Abstract
Early-onset Alzheimer’s disease (EOAD), defined as Alzheimer’s disease onset before 65 years of age, has been significantly less studied than the “classic” late-onset form (LOAD), although EOAD often presents with a more aggressive disease course, caused by variants in the APP, PSEN1, and PSEN2 genes. EOAD has significant differences from LOAD, including encompassing diverse phenotypic manifestations, increased genetic predisposition, and variations in neuropathological burden and distribution. Phenotypically, EOAD can be manifested with non-amnestic variants, sparing the hippocampi with increased tau burden. The aim of this article is to review the different genetic bases, risk factors, pathological mechanisms, and diagnostic approaches between EOAD and LOAD and to suggest steps to further our understanding. The comprehension of the monogenic form of the disease can provide valuable insights that may serve as a roadmap for understanding the common form of the disease.
1. Introduction
Dementia is any disorder in which a significant decline in a person’s previous level of cognition affects their ability to perform occupational, domestic, or social activities. In general, dementia should be viewed as an acquired syndrome with multiple potential causes rather than as a distinct disease on its own [1]. A traditional approach to understanding dementia is to divide it into two broad disease categories: neurodegenerative conditions (irreversible) and non-neurodegenerative conditions (reversible) [2]. Common causes of non-neurodegenerative mild cognitive impairment and dementia which can occur at any age include vitamin deficiencies such as B12 and thiamine, chronic alcohol abuse, chemotherapy-induced cognitive dysfunction, hypothyroidism, infections, traumatic brain injury, and other psychiatric conditions such as severe depression or anxiety. However, it is important to note that most cases of dementia in the elderly population are due to some degree of neurodegeneration. Common degenerative dementias in older people include Parkinson’s disease, dementia with Lewy bodies, vascular dementia, frontotemporal lobar degeneration, and, the most common of all, Alzheimer’s disease (AD) [3].
In 1907, Alois Alzheimer penned a case report describing the condition of Auguste Deter, who sought treatment at his clinic. Deter exhibited a variety of signs and symptoms, notably including severe language difficulties and behavioral disturbances, such as paranoid delusions and anxiety. By contemporary standards, this female would be classified as having an unusual variant of AD. Furthermore, Deter was diagnosed with the disease at the age of 51, indicating that she had an early-onset form of AD. After Auguste Deter’s death in 1906, Alois Alzheimer analyzed her brain and identified “neuritic” plaques and neurofibrillary tangles, which he linked to the patient’s condition. Following this groundbreaking discovery, thorough research has shed light on the complex characteristics of AD [4]. From its initial recognition as a rare disease, it has become a global public health concern. The incidence of AD has increased significantly with the aging of the population, solidifying its status as one of the most important health challenges of the 21st century [5,6].
Today, AD is considered a progressive neurodegenerative disease characterized by the gradual deterioration of cognitive capacities and memory functions. Within the spectrum of dementia pathologies, AD is a predominant subset, accounting for an estimated 60 to 70% of dementia cases. The global impact of AD is massive, with a prevalence of more than 50 million people worldwide. It has been projected that the prevalence of dementia will triple by 2050, leading to an increasing societal burden on a global scale; in addition, the reported deaths from AD increased by approximately 148% between 2000 and 2018 [7,8,9].
The age of onset (AoO) remains clinically important in defining the observable characteristics of the disease, and AD cases have traditionally been classified into two categories: early onset (EOAD) and late onset (LOAD). This classification is based on an arbitrary age threshold that aligns with the onset of clinical symptoms. On one hand, EOAD cases represent approximately 5–10% of all AD cases. Affected individuals develop symptoms before the age of 65 years with an estimated heritability of 92–100% [5]. Furthermore, they exhibit remarkable variability in their clinical and neurobiological characteristics, requiring different approaches to their management. Some researchers even propose that the dissimilarities are significant enough to establish a distinct form of the disorder [5,10,11,12].
On the other hand, the most common initial clinical presentation of LOAD is an episodic memory deficit accompanied by progressive impairment in other cognitive domains with a heritability rate of 60–80%. For every twenty people with AD, eighteen will have LOAD and two will have EOAD. Furthermore, for every twenty early-onset patients, one of them has an autosomal dominant variant [5].
2. Risk Factors: Early-Onset vs. Late-Onset Alzheimer’s Disease
2.1. Genetics Factors
The genetic basis of AD shows a significant divergence between EOAD and LOAD.
Between 35% and 60% of EOAD patients have at least one affected first-degree relative, and in 10% to 15% of those familial EOAD patients, the mode of inheritance is autosomal dominant transmission [13]. In a significant proportion of EOAD cases, mutations in specific genes are responsible for the early onset of the disease. The three most important genes are as follows. PSEN2 (Presenilin 2): Mutations in the PSEN2 gene are the least common but can lead to EOAD. PSEN1 (Presenilin 1) and APP (Amyloid Precursor Protein): Mutations in the PSEN1 and APP genes are the most common genetic causes of EOAD. These mutations affect the processing of amyloid precursor protein, resulting in increased production of beta-amyloid (Aβ) [14]. Individuals with mutations in any of these genes have a significantly higher risk of developing EOAD, often in their 30s to 50s. The familial nature of EOAD means that there is a higher likelihood of multiple affected family members, making genetic counseling and testing an important consideration [15]. Recent whole-genome and whole-exome studies have revealed a wealth of information regarding the genetic underpinnings of EOAD. These comprehensive analyses have identified over 20 loci associated with genes participating in various metabolic pathways, with each locus contributing a modest risk to the development of EOAD. Among these, notable genes include sortilin-related receptor A (SORLA), the anti-inflammatory microglial triggering receptor expressed on myeloid cells 2 (TREM2), ATP-binding cassette subfamily A member 7 (ABCA7), and others involved in endocytosis, endolysosomal transport, immunologic reactivity, and lipid metabolism, such as PLD3, PSD2, TCIRG1, RIN3, and RUFY1. Additionally, genes like GRN, MAPT, and C9ORF72 have also been implicated in the genetic architecture of EOAD [16,17].
A significant advancement in this field is the integration of these genetic variants into a polygenic risk score, which holds promise in predicting the likelihood of developing EOAD, with accuracies ranging from 72.9% to 75.5%. This approach allows for a comprehensive assessment of an individual’s genetic susceptibility to EOAD, facilitating early identification and intervention strategies for at-risk individuals [18].
LOAD: In contrast, LOAD does not have a single dominant genetic cause; the risk variants are frequently observed in the general population but impart minimal overall risk, thereby offering limited predictive value regarding the likelihood of developing LOAD. Nonetheless, they serve a crucial role in identifying enriched pathways, providing valuable insights that guide molecular cell biologists and biochemists toward a deeper understanding of cellular processes involved in AD pathogenesis. While the APOEε4 allele of the apolipoprotein E protein (ApoE) is a well-established risk factor for LOAD, it is nevertheless not deterministic. Having one copy of the APOEε4 allele increases the risk, and having two copies further increases the risk [19]. However, many individuals with LOAD do not carry the APOEε4 allele, and conversely, not all carriers of APOEε4 develop LOAD. In addition, LOAD is polygenic, with multiple genetic variants contributing to overall susceptibility. In fact, more than 600 Alzheimer’s susceptibility genes have been identified. Genome-wide association studies (GWASs) have identified several risk genes, including CLU, CR1, PICALM, BIN1, CD2AP, PICALM, PLD3, and TREM2. Together, these variants (but not all) contribute to the complex genetic architecture of LOAD [20,21].
2.2. Non-Genetic Factors
Environmental and lifestyle factors such as physical fitness and cognitive stimulation, among others, can play an essential role in AD, and their impact can vary between EOAD and LOAD. This can be explained by the Scaffolding Theory of Aging and Cognition (STAC), which suggests that as people age, their brain undergoes changes that impact cognitive performance. Also, compensatory brain processes can help preserve cognitive function in healthy older adults. For instance, such processes can compensate for mild white matter disease and age-related atrophy [22].
In individuals who have not developed cognitive dysfunction but have a genetic risk for Alzheimer’s disease, the compensatory response occurs at a faster rate. However, this response decreases quickly once cognitive dysfunction and hippocampal atrophy become noticeable. On the other hand, older people who have age-appropriate cognition and a lower risk of developing AD tend to use increased brain activation to maintain their functionality [23].
EOAD: While genetic factors often play a more dominant role, environmental and lifestyle factors can still contribute to the risk of the disease, especially in individuals with a genetic predisposition. In particular, head trauma has been associated with an increased risk of EOAD. This association may be more pronounced in individuals with pathogenic variants, due to the fact that traumatic brain injury may exacerbate neurodegenerative processes already initiated by genetic mutations [20,21,22,23,24].
LOAD is influenced by a wider range of environmental and lifestyle factors. Several cardiovascular risk factors have been associated with an increased risk, including hypertension. Chronic high blood pressure has been associated with an increased risk of LOAD. It may contribute to cerebrovascular changes that may exacerbate AD-related brain pathology [25]. Diabetes: Type 2 diabetes is considered a potential risk factor for LOAD. Insulin resistance and glucose dysregulation may adversely affect brain function and contribute to AD pathology [26]. Smoking is a significant risk factor for LOAD, and it is a factor that individuals can actively change. It is thought to promote oxidative stress and inflammation, processes that may exacerbate neurodegeneration [27]. Hypercholesterolemia: Elevated cholesterol levels in midlife have been associated with an increased risk of LOAD later in life. High cholesterol may contribute to the buildup of Aβ plaques in the brain [28]. Physical Activity and diet: Engaging in regular physical activity and maintaining a heart-healthy diet may help reduce the risk of LOAD. These lifestyle choices are associated with improved cardiovascular health, which, in turn, may benefit brain health [29].
2.3. Other Factors
It is essential to consider various contributing factors to the onset of AD. One such substantial element that has been extensively studied is non-coding RNAs. These can be categorized based on their length into short-chain ncRNAs that include circular RNAs, short-interfering RNAs, microRNAs, piwi-associated RNAs, and long ncRNA (lncRNA). Long non-coding RNA, as the name indicates, refers to species longer than 200 nucleotides in length. It is a critical player in many biological processes, including epigenetic control of chromatin modification, mRNA stability, and promoter-specific regulation of genes. MicroRNAs typically consist of 21–23 nucleotides and exert gene-regulatory functions by forming sequence-specific base pairs with mRNAs to repress translation and initiate mRNA degradation [30]. MicroRNAs and lncRNAs have been implicated in AD pathogenesis, offering hope as potential biomarkers for disease diagnosis and the progression of epigenetic modifications. MiR-455-3p is an interesting example, as its overexpression results in a significant decrease in full-length Amyloid Precursor Protein (APP) and improved mitochondrial biogenesis, fusion, and synaptic genes, promoting cell survival and proliferation [31]. These findings emphasize the vital role of miR-455-3p in regulating abnormal APP processing, mitochondrial dynamics, and synaptic plasticity. Therefore, it is a promising molecule for further research in AD therapy.
Regarding lncRNAs and the involvement of NEAT1, an lncRNA, it has been reported that its downregulation increased p-tau generation, enhanced Aβ levels, and generated neuronal damage, accelerating the progression of AD [32]. On the other hand, individuals who have been diagnosed with AD have increased levels of Brain-Derived Neurotrophic Factor Antisense (BDNF-AS) in their bloodstream. These higher levels of BDNF-AS are strongly associated with cognitive abilities. In vitro assays have indicated that BDNF-AS can potentially cause neurotoxic effects. Additionally, the induction of BDNF-AS resulted in increased expression of BACE1 by inhibiting miR-9-5p. This, in turn, promoted the formation of amyloid plaques [33]. Among other significant elements in AD development are epigenetic modifications, as evidenced by numerous studies examining DNA methylation, histone modifications, and non-coding RNAs. For instance, cross-tissue analysis revealed hypermethylation of the ANK1 gene in several brain regions of individuals with AD. In addition, differential methylation patterns were observed in genes associated with AD neuropathology, such as ABCA7 and BIN1. Furthermore, histone tail modifications, including acetylation and methylation, were found to mediate chromatin condensation and regulate gene expression associated with synaptic function. These findings provide meaningful insights into the complex relationship between epigenetic mechanisms and AD pathology [34,35,36,37,38].
