RNA Design. Genetics 2015, 199, 959–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, G.K.; Pei, W.; LaFave, M.C.; Idol, J.; Xu, L.; Gallardo, V.; Carrington, B.; Bishop, K.; Jones, M.; Li, M.; et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015, 25, 1030–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Ahkmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient Mutagenesis by Cas9 Protein-Mediated Oligonucleotide Insertion and Large-Scale Assessment of Single-Guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef] [PubMed]
- Hussain, W.; Khan, Y.D.; Rasool, N.; Khan, S.A.; Chou, K.C. SPalmitoylC-PseAAC: A sequence-based model developed via Chou’s 5-steps rule and general PseAAC for identifying S-palmitoylation sites in proteins. Anal. Biochem. 2019, 568, 14–23. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Fertility, G.R.U. Identifying Fertility-Related Proteins by Incorporating Deep-Gated Recurrent Units and Original Position-Specific Scoring Matrix Profiles. J. Proteome Res. 2019, 18, 3503–3511. [Google Scholar] [CrossRef]


{kind=link}
| Dataset | Original Sequences | Augmented Sequences |
|---|---|---|
| Chari | 1234 | 19,744 |
| Wang/Xu | 2076 | 33,216 |
| Doench mouse-EL4 | 951 | 15,216 |
| Doench A375 | 2333 | 37,328 |
| Hart | 4239 | 67,824 |
| Moreno-Mateos | 1020 | 16,320 |
| Gandhi | 72 | 1152 |
| Farboud | 50 | 800 |
| Varshney | 102 | 1632 |
| Gagnon | 111 | 1776 |
| N | Layer |
|---|---|
| 1 | Input: 4 × 23 × 1 |
| 2 | Convolution: 4 × 4 size, 50 filters, 1 stride |
| 3 | ReLU |
| 4 | Max pooling: 1 × 2 size, 1 stride |
| 5 | Flatten |
| 6 | Fully connected: 128 units |
| 7 | ReLU |
| 8 | Dropout: 0.3 dropout rate |
| 9 | Fully connected: 128 units |
| 10 | ReLU |
| 11 | Fully connected (output): 1 unit |
| 12 | Linear regression |
| Dataset | CRISPRLearner | DeepCas9 |
|---|---|---|
| Chari | 0.49 | 0.49 |
| Wang/Xu | 0.69 | 0.61 |
| Doench mouse-EL4 | 0.51 | 0.59 |
| Doench A375 | 0.23 | 0.38 |
| Hart | 0.55 | 0.41 |
| Moreno-Mateos | 0.19 | 0.23 |
| Gandhi | 0.36 | 0.32 |
| Farboud | 0.60 | 0.57 |
| Varshney | 0.35 | 0.3 |
| Gagnon | 0.35 | 0.25 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimauro, G.; Colagrande, P.; Carlucci, R.; Ventura, M.; Bevilacqua, V.; Caivano, D. CRISPRLearner: A Deep Learning-Based System to Predict CRISPR/Cas9 sgRNA On-Target Cleavage Efficiency. Electronics 2019, 8, 1478. https://doi.org/10.3390/electronics8121478
Dimauro G, Colagrande P, Carlucci R, Ventura M, Bevilacqua V, Caivano D. CRISPRLearner: A Deep Learning-Based System to Predict CRISPR/Cas9 sgRNA On-Target Cleavage Efficiency. Electronics. 2019; 8(12):1478. https://doi.org/10.3390/electronics8121478
Chicago/Turabian StyleDimauro, Giovanni, Pierpasquale Colagrande, Roberto Carlucci, Mario Ventura, Vitoantonio Bevilacqua, and Danilo Caivano. 2019. "CRISPRLearner: A Deep Learning-Based System to Predict CRISPR/Cas9 sgRNA On-Target Cleavage Efficiency" Electronics 8, no. 12: 1478. https://doi.org/10.3390/electronics8121478
APA StyleDimauro, G., Colagrande, P., Carlucci, R., Ventura, M., Bevilacqua, V., & Caivano, D. (2019). CRISPRLearner: A Deep Learning-Based System to Predict CRISPR/Cas9 sgRNA On-Target Cleavage Efficiency. Electronics, 8(12), 1478. https://doi.org/10.3390/electronics8121478