Mitochondrial trafficking to presynaptic terminals plays a crucial role in facilitating efficient synaptic vesicle release by regulating Ca2+ and ATP levels. However, pathological tau can disrupt this process by binding to kinesin and hindering the transport of mitochondria along microtubules. Consequently, this disruption may contribute to synaptic damage and eventual loss. Furthermore, pathological tau may also mislocalize to presynaptic terminals, where it can interfere with exocytosis by binding to synaptic vesicles [39,40] (risk factors are summarized in Figure 1).
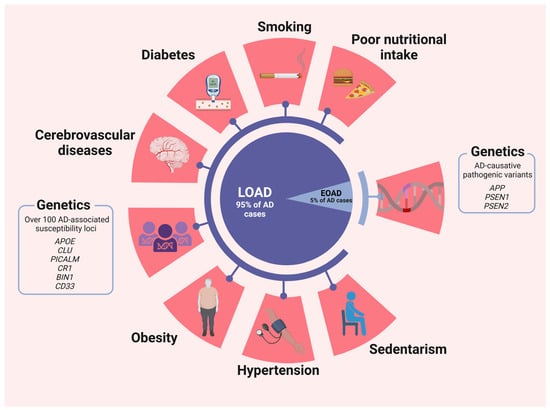
Figure 1.
Risk factors for LOAD and EOAD. Although some risk factors have a more prominent effect in one subtype than the other, it is important to clarify that they are not associated uniquely to one or another. Created with BioRender.com.
Endolysosomal dysfunction in AD is a hallmark at early and preclinical stages; abnormalities in this system lead to impaired clearance of various proteins, including Aβ and tau. One of the primary consequences of endolysosomal dysfunction is the inefficient clearance of Aβ peptides. Disruptions in endolysosomal function can alter the processing of APP, leading to increased production or impaired clearance of Aβ, resulting in its accumulation and subsequent aggregation into amyloid plaques [41].
Moreover, endolysosomal dysfunction affects the turnover of other proteins involved in AD pathology, such as tau. The degradation of abnormal tau species is also regulated by the endolysosomal system, and dysfunction in this pathway can contribute to tau accumulation and pathology. Recent research has highlighted the role of genetic risk factors associated with endolysosomal function in AD, such as mutations in genes encoding proteins involved in endosomal trafficking and lysosomal function [42,43].
Additionally, environmental factors, including oxidative stress and inflammation, can disrupt endolysosomal function and exacerbate AD pathology. Furthermore, disruptions in endoplasmic reticulum (ER) homeostasis can precipitate ER stress, a condition implicated in the initiation and progression of AD pathology. ER stress ensues when the folding capacity of the ER is overwhelmed by the accumulation of misfolded or unfolded proteins [44]. Within the context of AD, diverse factors contribute to ER stress, notably the buildup of Aβ peptides, which directly provoke ER stress by perturbing calcium homeostasis and impeding protein folding. Cells mount a response to ER stress through the unfolded protein response (UPR), a signaling pathway aimed at restoring ER equilibrium by bolstering its folding capacity, degrading misfolded proteins, and curtailing protein synthesis. Nevertheless, persistent or severe ER stress can culminate in neuronal dysfunction and apoptosis, exacerbating neurodegeneration in AD [45].
Variants in the PSEN1 gene also disrupt ER calcium homeostasis, further fueling ER stress. Dysregulated calcium signaling within the ER can precipitate mitochondrial dysfunction, oxidative stress, and ultimately neuronal apoptosis, thereby compounding the severity of AD pathology [46].
3. Pathological Mechanisms
Understanding the pathological mechanisms underlying AD is critical to the development of effective treatments and interventions. This section reviews the key pathological mechanisms, including the accumulation of Aβ plaques and neurofibrillary tangles, and examines how these mechanisms may differ between EOAD and LOAD [47].
The characteristic pathology of typical AD dementia includes the presence of extracellular amyloid plaques containing Aβ that are widely distributed throughout the cerebral cortex. Initially, Aβ accumulates in neocortical regions, followed by involvement of the limbic system, diencephalon, basal forebrain, and finally the cerebellum. In addition, inside neuron bodies, tau protein (which will be discussed in Section 3.2) undergoes abnormal chemical modifications (hyperphosphorylation), forming what are known as neurofibrillary tangles (NFTs). These tangles appear first in the medial temporal lobe and subsequently develop in isocortical regions of the temporal, parietal, and frontal lobes and interfere with normal cellular processes, disrupt neuronal function, and eventually, lead to cell death [48,49].
The receptors on the cell surface for ApoE are heparan sulfate proteoglycans (HSPGs), which serve as integral components in the orchestration of numerous intricate cellular mechanisms, including but not limited to cell adhesion, signaling cascades, and the effective clearance of Aβ peptides. Importantly, ApoE4 exhibits a binding affinity to HSPGs approximately three times higher than ApoE2 or ApoE3, indicating that the APOEε4 allele, associated with AD risk, binds more strongly to HSPGs [50]. This interaction may affect the binding, uptake, clearance, and aggregation of Aβ peptides by neurons and microglia, leading to the accumulation of Aβ plaques in the brain, a hallmark of AD pathology [51].
Furthermore, the presence of ApoE4 may disrupt the normal function of HSPG in the brain, impairing synaptic plasticity and neuronal integrity. HSPG has been proposed to be critically involved in all phases of tau diffusion, hypothesized to mimic prion-like spread, including tau release from intracellular space to extracellular environment, cell surface binding, internalization, and aggregation. This disruption in the molecular interactions between HSPG and ApoE4 may contribute to the synaptic dysfunction, neuronal loss, and cognitive decline observed in AD (Figure 2 emphasize the central role of ApoE4 in AD development) [52].
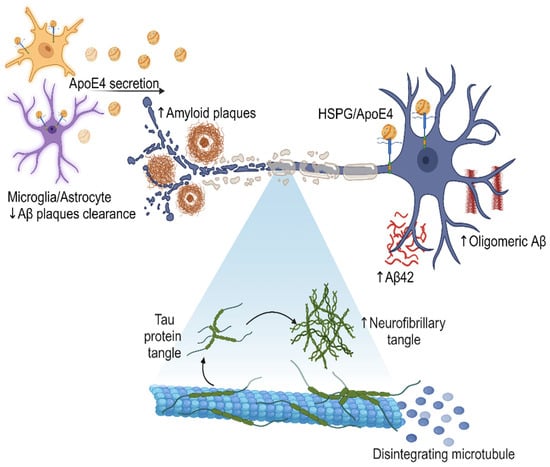
Figure 2.
Secretion of ApoE4 by microglia and astrocytes in the brain, leading to an increase in the amount of Aβ, oligomeric Aβ, and amyloid plaques. The presence of ApoE4 also correlates with the formation of neurofibrillary tangles, which disintegrate microtubules and contribute to neurodegeneration. The effect of the interaction between ApoE4 and HSPG may further modulate amyloid pathology and disease progression. This visual representation illustrates the molecular mechanisms and cellular interactions involved in the AD pathological cascade, highlighting the central role of ApoE4 in this process. Created with BioRender.com.
3.1. Amyloid Plaques
The production of the Aβ peptide arises from the enzymatic processing of APP. APP is identified as a type I integral transmembrane glycoprotein characterized by a small intracellular C-terminal domain, an Aβ peptide region, and a large extracellular N-terminal domain [53]. APP undergoes proteolytic processing through two distinct pathways:
- Non-amyloidogenic pathway: In this pathway, APP is cleaved by α-secretases, such as ADAM10, resulting in the generation of a soluble fraction of alpha-APP (sAPPα) and a carboxy-terminal fragment (CTF-α). Subsequently, γ-secretase also acts on CTF-α, producing the P3 fragment. Importantly, P3 is a soluble peptide that lacks the propensity to aggregate, unlike Aβ [54].
- Amyloidogenic pathway: APP can also be cleaved at the β-site by a β-secretase (BACE1), resulting in the production of a soluble beta-amyloid precursor protein (sAPPβ) and a carboxy-terminal fragment composed of 99 amino acids (known as CTF-β or C99). Subsequently, γ-secretase acts on CTF-β, releasing the Aβ, which can vary in size (primarily Aβ40, Aβ42, and Aβ43), depending on enzymatic cleavage. The pathway described above leads to the accumulation of Aβ protein and, consequently, the formation of amyloid plaques. Although the 42-amino acid form (Aβ42) is suspected to be a causative agent in AD, the molecular basis of its neurotoxicity remains unknown. However, amyloid plaques can induce synaptic dysfunction and an inflammatory response that ultimately triggers neurodegeneration. In a healthy individual, the non-amyloidogenic pathway predominates, while in a patient with AD, Aβ clearance is lower than its production through the amyloidogenic pathway [55,56].
In EOAD, the presence of pathogenic variants in the APP gene can trigger an excessive production of Aβ protein, and variants in PSEN1 and PSEN2 may precipitate a reduction in γ-cleavage, fostering the production of longer and more toxic forms of Aβ, contributing to the onset of AD. Alternatively, in LOAD, the mechanisms underlying the accumulation of Aβ are more complex. While individuals carrying the APOEε4 allele face an elevated risk of LOAD, it does not directly initiate the overproduction of Aβ. However, the presence of the APOEε4 allele correlates with pronounced disruption in white matter integrity (Figure 3 summarize the pathological mechanism) [11,57].
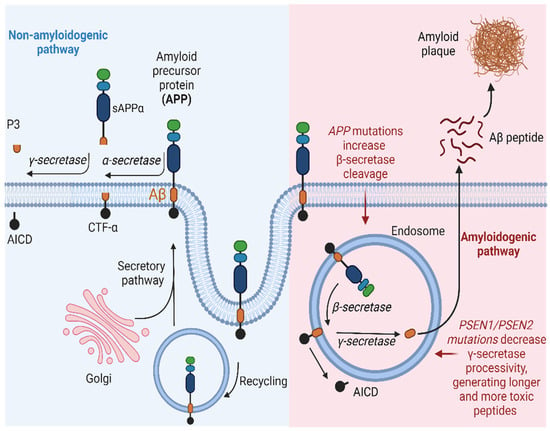
Figure 3.
Schematic representation of the amyloidogenic and non-amyloidogenic pathways. (Left) APP undergoes proteolytic cleavage by α-secretase, generating sAPPα and CTFα. Subsequent processing by γ-secretase yields p3 and AICD. This non-amyloidogenic pathway occurs primarily at the plasma membrane, with APP recycling in endosomes facilitated by interaction with low-density lipoprotein receptors. (Right) In the amyloidogenic pathway, APP is cleaved by β-secretase, releasing sAPPβ and CTFβ. Further cleavage by γ-secretase results in the release of Aβ and AICD. This pathway predominantly occurs within endosomes. AICD translocates to the nucleus, functioning as a transcriptional factor, while extracellular Aβ monomers aggregate, forming oligomers or fibrils/plaques associated with AD. Created with BioRender.com.
3.2. Neurofibrillary Tangles
NFTs, consisting of aggregated tau protein, are another hallmark of AD pathology. Tau is a microtubule-associated protein that is typically found in the cytoplasm of axons, where it also plays a role in stabilizing microtubules, thereby supporting their structural integrity and essential cellular functions. However, it is also found in both presynaptic and postsynaptic areas and is associated with the nuclear envelope [58,59].
In AD and similar tau-related diseases, tau protein undergoes abnormal changes. It becomes excessively phosphorylated, leading to its detachment from microtubules and its assembly into paired helical filaments and straight filaments, which constitute the central component of NFTs. These accumulations of tau disrupt the structural stability of neurons and hinder intracellular transport processes [60,61].
Interestingly, recent investigations propose that the spread of tau pathology in AD may follow a mechanism akin to prions. In contrast to prion diseases, human tauopathies are not considered to be transmissible; however, the prion-like behavior of tau is still under study. The propagation process of tau protein likely involves multiple stages. Initially, tau aggregates, followed by its release from donor neurons. Subsequently, tau is taken up by specific recipient neurons, leading to the induction of tau aggregation within neurons, although the sequence of these events remains uncertain. Within this mechanism, the presence of abnormal tau aggregates in one neuron could trigger the alteration of normally folded tau proteins in nearby neurons, subsequently resulting in the dissemination of tau pathology across the entire brain. This spread phenomenon is thought to be a driving factor in the gradual progression of AD and the eventual involvement of different brain regions as the disease progresses [62,63,64,65,66].
The propagation of tau pathology frequently occurs via synapses, which are the junctions for communication between neurons. Anomalous tau aggregates have the ability to traverse from one neuron’s axon to the dendrites of a connected neuron, potentially triggering the formation of new tau aggregates within the receiving neuron. This mechanism could play a role in the dissemination of tau pathology within neural networks [67]. Recent research suggests that neurons have the ability to release tau aggregates into the extracellular environment. These tau aggregates in the extracellular space can subsequently be taken up by neighboring neurons or glial cells, either via endocytic pathways involving engulfment by the cell membrane, receptor-mediated or exosome-mediated mechanisms, or lipid raft-dependent mechanisms, among other proposed mechanisms. After internalization, tau undergoes intracellular trafficking where a portion is degraded through the endosome–lysosome route, but the remainder (non-degraded tau) can initialize pathological processes [68,69].
3.3. Differences of Pathological Mechanisms between EOAD and LOAD
The extent to which the neuropathologic features underlying EOAD differ from those of LOAD remains a subject of ongoing investigation, and certain differences have been observed. EOAD cases tend to have a higher burden of amyloid plaques and NFTs in the frontal cortex when compared to LOAD patients. In particular, the neocortex, specifically the parietal and occipital parietal regions, appears to bear a similar burden in both EOAD and LOAD cases [70].
In addition, individuals who develop AD at a younger age often have a neuropathological profile characterized by a “purer” AD pathology (plaques and tangles), with fewer concurrent neuropathological changes compared to older AD patients. This can be explained in great proportion by the intervention of genetic factors, which have a stronger effect or as the primary cause of the disease in younger cases, and the young age, at which comorbidities are less frequent. Older AD patients often have a range of coexisting pathologies alongside the typical AD inclusions, including Lewy bodies and vascular pathology. Tau pathology primarily initiates within the entorhinal cortex and hippocampus, leading to early-stage neurofibrillary degeneration, synaptic and neuronal loss, as well as regional atrophy. Subsequently, in the early to intermediate stages of the disease, tau pathology extends to affect the locus coeruleus, basal forebrain, and associated neocortical regions, culminating with the involvement of the primary sensory cortex [9,13,71,72].
4. Diagnosis and Assessment
Accurate and timely diagnosis of AD is critical for effective management and intervention. This section provides a comprehensive comparison of the diagnostic and assessment strategies used in both EOAD and LOAD, taking into account various factors, including neuroimaging techniques, biomarkers, and cognitive assessments [73,74].
4.1. Neuroimaging
Neuroimaging plays a central role in the diagnosis and understanding of AD, providing valuable insight into the structural and functional changes that occur in the brain. While there is considerable overlap in the use of neuroimaging techniques for EOAD and LOAD, there are subtle differences that merit exploration [75].
4.1.1. Positron Emission Tomography (PET)
PET imaging has revolutionized our ability to visualize molecular and metabolic processes in the brain, making it an invaluable tool in the diagnosis and characterization of AD. Here, we take a closer look at how PET imaging is used in EOAD and LOAD [76].
Amyloid PET imaging uses radiotracers such as Pittsburgh compound B (PiB) and florbetapit (18F-AV-45) to detect the presence and distribution of amyloid plaques in the brain. This technique is particularly relevant in EOAD and LOAD, as the accumulation of AB is a hallmark. However, the extent and pattern of amyloid deposition can vary. As NFTs are also a hallmark of AD, this technique can provide valuable insight into the distribution and severity of tau pathology [77,78].
EOAD: These patients often have more aggressive and widespread deposition of amyloid plaques, which are easier to detect with PiB PET scans. The distinctive amyloid burden in EOAD cases helps distinguish it from other neurodegenerative diseases. They also have more extensive and severe tau pathology, and tau PET scans can show the widespread distribution of tau aggregates in the brain. This information helps to confirm the diagnosis and assess disease progression [79].
LOAD: These patients also benefit from amyloid PET scans, especially when the diagnosis is difficult due to comorbidities or overlapping symptoms with other dementias. While LOAD generally presents with amyloid plaques, the pattern may be less aggressive than in EOAD cases. Tau PET imaging can also be useful in LOAD, particularly when there is a need to differentiate AD from other neurodegenerative conditions that may exhibit different patterns of tau pathology [80,81,82].
4.1.2. Magnetic Resonance Imaging (MRI)
MRI is a versatile imaging modality that provides detailed structural information about the brain. MRI is instrumental in the diagnosis and characterization of EOAD and LOAD, providing valuable insight into the extent and pattern of structural abnormalities [83].
In EOAD, MRI often shows prominent structural changes. These changes may include significant cortical atrophy, particularly in the frontal and temporal lobes, which are commonly affected in this subtype. The degree of atrophy observed in EOAD may be more severe and widespread than in LOAD. MRI can precisely delineate these structural changes, helping to differentiate AD from other neurodegenerative diseases [84,85].
LOAD also has structural abnormalities that can be detected by MRI. Although the pattern of atrophy may be different from EOAD, MRI remains a valuable tool for assessing regional brain atrophy. The medial temporal lobe, including the hippocampus, is often one of the first areas affected in LOAD, and MRI can provide detailed images of these changes. In addition, LOAD may have more widespread cortical thinning, which can be detected by MRI [85].
4.2. Cognitive Assessment
Cognitive or neuropsychological assessment for AD is essentially the same as those used for dementia in general. It involves tests and evaluations designed to assess various aspects of cognitive function (attention, memory, language, visuospatial skills, executive function) to determine the patient’s capacity and monitor their progression over time [86].
Among the most commonly used is the Mini-Mental State Examination (MMSE), which is the most widely used and validated neuropsychological tool for assessing an individual’s cognitive functional status [87,88]. It consists of a 30-point questionnaire, in which scores of 24 or below indicate cognitive impairment, used to assess spatial–temporal orientation, short-term verbal memory, delayed recall, linguistic ability, numerical calculation, and construction of simple figures in individuals suspected of having a neuropsychological problem [86]. Another neuropsychological tool is the Clinical Dementia Rating (CDR) [89]. It is a semi-structured interview conducted with the patient (affected or suspected to be) and their caregiver (informant). The questions are directed to determine whether the patient exhibits memory problems, judgment issues, difficulty with problem solving, daily and social activities, etc., from the informant and patient point of view. The results can range from 0 to 3: no dementia (CDR = 0) to severe cognitive impairment (CDR = 3). Although this test can discriminate between mild cognitive impairment and a patient without impairment [90], the CDR is biased in identifying early-stage dementia [91]. A final example would be the Clock Drawing Test (CDT). This test includes activities aimed to assess attention, mathematical ability, comprehension, construction, naming, orientation, recall, repetition, spelling, and writing, all necessary skills for drawing a clock, including the figure outline, clock hands, and determining the hours [86].
All cognitive assessment must be conducted by trained healthcare professionals like neurologists, neuropsychologists, or geriatricians, and interpreted in the context of the individual’s medical history, clinical symptoms, other diagnostic tests (e.g., neuroimaging), educational achievement, and standardized scores for specific populations [86].
4.3. Biomarkers
Biomarkers in AD have significant diagnostic and prognostic utility, providing valuable information for early diagnosis and prediction of disease progression. Understanding their utility is essential for improved patient care and the development of targeted therapies. Current AD biomarkers include identification, in cerebrospinal fluid (CSF), of pathogenic hallmarks Aβ40, Aβ42, total-tau and p-tau, neurodegeneration markers (neurofilament light (NFL), and inflammation markers like interleukin 1α (IL-1α), IL-1β, IL-6, IL-8, IL-33, intercellular adhesion molecule 1 (ICAM-1), progranulin, SDF-1, soluble interleukin 1 receptor-like (sST2), and vascular cell adhesion protein 1 (VCAM-1). For EOAD cases, genetic biomarkers such as APP, PSEN1, and PSEN2 mutations are added to the list. The existence of mutations on one of these genes changes the expected values of certain biomarkers. For example, it has been described that PSEN1 mutation carriers show decreased Aβ40 levels and increased Aβ42/Aβ40 ratio levels, while PSEN2 mutation carriers show increased Aβ42 levels and increased Aβ42/Aβ40 ratio levels. Nevertheless, biomarker profiles between EOAD and LOAD, through biomarker analyses only, seem far from being reached [92]. Emerging biomarkers in the field of AD research offer promising avenues for early diagnosis, disease monitoring, and potential therapeutic interventions [93].
The need to expand the range of cerebrospinal fluid (CSF) biomarkers used to characterize different aspects of AD pathophysiology is underscored by the diversity of pathological presentations observed in LOAD [94]. The inclusion of neurogranin, a synaptic protein with AD specificity and predictive value for the future rate of cognitive decline, is a potential and promising addition to the AD CSF biomarker toolkit [95,96].
While CSF- and PET-based assessments are available, their use remains limited due to limitations such as high cost and perceived invasiveness [97]. However, rapid advances in the development of highly sensitive assays have opened the possibility of measuring levels of pathological brain-related and AD-associated proteins in the blood, and recent research has shown encouraging results in this regard. Among these, plasma p-tau appears to be emerging as a prime candidate marker, particularly in cases of symptomatic AD, including prodromal AD and AD dementia, when combined with the Aβ42/40 ratio. Although not an exclusive indicator of AD, blood neurofilament light chain (NfL) is a promising marker of neurodegeneration and could serve as a tool for monitoring the effect of disease-modifying therapies [93,98,99].
5. Treatments and Therapeutic Approaches
Several treatment options have been developed and new therapeutic approaches are being explored as research into the disease continues. The choice of appropriate therapy may vary depending on the onset of the disease, whether EOAD or LOAD.
5.1. Conventional Treatments
5.1.1. Cholinesterase Inhibitors
Cholinesterase inhibitors (CIs) are a class of drugs commonly used in the treatment of AD. These drugs play a critical role in managing the cognitive and behavioral symptoms of AD, especially in its early and moderate stages. CI works by increasing acetylcholine levels in the brain [100]. Acetylcholine is a neurotransmitter that plays a key role in cognitive functions such as memory, learning, and decision making. In AD, acetylcholine is reduced due to excessive breakdown by the enzyme cholinesterase. Cholinergic neurons are a type of nerve cell that utilizes acetylcholine as its primary neurotransmitter. These neurons are found in several regions of the brain and the peripheral nervous system. In AD, there is a selective degeneration or death of these cholinergic neurons [100,101].
CI may help improve memory, attention, and other cognitive functions in patients with AD. While these effects do not reverse the disease, they can lead to improved quality of life and greater functional independence. In addition to their cognitive effects, cholinesterase inhibitors may also help reduce behavioral and psychological symptoms associated with AD, such as agitation, depression, and aggression [102].
The most commonly used CIs in the treatment of AD include donepezil, rivastigmine, and galantamine. These drugs have slightly different efficacy and side effect profiles, allowing physicians to choose the most appropriate drug for each patient [103,104].
CIs are typically used in the early and moderate stages of AD, when cognitive symptoms are most pronounced. These drugs have been shown to be most effective in these stages, as they work to preserve the remaining cognitive capacity and slow its decline. In the advanced stages of AD, cholinesterase inhibitors may become less effective as the disease progresses and behavioral and physical symptoms become more pronounced. Donepezil has been approved to treat all AD stages, while galantamine and rivastigmine have been approved for mild and moderate stages. However, in some cases, these drugs may still provide benefit in symptom management [105,106,107].
Although CIs are generally safe, they can cause side effects such as nausea, vomiting, and diarrhea. Doctors closely monitor patients taking these drugs to minimize these effects [108].
5.1.2. NMDA Receptor Inhibitor
Memantine, a pharmacological agent utilized in the management of AD, falls under the category of NMDA receptor antagonists. Its mechanism of action revolves around modulating the activity of glutamate, a neurotransmitter crucial in memory and learning processes; normally, NMDA receptors are activated by glutamate, leading to the influx of calcium ions into the postsynaptic neuron. This calcium influx is crucial for synaptic plasticity and memory formation. In the context of AD, glutamate activity becomes dysregulated, and Aβ oligomers can bind and activate NMDA receptors. However, excessive or dysregulated activation of NMDA receptors, such as that caused by Aβ oligomers, can lead to excitotoxicity, neuronal damage, and ultimately cell death [109,110].
The chronic activation of NMDA receptors by Aβ oligomers disrupts the delicate balance of synaptic transmission and plasticity, contributing to the cognitive impairments observed in AD. Understanding this mechanism is important for developing potential therapies aimed at mitigating the toxic effects of Aβ oligomers on synaptic function and preserving cognitive function. By interfering with NMDA receptors, memantine helps restore glutamate balance, mitigating the excitotoxic repercussions associated with excessive stimulation. This action serves to safeguard nerve cells from damage and decelerate the progression of AD symptoms. It is worth highlighting that memantine is typically administered for moderate to severe AD cases, often complemented with other drugs like CI. While memantine exhibits potential in enhancing cognitive function and delaying symptom exacerbation in some patients, its efficacy varies among individuals, necessitating vigilant monitoring by healthcare providers (the mechanisms of action of both CI and memantine are summarized in Figure 4) [109,110].
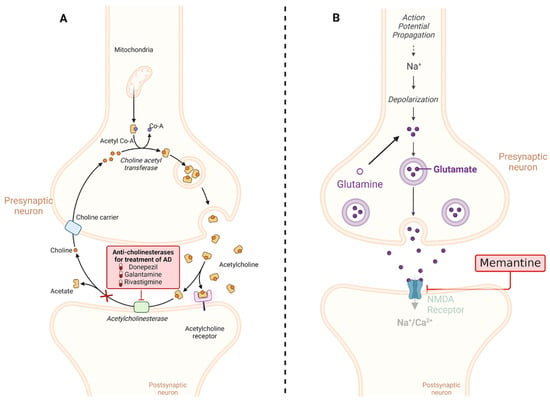
Figure 4.
Mechanisms of action of the most common drugs used in AD. (A) CIs such as donepezil, rivastigmine, and galantamine work by blocking the activity of acetylcholinesterase, an enzyme that breaks down acetylcholine. By inhibiting acetylcholinesterase, these medications increase the levels of acetylcholine in the brain, which can help improve cognitive function in AD patients. (B) Memantine is an NMDA receptor antagonist that regulates glutamate activity in the brain. By blocking excessive activation of NMDA receptors, memantine helps to protect neurons from excitotoxicity, which is associated with neurodegenerative processes in AD. This mechanism helps to stabilize neuronal activity and may slow the progression of cognitive decline in AD patients. This diagram illustrates the distinct mechanisms of action of cholinesterase inhibitors and memantine in the treatment of AD. Created with BioRender.com.
5.2. Emerging Treatments
5.2.1. Monoclonal Antibodies
The development of safe and effective drugs for treating AD has always been the goal. Currently, anti-amyloid therapies are showing promising outcomes. Monoclonal antibodies (mAbs) against tau proteins are designed to bind to extracellular tau proteins. This action slows down or prevents their distribution between cells, inhibiting tau protein aggregation and the formation of neurofibrillary tangles. Additionally, these antibodies help regulate the clearance and production of Aβ. Several anti-Aβ monoclonal antibodies such as Solanezumab, Bapineuzumab, Gantenerumab, Aducanumab, Lecanemab, and Donanemab are currently undergoing randomized clinical trials (RCTs) with varying results. Solanezumab was among the first anti-Aβ monoclonal antibodies to be tested in preclinical and clinical AD studies. It was found that solanezumab reduced brain Aβ levels by targeting soluble monomeric forms of Aβ peptide but had little effect on deposits. However, this compound, unfortunately, accelerated cognitive decline in both asymptomatic and symptomatic trial participants [111,112].
In contrast, Aducanumab captures aggregated Aβ and indirectly supports clearance of Aβ, most likely by presenting it to microglia for phagocytosis, thereby increasing the clearance of Aβ [113]. Results from the EMERGE Phase III clinical trial in EOAD patients treated with high-dose Aducanumab showed a significant reduction in clinical decline of CDR-SB scores at 78 weeks compared to baseline [114]. Individuals who received Aducanumab showed reduced clinical decline and had significant cognitive and functional benefits, such as improved orientation, language, and memory [115]. Lecanemab, a mAB against Aβ, is in phase III trials. It has been shown to reduce markers of amyloid in EOAD and resulted in a moderate decline in measures of cognition and function [116].
Studies have shown that Donanemab has the most significant impact on slowing down the progression of tau in individuals who have completely cleared amyloid, specifically in the brain regions that are in the advanced stages of the disease. It is important to further study these findings to understand how individuals with APOEε4 status respond to this treatment [117].
Emerging therapeutic approaches for AD have focused on leveraging the immune system to target Aβ (Figure 5). Among these strategies, activating antibodies targeting TREM2 and the Syncytin-1 protein (SYNK) has shown promise. TREM2 is expressed on microglia and is involved in the regulation of the immune response in the brain. Activation of TREM2 can enhance microglial phagocytosis and clearance of Aβ plaques. Similarly, SYNC, a protein involved in syncytin-mediated cell–cell fusion, has been identified as a potential target for antibody therapy due to its role in Aβ degradation. Conversely, inhibitory antibodies targeting Cluster of Differentiation 33 (CD33) and Src homology region 2 domain-containing phosphatase-1 (SHP1) aim to dampen the inflammatory response and promote Aβ clearance. CD33 is expressed on microglia and acts as an inhibitory receptor, while SHP1 negatively regulates immune signaling pathways. By blocking these inhibitory pathways, the goal is to enhance microglial activity and facilitate the removal of Aβ deposits. On one hand, microglial activation can be beneficial, as it contributes to the clearance of Aβ plaques, as activated microglia are capable of phagocytosing and clearing Aβ from the brain. This process is crucial for reducing the burden of Aβ oligomers, and microglia can also recognize and engulf Aβ through various receptors. On the other hand, prolonged or dysregulated microglial activation can lead to a state of chronic neuroinflammation. In this state, microglia release pro-inflammatory cytokines, chemokines, and reactive oxygen species, which can damage neurons and exacerbate neurodegeneration. Therefore, novel immunotherapeutic approaches offer promising avenues for the treatment of AD by harnessing the body’s immune system to target and degrade pathological protein aggregates implicated in disease progression [118,119].
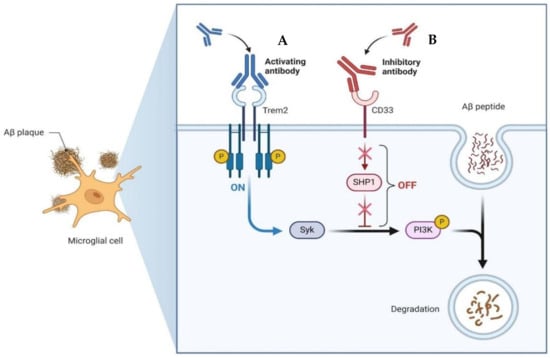
Figure 5.
Example of the mechanisms of action of an emerging treatment with antibodies for AD. (A) Activating antibody for TREM2. This antibody targets TREM2 and interacts with Syk. By activating TREM2 signaling pathways via Syk, this antibody enhances microglial function and promotes the clearance of Aβ plaques from the brain. (B) Inhibitory antibody for CD33. By interacting with SHP1, the antibody inhibits the inhibitory signaling pathways downstream of CD33. This results in enhanced microglial phagocytic activity and degradation of Aβ. Both antibodies offer promising therapeutic approaches for AD by targeting key immune regulatory proteins involved in the clearance of Aβ plaques. Created with BioRender.com.
5.2.2. Hybrid Molecules with Dual Affinity
Hybrid molecules with dual affinity represent a novel approach in drug design aimed at addressing complex neurological and psychiatric disorders. By targeting two receptors simultaneously, these hybrid molecules aim to modulate two distinct but interconnected pathways implicated in conditions such as AD. The rationale behind this approach lies in the potential synergistic effects of targeting multiple molecular targets involved in the pathophysiology of these complex disorders. The reliance on highly selective drugs targeting specific aspects of AD pathology has proven not so effective, necessitating the use of combination therapies to address the array of symptoms associated with disease progression [120].
An effective therapeutic strategy for AD may hinge on the ability of newly developed agents to interact with and regulate multiple molecular targets, thereby concurrently modulating numerous interconnected pathogenic pathways.
Within this realm, numerous compounds with multitarget profiles have emerged, demonstrating intriguing and promising pharmacological characteristics that position them as prospective therapeutic candidates. Hybrid therapeutic complexes, developed through specific combinations of two bioactive pharmacophores, yield homo- and heterodimers with enhanced affinity, therapeutic efficacy, biological profile, and complementary effects. Consequently, many disease-modifying hybrids hold promise for advancement into next-generation AD medicines (examples are shown in Figure 6).
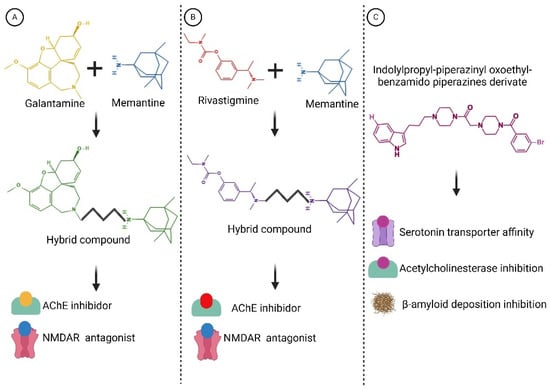
Figure 6.
Structures of hybrid compounds and indolylpropyl benzamidopiperazine derivate. (A) Galantamine and memantine. (B) Rivastigmine and memantine. (C) indolylpropyl benzamidopiperazine derivate. The hybrid compounds are shown by the color combination of the drugs. Functions are indicated below. These compounds represent a promising class of multitarget drugs, potentially leading to a reduction in its progression. Created with BioRender.com.
Designing hybrid multitargeted therapeutic compounds represents a promising approach in the pursuit of effective AD treatments. However, several challenges must be addressed, and further research is warranted to develop hybrid therapeutic compounds suitable for clinical use while mitigating off-target adverse effects [121,122,123].
5.2.3. Gamma-Secretase Modulators
Gamma-secretase modulators (GSMs) regulate the action of gamma-secretase enzymes, thereby reducing the production of Aβ. It has been observed that the ratio of Aβ37 or Aβ38 to Aβ42 or Aβ43 levels in CSF has a correlation with AoO as well as scores on the MMSE in individuals with fAD mutations [124]. There is also a link between the proportion of shorter peptides (Aβ37 + 38 + 40) and AoO in familial fAD caused by different PSEN1 mutations [125].
Pfizer conducted a promising phase 1 trial with a drug referred to as PF-06648671, which showed reductions in Aβ42 and Aβ40 levels, along with increases in Aβ37 levels, in healthy volunteers [126]. Therefore, Gamma-secretase modulators do not affect the overall production of Aβ peptides but instead modify the balance between longer and shorter Aβ forms. Thus, the potential signaling role of Aβ in the presence of GSMs may remain intact. As discussed above, the initial increase in Aβ pathology, detected through PET scans, marks the first pathological alteration in AD, which subsequently triggers tau pathology. Aβ peptides have significant physiological functions in normal cellular processes. Thus, using a GSM before the rapid escalation of tau pathology driven by Aβ could present an alternative strategy for treatment for the early stages of AD [127,128].
6. The Effect of ApoE
The gene for APOE, located at 19q13.32, encoding for ApoE in humans, is a 299-amino acid glycoprotein with a molecular weight of 34 kDa, primarily produced by astrocytes and activated microglia in the brain and hepatocytes in the liver. ApoE is composed of two primary structural segments linked by a flexible region. The first segment, spanning residues 1 to 167, encompasses the area responsible for receptor binding. Conversely, the second segment, spanning residues 206 to 299, houses the region responsible for lipid binding. ApoE plays a crucial role in lipid metabolism and transport, particularly in the central nervous system (CNS). In the brain, ApoE facilitates the transport of cholesterol and other lipids to neurons through interactions with cell surface receptors and also plays a key role in several brain functions, including lipid metabolism, processing of Aβ peptides, neural growth, and immunoregulation. ApoE exists in three main isoforms: ApoE2, ApoE3, and ApoE4, which are encoded by different alleles of the APOE gene. The structural differences between these isoforms arise from amino acid substitutions at positions 112 and 158. ApoE2 has cysteine residues at both positions, ApoE3 has a cysteine at position 112 and an arginine at position 158, while ApoE4 has arginine residues at both positions [129,130]. In the context of AD, the APOEε4 allele is a well-studied risk factor for developing the disease and other dementia [131]. On the other hand, APOEε2 is considered a protective allele [132], while APOEε3 is considered neutral in the risk of developing AD, not considering APOEε3 rare variants like Christchurch (APOE-R136S), which exerts a protective effect [133]. Nonetheless, this risk is relative, changing according to population characteristics. It has been described that the risk conferred by the APOEε4 allele is lower on Latin American, African, and other non-white populations due to ethnic origin in comparison to Caucasians [134].
Besides the well-studied effect of ApoE on physiological matters, it is debated whether it may also modulate the cognitive and structural phenotype of the disease and if its effect changes relative to LOAD or EOAD.
6.1. Effects of ApoE on LOAD
6.1.1. Effect of ApoE on Cognition
In previous research, it has been described that the effect of APOEε4 on the LOAD population reduces the age of symptom onset [132], increases disease progression, impairs cognitive function, especially episodic memory [135], is related to an increase in the regional density of Aβ and tau aggregates [136], decreases hippocampal volume [135], increases levels of biomarkers in cerebrospinal fluid, and decreases glucose metabolism [137], among other effects.
Morgen et al. 2013 [138], through neuroimaging techniques, showed that the presence of APOEε4 was related to a decrease in temporal medial lobe volume, a crucial structure for memory processing, while APOEε4 non-carriers showed more cortically generalized damage. This structural damage of the temporal medial lobe, for APOEε4 carriers, is reflected in results of neuropsychological research showing the appearance of amnestic presentations, meaning that memory is the main cognitive domain affected, that would account for typical AD presentations. On the other hand, non-carriers showed lower performances in executive functioning evaluation (trail making test, part B) and cognitive function related to cortical and prefrontal areas. On the contrary, the absence of the APOEε4 allele in LOAD patients was related to slow progression of cognitive decline, decreasing 1.3 points per year on MMSE in comparison with EOAD patients (discussed below) [139,140,141].
6.1.2. Effect of ApoE on Aβ
The differential impact of ApoE isoforms on AD risk involves ApoE4’s detrimental effect and ApoE2’s protective role regarding Aβ severity, plaque burden, and cerebral amyloid angiopathy (CAA) [142]. The tertiary conformation differences across ApoE isoforms affect their interaction affinity with Aβ and propensity for enzymatic cleavage, potentially generating toxic fragments. The interaction between ApoE and pathological Aβ deposition is central to AD risk, influencing plaque morphology and fibril formation [143,144]. ApoE also plays a role in Aβ clearance via receptor-mediated mechanisms, with LRP1 receptors facilitating Aβ/ApoE complex uptake. However, the less-effective lipid transport of ApoE4 compromises Aβ clearance, contributing to reduced Aβ elimination [145]. These findings suggest that targeting the interaction between ApoE and Aβ may offer therapeutic intervention opportunities at early disease stages, emphasizing the importance of understanding the differential effects of ApoE isoforms on AD pathogenesis (Figure 6) [146].
6.1.3. Effect of ApoE on Neuroinflammation
Microglial function appears to be influenced by isoform-specific effects, particularly regarding the response to Aβ pathology. Recent findings indicate that ApoE3 is more effective than ApoE4 in stimulating microglial responses to Aβ injection, potentially mediated by the triggering TREM2 [147]. TREM2, expressed by microglia in the brain, exhibits a high affinity for ApoE and modulates microglial responses, suggesting their involvement in chemotaxis towards plaques [148]. These insights underscore the intricate interplay between ApoE, TREM2, and microglial responses in the context of AD pathology, highlighting potential avenues for therapeutic targeting to enhance neuroprotection. sTREM2 is a soluble form of the TREM2 receptor that is released into the extracellular space. Although the exact function of sTREM2 is not completely clear, it is known to be involved in several cellular processes in astrocytes, neurons, and microglia [149].
ApoE4 leads to reduced levels of ApoE in both microglia and astrocytes. This reduction in ApoE levels makes the brain more susceptible to inflammation, which is indicated by higher levels of TNFα even under resting conditions. ApoE plays a crucial role in modulating microglial response to inflammation. Microglia can release TNFα, with increased ApoE secretion observed in ApoE2 and ApoE3 but not in ApoE4 microglia. In astrocytes, inflammatory stimuli leave ApoE2 and ApoE3 levels unaffected, while ApoE4 is diminished [150,151,152].
6.1.4. Effect of APOE on Blood–Brain Barrier
APOE has been linked to a more severe CAA, resulting in a heightened risk of lobar intracerebral hemorrhage, as well as other cerebrovascular complications. ApoE4 has been found to independently increase BBB permeability compared to ApoE3, as demonstrated in knock-in mouse models and confirmed through dynamic contrast-enhanced MRI in individuals with different cognitive statuses [153,154]. Proposed mechanisms underlying this effect include the activation of cyclophilin A, leading to elevated levels of MMP9 and pericyte injury, as well as disruption of the capillary basement membrane [155]. Mounting evidence suggests that BBB breakdown could be one of the earliest events in the pathogenesis of cognitive decline and AD pathology [156]. ApoE has been implicated in modulating cerebrovascular tight junction integrity, independent of CAA presence in AD brains. Additionally, ApoE4 disrupts endothelial BBB integrity by influencing various cellular processes, including extracellular matrix regulation, cell adhesion, cytoskeleton stability, and translation in brain endothelium. Moreover, studies have indicated that BBB breakdown contributes to cognitive decline in APOEε4 carriers, irrespective of Aβ or tau pathology [157,158,159]. Vascular endothelial cells, forming tight junctions to create a functional barrier, are a crucial component of the BBB. The progressive BBB breakdown associated with ApoE4 presence leads to synaptic dysfunction and behavioral deficits. Dysfunction in pericytes, which regulate the BBB by influencing gene expression in endothelial cells and inducing polarization of astrocytic end-feet, further contributes to BBB impairment [160]. Pericytes expressing ApoE4 exhibit reduced capacity to support endothelial cell function, thereby compromising BBB integrity and heightening susceptibility to cognitive decline [153].
6.1.5. Effect of ApoE on Tau
ApoE’s role in influencing tau neuropathological changes in AD brains is multifaceted. While ApoE3 may inhibit abnormal hyperphosphorylation and destabilization of the neuronal cytoskeleton in AD, the C-terminal-truncated form of ApoE4 is neurotoxic and stimulates tau phosphorylation, contributing to pre-NFTs [161,162]. Although there is little direct interaction between ApoE and tau in vivo, studies in transgenic mice suggest that ApoE isoforms indirectly affect tau pathology through their influence on microglia phenotype, with ApoE4 promoting tau-induced neurodegeneration compared to ApoE3 [163,164]. LRP1 has been identified as a receptor for tau uptake by neurons, and ApoE affects tau’s ability to bind LRP1, potentially influencing tau intracellular trafficking in an isoform-dependent manner [165]. The involvement of different receptors in tau uptake across various cell types remains unclear, particularly regarding how tau escapes to the cytoplasm for interaction with endogenous tau or accumulation as glial fibrillary tangles. Nonetheless, ApoE likely influences tau intracellular trafficking in an isoform-specific manner, suggesting a complex interplay between ApoE and tau pathology (the effects of ApoE are summarized in Figure 7) [166].
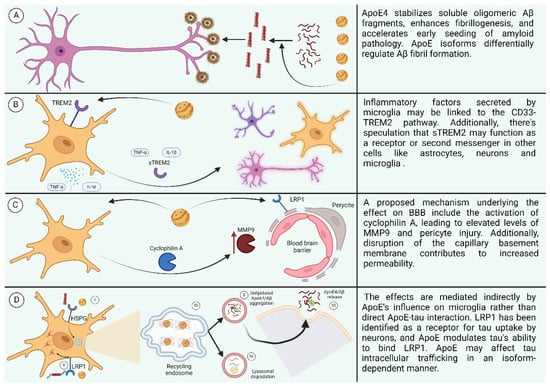
Figure 7.
(A) Effect of ApoE4 on AB. (B) Effect of ApoE4 on neuroinflammation. (C) Effect of ApoE4 on BBB. (D) Effect of ApoE on receptors. (I: ApoE4 and Aβ oligomers compete for binding to HSPGs. II: LRP1 interacts with lipidated ApoE4, leading to its internalization. III: Recycling endosomes serve as sites for Aβ generation. IV: Aβ and lipidated ApoE4 undergo degradation in lysosomes. V: ApoE4 stimulates the aggregation of Aβ. VI: Aggregated ApoE4 and Aβ are released into the extracellular space.) Created with BioRender.com.
6.2. Effect of ApoE on EOAD
As much as it is clear that APOEε4 is a risk factor, it is not fully understood whether it represents a risk factor for EOAD cases, mainly because the literature on its effect over AoO, clinical features, and cognitive phenotype is not as extensive as for LOAD cases. In general, a lower age at onset (EOAD cases) seems to be associated with faster progression of the disease [140]. However, articles show great heterogeneity in their results. Regarding AoO, De Luca et al., 2016 and Valdez-Gaxiola et al., 2023 [167,168] showed that the presence of APOEε4 retards the appearance of EOAD symptoms, while studies like Langella et al., 2023 [169] suggest that it decreases AoO and accelerates cognitive decline in PSEN1 E280A carriers (related to fAD). Nonetheless, Ryan et al., 2016 [170], studying a fAD population, found no effect of APOEε4 over AoO nor cognitive symptoms.
About ApoE genotype over cognition, the study by Van der vlies et al., 2007 [171] showed more rapid global cognitive decline in early-onset patients, especially prominent in APOEε4-negative patients. Smits et al., 2015 [172] went further, evaluating all cognitive functions, to discover the effect of the allele on each. They observed that younger AD patients, especially when they are APOEε4-negative, show faster decline in language, attention, executive and visuo-spatial functioning, all non-memory domains. The majority of APOEε4-positive EOAD patients had worse results for memory tasks than non-carriers of the APOEε4 allele, while APOEε4-negative AD patients declined fastest on non-amnestic domains, which is congruent with amnestic and non-amnestic phenotypes seen in LOAD [173,174]. Recent results from a Swedish population carrying PSEN1 and APP variants related to fAD showed that APOEε4 did not have an effect as a main predictor over cognition, but it did when interacting with other factors. Episodic memory was significantly affected by the interaction between APOEε4 and APP/PSEN1 mutations, showing favorable performance in the absence of APOEε4 in PSEN1 compared to APP mutation carriers [175,176]. Also, interactions between APOEε4 and AoO showed differential effects depending on whether it was APP or PSEN1 mutation carriers; in the first group, verbal abilities and attention were maintained in comparison with those who lacked the APOEε4 allele. In the latter, a detrimental effect of the APOEε4–AoO interaction was observed in visuospatial and verbal abilities [175].
Regarding global cognitive decline, Van der vlies et al. 2007 [141] showed that EOAD patients without the APOEε4 allele had rapid disease progression, losing approximately 2.4 MMSE points per year (in comparison with the 1.3 points lost in LOAD cases). This demonstrates the existence of an opposite effect of APOEε4 over cognition between EOAD and LOAD patients. Authors have addressed this heterogeneity in disease progression, rate of cognitive decline, and the cognitive domains that are affected in patients with early-onset versus late-onset AD, as well as epistasis or other complex genetic interactions when EOAD is caused by pathogenic mutations [175,176], antagonist pleiotropic effect of APOEε4 heterozygosity over the adult life course [177], the modulation of educational attainment [169], or the interaction of environmental factors yet to be discovered. Meanwhile, this heterogeneity in results means that the effect of APOEε4 cannot be or must not be generalized when talking about EOAD. Focusing on ApoE and its differential impact on EOAD and LOAD provides an opportunity to unravel the complex genetic and molecular mechanisms underlying AD. This knowledge is essential for developing effective therapeutic interventions that address the specific pathological processes associated with each disease subtype (All the differences between LOAD and EOAD are summarized in Table 1).

Table 1.
Mainly differences between EOAD and LOAD.
7. Future Perspectives
By 2025, Alzheimer disease will affect more than 150 million people, placing increasing strain on healthcare systems and caregivers. That is why it is crucial to understand risk factors, disease progression, and potential interventions. In the above, we provide a comprehensive review of the majority of points that encompass the AD umbrella, offering insights into the unique challenges and considerations of its traditional classification according to AoO. Regarding the information available on the disease, we highlight the need for increased awareness, not only in research, but also in social contexts, and resources to support affected individuals and their families, especially for EOAD, which tends to receive less attention in terms of public discourse and awareness campaigns. AD is often associated with older age, and the focus may disproportionately center on the more common late-onset form. However, in the research field, most of the experimental models are based on EOAD. This can result in less visibility and advocacy for individuals and families affected by EOAD compared to LOAD, yet its impact can be equally devastating, considering that it affects adults in their midlife, a period of time in which they can become parents, achieve professional goals, and find an economic balance, among other things.
Regarding diagnosis, proper patient classification will provide appropriate and on-time diagnosis, treatment, interventions, and support. Nevertheless, the latter tends to be more difficult for LOAD cases due to the multifactorial origin, and a not-so-clear preclinical stage. Emerging biomarkers in the field of AD research offer promising avenues for early diagnosis, disease monitoring, and potential therapeutic interventions. The need to expand the range of CSF biomarkers used to characterize different aspects of AD pathophysiology is underscored by the diversity of pathological presentations observed in LOAD. The inclusion of neurogranin is a potential and promising addition to the AD CSF biomarker toolkit. Advocacy efforts may focus on a more multifaceted approach. This includes genetic and medical analysis, genetic counseling, psychological accompaniment, and the caregiver perspective. This integrative approach encompasses not only the medical part of the disease, but also its personal and social implications. Emerging therapeutic approaches focus on leveraging the immune system to target Aβ. Among these strategies, activating antibodies targeting TREM2 and SYNK has shown promising results. Furthermore, given the broad impact of ApoE on various systems and parts of the body, it may exert its influence or have effects in a variety of ways, manifesting itself in multiple physiological functions and biological processes. While the APOEε4 allele is the primary risk factor for AD, finding a way to alter the mechanism of action of ApoE or its receptors could be an effective treatment for the prevention and/or treatment of AD. However, considering the inverse effects observed in EOAD and LOAD, it may be necessary to first identify the cause.
Additionally, another promising avenue for therapy involves altering the structure of ApoE through genetic engineering or small molecule intervention. The CRISPR/Cas9 system holds promise for directly converting ApoE4 to ApoE3 or ApoE2. Similarly, an AAV system could be employed to stimulate the expression of ApoE2. Alternatively, small molecule inhibitors could be utilized to disrupt interdomain interactions, leading to structural modifications of ApoE and alterations in its functionality, leading to a better clearance of Aβ [179]. Pharmaceutical therapies can be complemented by non-pharmacological interventions. For example, new research is focusing on the potential benefits of nutraceuticals, fortified foods with health-promoting ingredients like curcumin, omega-3 fatty acids, flavonoids, and certain vitamins, microbiota interactions, and fasting, which have shown promise in preclinical studies and early clinical trials for their potential to mitigate AD pathology or symptoms. Also, other lifestyle modifications, including exercise, cognitive stimulation, and social engagement, are gaining popularity and are being consolidated as important components of the management of AD [181].
As science continues to extend the average human lifespan, the prevalence of AD is expected to increase, and so typical classification parameters such as AoO will definitely change. Improving access to specialized care worldwide, promoting and funding research, from basic to specialized, making efforts to destigmatize the condition through scientific diffusion, proper recruiting of patients for clinical trials, creation of public policies, and social initiatives, on the whole, will help ensure earlier diagnosis and interventions to enhance the quality of life for both patients and their families.
8. Conclusions
AD is a major global health challenge due to its complex etiology. The conventional understanding of AD may oversimplify its nature, especially concerning late-onset cases. LOAD is influenced by a myriad of factors, including genetics, environment, medical conditions, and lifestyle choices, rendering it difficult to classify as a singular pathological entity. Recent studies focusing on Aβ have highlighted the limitations of a uni-dimensional therapeutic approach, highlighting the necessity for a holistic strategy to effectively prevent and manage AD. Central to this approach is a thorough understanding of the heterogeneous nature of AD and the intricate interplay of different underlying pathological mechanisms. In addition, it is imperative to recognize the potential impact of protective factors on the clinical manifestation of the disease. While significant progress has been made in unraveling the molecular and genetic basis of AD, numerous questions, particularly regarding EOAD and its distinct genetic and clinical features, remain unanswered. Filling these knowledge gaps will require collaborative endeavors and the inclusion of diverse populations in research endeavors. Looking ahead, the prospect of personalized medicine, guided by comprehensive genetic and clinical profiling, holds immense potential in devising more efficacious strategies for the prevention and treatment of AD.
Author Contributions
Conceptualization, C.A.V.-G., F.R.-L. and L.E.F.; methodology, C.A.V.-G. and F.R.-L.; software, C.A.V.-G., F.R.-L. and A.G.-R.; investigation, C.A.V.-G., F.R.-L., L.E.F. and A.G.-R.; writing—original draft preparation, C.A.V.-G., F.R.-L., L.E.F. and A.G.-R.; writing—review and editing C.A.V.-G., F.R.-L., L.E.F., A.G.-R. and J.M.M.-O.; supervision, L.E.F. and J.M.M.-O. All authors have read and agreed to the published version of the manuscript.
Funding
The APC was funded by Universidad de Guadalajara, Guadalajara, Jalisco, Mexico.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The Global Prevalence of Dementia: A Systematic Review and Metaanalysis. Alzheimers Dement. J. Alzheimers Assoc. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed Brain Pathologies Account for Most Dementia Cases in Community-Dwelling Older Persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English Translation of Alzheimer’s 1907 Paper, “Über Eine Eigenartige Erkankung Der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Hasana, S.; Hossain, M.F.; Islam, M.S.; Behl, T.; Perveen, A.; Hafeez, A.; Ashraf, G.M. Molecular Genetics of Early- and Late-Onset Alzheimer’s Disease. Curr. Gene Ther. 2021, 21, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Rosa-Neto, P.; Morais, J.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia. 2021. Available online: https://cdn.alzheimers.org.nz/wp-content/uploads/2021/09/World-Alzheimer-Report-2021-Web.pdf (accessed on 3 April 2024).
- Langa, K.M. Is the Risk of Alzheimer’s Disease and Dementia Declining? Alzheimers Res. Ther. 2015, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Skaria, A.P. The Economic and Societal Burden of Alzheimer Disease: Managed Care Considerations. Am. J. Manag. Care 2022, 28 (Suppl. S10), S188–S196. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The Genetic Landscape of Alzheimer Disease: Clinical Implications and Perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef]
- Larner, A.J. Presenilin-1 Mutations in Alzheimer’s Disease: An Update on Genotype-Phenotype Relationships. J. Alzheimers Dis. 2013, 37, 653–659. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-Onset Alzheimer’s Disease: Nonamnestic Subtypes and Type 2 AD. Arch. Med. Res. 2012, 43, 677–685. [Google Scholar] [CrossRef]
- Ayodele, T.; Rogaeva, E.; Kurup, J.T.; Beecham, G.; Reitz, C. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Curr. Neurol. Neurosci. Rep. 2021, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular Genetics of Early-onset Alzheimer’s Disease Revisited. Alzheimers Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 Mutations in Early-Onset Alzheimer Disease: A Genetic Screening Study of Familial and Sporadic Cases. PLOS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.S.; Hou, C.E. Early-Onset Alzheimer Disease: When Is Genetic Testing Appropriate? Alzheimer Dis. Assoc. Disord. 2004, 18, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, K.N.H.; Jackson, T.; Rumbaugh, M.; Eloyan, A.; Abreu, M.; Dage, J.L.; Snoddy, C.; Faber, K.M.; Foroud, T.; Hammers, D.B.; et al. Pathogenic Variants in the Longitudinal Early-onset Alzheimer’s Disease Study Cohort. Alzheimers Dement. 2023, 19 (Suppl. S9), S64–S73. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to Alzheimer’s Disease Risk of Rare Variants in TREM2, SORL1, and ABCA7 in 1779 Cases and 1273 Controls. Neurobiol. Aging 2017, 59, 220.e1–220.e9. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Patel, T.; Barber, I.S.; Guetta-Baranes, T.; Brookes, K.J.; Chappell, S.; Turton, J.; Guerreiro, R.; Bras, J.; Hernandez, D.; et al. Polygenic Risk Score in Postmortem Diagnosed Sporadic Early-Onset Alzheimer’s Disease. Neurobiol. Aging 2018, 62, 244.e1–244.e8. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Brookhouser, N.; Brafman, D.A. Using Human Induced Pluripotent Stem Cells (hiPSCs) to Investigate the Mechanisms by Which Apolipoprotein E (APOE) Contributes to Alzheimer’s Disease (AD) Risk. Neurobiol. Dis. 2020, 138, 104788. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of Risk Loci from Genome-Wide Association Studies of Alzheimer’s Disease. Lancet Neurol. 2020, 19, 326–335. [Google Scholar] [CrossRef]
- Van Acker, Z.P.; Bretou, M.; Annaert, W. Endo-Lysosomal Dysregulations and Late-Onset Alzheimer’s Disease: Impact of Genetic Risk Factors. Mol. Neurodegener. 2019, 14, 20. [Google Scholar] [CrossRef]
- Park, D.C.; Reuter-Lorenz, P. The Adaptive Brain: Aging and Neurocognitive Scaffolding. Annu. Rev. Psychol. 2009, 60, 173–196. [Google Scholar] [CrossRef]
- Rao, S.M.; Bonner-Jackson, A.; Nielson, K.A.; Seidenberg, M.; Smith, J.C.; Woodard, J.L.; Durgerian, S. Genetic Risk for Alzheimer’s Disease Alters the Five-Year Trajectory of Semantic Memory Activation in Cognitively Intact Elders. NeuroImage 2015, 111, 136–146. [Google Scholar] [CrossRef]
- Guo, Z.; Cupples, L.A.; Kurz, A.; Auerbach, S.H.; Volicer, L.; Chui, H.; Green, R.C.; Sadovnick, A.D.; Duara, R.; DeCarli, C.; et al. Head Injury and the Risk of AD in the MIRAGE Study. Neurology 2000, 54, 1316–1323. [Google Scholar] [CrossRef]
- Yaffe, K.; Vittinghoff, E.; Hoang, T.; Matthews, K.; Golden, S.H.; Zeki Al Hazzouri, A. Cardiovascular Risk Factors Across the Life Course and Cognitive Decline: A Pooled Cohort Study. Neurology 2021, 96, e2212–e2219. [Google Scholar] [CrossRef]
- Diniz Pereira, J.; Gomes Fraga, V.; Morais Santos, A.L.; Carvalho, M.D.G.; Caramelli, P.; Braga Gomes, K. Alzheimer’s Disease and Type 2 Diabetes Mellitus: A Systematic Review of Proteomic Studies. J. Neurochem. 2021, 156, 753–776. [Google Scholar] [CrossRef]
- Durazzo, T.C.; Mattsson, N.; Weiner, M.W. Alzheimer’s Disease Neuroimaging Initiative. Smoking and Increased Alzheimer’s Disease Risk: A Review of Potential Mechanisms. Alzheimers Dement. 2014, 10 (Suppl. S3), S122–S145. [Google Scholar] [CrossRef]
- Xu, C.; Apostolova, L.G.; Oblak, A.L.; Gao, S. Association of Hypercholesterolemia with Alzheimer’s Disease Pathology and Cerebral Amyloid Angiopathy. J. Alzheimers Dis. 2020, 73, 1305–1311. [Google Scholar] [CrossRef]
- Di Liegro, C.M.; Schiera, G.; Proia, P.; Di Liegro, I. Physical Activity and Brain Health. Genes 2019, 10, 720. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Holsinger, R.M.D. Roles of Non-Coding RNA in Alzheimer’s Disease Pathophysiology. Int. J. Mol. Sci. 2023, 24, 12498. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, A.P.; Yin, X.; Reddy, P.H. Novel MicroRNA-455-3p and Its Protective Effects against Abnormal APP Processing and Amyloid Beta Toxicity in Alzheimer’s Disease. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2019, 1865, 2428–2440. [Google Scholar] [CrossRef]
- Ke, S.; Yang, Z.; Yang, F.; Wang, X.; Tan, J.; Liao, B. Long Noncoding RNA NEAT1 Aggravates Aβ-Induced Neuronal Damage by Targeting miR-107 in Alzheimer’s Disease. Yonsei Med. J. 2019, 60, 640. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Luan, W.; Shen, X.; Wang, Z.; Cao, Y. LncRNA BDNF-AS as ceRNA Regulates the miR-9-5p/BACE1 Pathway Affecting Neurotoxicity in Alzheimer’s Disease. Arch. Gerontol. Geriatr. 2022, 99, 104614. [Google Scholar] [CrossRef]
- Smith, R.G.; Hannon, E.; De Jager, P.L.; Chibnik, L.; Lott, S.J.; Condliffe, D.; Smith, A.R.; Haroutunian, V.; Troakes, C.; Al-Sarraj, S.; et al. Elevated DNA Methylation across a 48-kb Region Spanning the HOXA Gene Cluster Is Associated with Alzheimer’s Disease Neuropathology. Alzheimers Dement. 2018, 14, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s Disease: Early Alterations in Brain DNA Methylation at ANK1, BIN1, RHBDF2 and Other Loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Marzi, S.J.; Leung, S.K.; Ribarska, T.; Hannon, E.; Smith, A.R.; Pishva, E.; Poschmann, J.; Moore, K.; Troakes, C.; Al-Sarraj, S.; et al. A Histone Acetylome-Wide Association Study of Alzheimer’s Disease Identifies Disease-Associated H3K27ac Differences in the Entorhinal Cortex. Nat. Neurosci. 2018, 21, 1618–1627. [Google Scholar] [CrossRef]
- Chen, L.; Guo, X.; Li, Z.; He, Y. Relationship between Long Non-Coding RNAs and Alzheimer’s Disease: A Systematic Review. Pathol.-Res. Pract. 2019, 215, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Takousis, P.; Sadlon, A.; Schulz, J.; Wohlers, I.; Dobricic, V.; Middleton, L.; Lill, C.M.; Perneczky, R.; Bertram, L. Differential Expression of microRNAs in Alzheimer’s Disease Brain, Blood, and Cerebrospinal Fluid. Alzheimers Dement. 2019, 15, 1468–1477. [Google Scholar] [CrossRef]
- Ivannikov, M.V.; Sugimori, M.; Llinás, R.R. Synaptic Vesicle Exocytosis in Hippocampal Synaptosomes Correlates Directly with Total Mitochondrial Volume. J. Mol. Neurosci. 2013, 49, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.-C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau Association with Synaptic Vesicles Causes Presynaptic Dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef]
- Peric, A.; Annaert, W. Early Etiology of Alzheimer’s Disease: Tipping the Balance toward Autophagy or Endosomal Dysfunction? Acta Neuropathol. 2015, 129, 363–381. [Google Scholar] [CrossRef]
- Vaz-Silva, J.; Gomes, P.; Jin, Q.; Zhu, M.; Zhuravleva, V.; Quintremil, S.; Meira, T.; Silva, J.; Dioli, C.; Soares-Cunha, C.; et al. Endolysosomal Degradation of Tau and Its Role in Glucocorticoid-driven Hippocampal Malfunction. EMBO J. 2018, 37, e99084. [Google Scholar] [CrossRef] [PubMed]
- Giovedì, S.; Ravanelli, M.M.; Parisi, B.; Bettegazzi, B.; Guarnieri, F.C. Dysfunctional Autophagy and Endolysosomal System in Neurodegenerative Diseases: Relevance and Therapeutic Options. Front. Cell. Neurosci. 2020, 14, 602116. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic Reticulum Stress: Molecular Mechanism and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER Stress and UPR in Alzheimer’s Disease: Mechanisms, Pathogenesis, Treatments. Cell Death Dis. 2022, 13, 706. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Sanjo, N.; Uchihara, T.; Watabe, K.; George-Hyslop, P.S.; Fraser, P.E.; Mizusawa, H. Presenilin-1 Holoprotein Is an Interacting Partner of Sarco Endoplasmic Reticulum Calcium-ATPase and Confers Resistance to Endoplasmic Reticulum Stress. J. Alzheimers Dis. 2010, 20, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Hyman, B.T.; Flory, J.; Damasio, A.R.; Van Hoesen, G.W. The Topographical and Neuroanatomical Distribution of Neurofibrillary Tangles and Neuritic Plaques in the Cerebral Cortex of Patients with Alzheimer’s Disease. Cereb. Cortex 1991, 1, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease: A Practical Approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Raskin, J.; Cummings, J.; Hardy, J.; Schuh, K.; Dean, R. Neurobiology of Alzheimer’s Disease: Integrated Molecular, Physiological, Anatomical, Biomarker, and Cognitive Dimensions. Curr. Alzheimer Res. 2015, 12, 712–722. [Google Scholar] [CrossRef]
- Snow, A.D.; Sekiguchi, R.T.; Nochlin, D.; Kalaria, R.N.; Kimata, K. Heparan Sulfate Proteoglycan in Diffuse Plaques of Hippocampus but Not of Cerebellum in Alzheimer’s Disease Brain. Am. J. Pathol. 1994, 144, 337–347. [Google Scholar]
- Fukuchi, K. Alzheimer s Disease and Heparan Sulfate Proteoglycan. Front. Biosci. 1998, 3, d327–d337. [Google Scholar] [CrossRef]
- Zhu, Y.; Gandy, L.; Zhang, F.; Liu, J.; Wang, C.; Blair, L.J.; Linhardt, R.J.; Wang, L. Heparan Sulfate Proteoglycans in Tauopathy. Biomolecules 2022, 12, 1792. [Google Scholar] [CrossRef]
- Coronel, R.; Bernabeu-Zornoza, A.; Palmer, C.; Muñiz-Moreno, M.; Zambrano, A.; Cano, E.; Liste, I. Role of Amyloid Precursor Protein (APP) and Its Derivatives in the Biology and Cell Fate Specification of Neural Stem Cells. Mol. Neurobiol. 2018, 55, 7107–7117. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted Amyloid β–Protein Similar to That in the Senile Plaques of Alzheimer’s Disease Is Increased in Vivo by the Presenilin 1 and 2 and APP Mutations Linked to Familial Alzheimer’s Disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gao, Y.; Winblad, B.; Tjernberg, L.O.; Schedin-Weiss, S. A Super-Resolved View of the Alzheimer’s Disease-Related Amyloidogenic Pathway in Hippocampal Neurons. J. Alzheimers Dis. 2021, 83, 833–852. [Google Scholar] [CrossRef]
- Hatters, D.M.; Peters-Libeu, C.A.; Weisgraber, K.H. Apolipoprotein E Structure: Insights into Function. Trends Biochem. Sci. 2006, 31, 445–454. [Google Scholar] [CrossRef]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. In Tau Biology; Takashima, A., Wolozin, B., Buee, L., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1184, pp. 187–203. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940.e7. [Google Scholar] [CrossRef]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgräfe, K.; Mandelkow, E.-M.; et al. Neuronal Activity Regulates Extracellular Tau in Vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tan, L.; Yu, J.-T.; Tan, L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.; et al. Neuronal Activity Enhances Tau Propagation and Tau Pathology In Vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, M.; Wang, D. The Propagation Mechanisms of Extracellular Tau in Alzheimer’s Disease. J. Neurol. 2022, 269, 1164–1181. [Google Scholar] [CrossRef]
- Annadurai, N.; De Sanctis, J.B.; Hajdúch, M.; Das, V. Tau Secretion and Propagation: Perspectives for Potential Preventive Interventions in Alzheimer’s Disease and Other Tauopathies. Exp. Neurol. 2021, 343, 113756. [Google Scholar] [CrossRef]
- Vogels, T.; Leuzy, A.; Cicognola, C.; Ashton, N.J.; Smolek, T.; Novak, M.; Blennow, K.; Zetterberg, H.; Hromadka, T.; Zilka, N.; et al. Propagation of Tau Pathology: Integrating Insights From Postmortem and In Vivo Studies. Biol. Psychiatry 2020, 87, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Avila, J.; Hernández, F. Propagation of Tau via Extracellular Vesicles. Front. Neurosci. 2019, 13, 698. [Google Scholar] [CrossRef]
- Leake, I. Oligomeric Tau Might Spread Trans-Synaptically in Alzheimer Disease. Nat. Rev. Neurosci. 2023, 24, 393. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, H.; Tang, X. Tau Internalization: A Complex Step in Tau Propagation. Ageing Res. Rev. 2021, 67, 101272. [Google Scholar] [CrossRef]
- Fleeman, R.M.; Proctor, E.A. Astrocytic Propagation of Tau in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 645233. [Google Scholar] [CrossRef]
- Sirkis, D.W.; Bonham, L.W.; Johnson, T.P.; La Joie, R.; Yokoyama, J.S. Dissecting the Clinical Heterogeneity of Early-Onset Alzheimer’s Disease. Mol. Psychiatry 2022, 27, 2674–2688. [Google Scholar] [CrossRef]
- Middleton, L.E.; Grinberg, L.T.; Miller, B.; Kawas, C.; Yaffe, K. Neuropathologic Features Associated with Alzheimer Disease Diagnosis: Age Matters. Neurology 2011, 77, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Palasí, A.; Gutiérrez-Iglesias, B.; Alegret, M.; Pujadas, F.; Olabarrieta, M.; Liébana, D.; Quintana, M.; Álvarez-Sabín, J.; Boada, M. Differentiated Clinical Presentation of Early and Late-Onset Alzheimer’s Disease: Is 65 Years of Age Providing a Reliable Threshold? J. Neurol. 2015, 262, 1238–1246. [Google Scholar] [CrossRef]
- Squire, L.R.; Genzel, L.; Wixted, J.T.; Morris, R.G. Memory Consolidation. Cold Spring Harb. Perspect. Biol. 2015, 7, a021766. [Google Scholar] [CrossRef]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically Defined Subtypes of Alzheimer’s Disease with Distinct Clinical Characteristics: A Retrospective Study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.; Lin, C.-L.; Chiang, M.-C. Exploring the Frontiers of Neuroimaging: A Review of Recent Advances in Understanding Brain Functioning and Disorders. Life 2023, 13, 1472. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.A.; Iaccarino, L.; Edwards, L.; Asken, B.M.; Gorno-Tempini, M.L.; Kramer, J.H.; Pham, J.; Perry, D.C.; Possin, K.; Malpetti, M.; et al. Amyloid, Tau and Metabolic PET Correlates of Cognition in Early and Late-Onset Alzheimer’s Disease. Brain 2022, 145, 4489–4505. [Google Scholar] [CrossRef]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum. Med. Clin. North Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jeong, M.; Stiles, W.R.; Choi, H.S. Neuroimaging Modalities in Alzheimer’s Disease: Diagnosis and Clinical Features. Int. J. Mol. Sci. 2022, 23, 6079. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.M.; Murray, A.D. Alzheimer’s Dementia: The Emerging Role of Positron Emission Tomography. Neurosci. 2022, 28, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Rowley, P.A.; Samsonov, A.A.; Betthauser, T.J.; Pirasteh, A.; Johnson, S.C.; Eisenmenger, L.B. Amyloid and Tau PET Imaging of Alzheimer Disease and Other Neurodegenerative Conditions. Semin. Ultrasound CT MRI 2020, 41, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Brier, M.R.; Gordon, B.; Friedrichsen, K.; McCarthy, J.; Stern, A.; Christensen, J.; Owen, C.; Aldea, P.; Su, Y.; Hassenstab, J.; et al. Tau and Aβ Imaging, CSF Measures, and Cognition in Alzheimer’s Disease. Sci. Transl. Med. 2016, 8, 338ra66. [Google Scholar] [CrossRef]
- Mosconi, L.; McHugh, P.F. FDG- and Amyloid-PET in Alzheimer’s Disease: Is the Whole Greater than the Sum of the Parts? Q. J. Nucl. Med. Mol. Imaging 2011, 55, 250–264. [Google Scholar]
- Meysami, S.; Raji, C.A.; Merrill, D.A.; Porter, V.R.; Mendez, M.F. Quantitative MRI Differences Between Early versus Late Onset Alzheimer’s Disease. Am. J. Alzheimers Dis. Dement. 2021, 36, 153331752110553. [Google Scholar] [CrossRef] [PubMed]
- Filley, C.M.; Kelly, J.; Heaton, R.K. Neuropsychologic Features of Early- and Late-Onset Alzheimer’s Disease. Arch. Neurol. 1986, 43, 574–576. [Google Scholar] [CrossRef] [PubMed]
- for the Alzheimer’s Disease Neuroimaging Initiative; Chandra, A.; Dervenoulas, G.; Politis, M. Magnetic Resonance Imaging in Alzheimer’s Disease and Mild Cognitive Impairment. J. Neurol. 2019, 266, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.K. Clinical Diagnosis of Alzheimer’s Disease. In Biomarkers in Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 2016; pp. 27–48. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. Mini-Mental State. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Molloy, D.W.; Alemayehu, E.; Roberts, R. Reliability of a Standardized Mini-Mental State Examination Compared with the Traditional Mini-Mental State Examination. Am. J. Psychiatry 1991, 148, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Ernesto, C.; Schafer, K.; Coats, M.; Leon, S.; Sano, M.; Thal, L.J.; Woodbury, P. Clinical Dementia Rating Training and Reliability in Multicenter Studies: The Alzheimer’s Disease Cooperative Study Experience. Neurology 1997, 48, 1508–1510. [Google Scholar] [CrossRef] [PubMed]
- Duara, R.; Loewenstein, D.A.; Greig-Custo, M.T.; Raj, A.; Barker, W.; Potter, E.; Schofield, E.; Small, B.; Schinka, J.; Wu, Y.; et al. Diagnosis and Staging of Mild Cognitive Impairment, Using a Modification of the Clinical Dementia Rating Scale: The mCDR. Int. J. Geriatr. Psychiatry 2010, 25, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, K.; Strang, D.; MacKnight, C.; Downer, R.; Morris, J.C. Interrater Reliability of the Clinical Dementia Rating in a Multicenter Trial. J. Am. Geriatr. Soc. 2000, 48, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Nystuen, K.L.; McNamee, S.M.; Akula, M.; Holton, K.M.; DeAngelis, M.M.; Haider, N.B. Alzheimer’s Disease: Models and Molecular Mechanisms Informing Disease and Treatments. Bioengineering 2024, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Zetterberg, H. The Past and the Future of Alzheimer’s Disease Fluid Biomarkers. J. Alzheimers Dis. 2018, 62, 1125–1140. [Google Scholar] [CrossRef]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Cerebrospinal Fluid Biomarker Signature in Alzheimer’s Disease Neuroimaging Initiative Subjects. Ann. Neurol. 2009, 65, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Nazir, F.H.; Brinkmalm, G.; Camporesi, E.; Kvartsberg, H.; Portelius, E.; Boström, M.; Kalm, M.; Höglund, K.; Olsson, M.; et al. Alzheimer-Associated Cerebrospinal Fluid Fragments of Neurogranin Are Generated by Calpain-1 and Prolyl Endopeptidase. Mol. Neurodegener. 2018, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Willemse, E.A.J.; De Vos, A.; Herries, E.M.; Andreasson, U.; Engelborghs, S.; Van Der Flier, W.M.; Scheltens, P.; Crimmins, D.; Ladenson, J.H.; Vanmechelen, E.; et al. Neurogranin as Cerebrospinal Fluid Biomarker for Alzheimer Disease: An Assay Comparison Study. Clin. Chem. 2018, 64, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Engelborghs, S.; Niemantsverdriet, E.; Struyfs, H.; Blennow, K.; Brouns, R.; Comabella, M.; Dujmovic, I.; Van Der Flier, W.; Frölich, L.; Galimberti, D.; et al. Consensus Guidelines for Lumbar Puncture in Patients with Neurological Diseases. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2017, 8, 111–126. [Google Scholar] [CrossRef]
- Simrén, J.; Elmgren, A.; Blennow, K.; Zetterberg, H. Fluid Biomarkers in Alzheimer’s Disease. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2023; Volume 112, pp. 249–281. [Google Scholar] [CrossRef]
- Moscoso, A.; Grothe, M.J.; Ashton, N.J.; Karikari, T.K.; Lantero Rodríguez, J.; Snellman, A.; Suárez-Calvet, M.; Blennow, K.; Zetterberg, H.; Schöll, M.; et al. Longitudinal Associations of Blood Phosphorylated Tau181 and Neurofilament Light Chain with Neurodegeneration in Alzheimer Disease. JAMA Neurol. 2021, 78, 396. [Google Scholar] [CrossRef] [PubMed]
- Tabet, N. Acetylcholinesterase Inhibitors for Alzheimer’s Disease: Anti-Inflammatories in Acetylcholine Clothing! Age Ageing 2006, 35, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The Cholinergic Hypothesis of Geriatric Memory Dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase Inhibitors as Alzheimer’s Therapeutics (Review). Mol. Med. Rep. 2019, 2, 1479–1487. [Google Scholar] [CrossRef]
- Birks, J.S. Cholinesterase Inhibitors for Alzheimer’s Disease. Cochrane Database Syst. Rev. 2006, 2016, CD005593. [Google Scholar] [CrossRef]
- Ogura, H.; Kosasa, T.; Kuriya, Y.; Yamanishi, Y. Comparison of Inhibitory Activities of Donepezil and Othercholinesterase Inhibitors on Acetylcholinesterase Andbutylcholinesterase in Vitro. Methods Find. Exp. Clin. Pharmacol. 2000, 22, 609. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.A.; Sabbagh, M.N. Donepezil: Potential Neuroprotective and Disease-Modifying Effects. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, P.; Millard, C.B.; Harel, M.; Dvir, H.; Enz, A.; Sussman, J.L.; Silman, I. Kinetic and Structural Studies on the Interaction of Cholinesterases with the Anti-Alzheimer Drug Rivastigmine. Biochemistry 2002, 41, 3555–3564. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B. A Review on Cholinesterase Inhibitors for Alzheimer’s Disease. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Matsunaga, S.; Oya, K.; Nomura, I.; Ikuta, T.; Iwata, N. Memantine for Alzheimer’s Disease: An Updated Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2017, 60, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Balázs, N.; Bereczki, D.; Kovács, T. Cholinesterase Inhibitors and Memantine for the Treatment of Alzheimer and Non-Alzheimer Dementias. Ideggyógy. Szle. 2021, 74, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Day, G.S. Anti-Amyloid Therapies for Alzheimer Disease: Finally, Good News for Patients. Mol. Neurodegener. 2023, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, M.; Dibello, V.; Sardone, R.; Castellana, F.; Zupo, R.; Lampignano, L.; Bortone, I.; Stallone, R.; Altamura, M.; Bellomo, A.; et al. Lessons Learned from the Failure of Solanezumab as a Prospective Treatment Strategy for Alzheimer’s Disease. Expert Opin. Drug Discov. 2024, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; Von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef]
- Howard, R.; Liu, K.Y. Questions EMERGE as Biogen Claims Aducanumab Turnaround. Nat. Rev. Neurol. 2020, 16, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Shcherbinin, S.; Evans, C.D.; Lu, M.; Andersen, S.W.; Pontecorvo, M.J.; Willis, B.A.; Gueorguieva, I.; Hauck, P.M.; Brooks, D.A.; Mintun, M.A.; et al. Association of Amyloid Reduction After Donanemab Treatment with Tau Pathology and Clinical Outcomes: The TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol. 2022, 79, 1015. [Google Scholar] [CrossRef] [PubMed]
- Biber, K.; Bhattacharya, A.; Campbell, B.M.; Piro, J.R.; Rohe, M.; Staal, R.G.W.; Talanian, R.V.; Möller, T. Microglial Drug Targets in AD: Opportunities and Challenges in Drug Discovery and Development. Front. Pharmacol. 2019, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Jhee, S.; Shiovitz, T.; Crawford, A.W.; Cutler, N.R. β-Amyloid Therapies in Alzheimer’s Disease. Expert Opin. Investig. Drugs 2001, 10, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Rampa, A.; Belluti, F.; Gobbi, S.; Bisi, A. Hybrid-Based Multi-Target Ligands for the Treatment of Alzheimer’s Disease. Curr. Top. Med. Chem. 2011, 11, 2716–2730. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Kumar, A.; Panda, G. Anti-Cholinesterase Hybrids as Multi-Target-Directed Ligands against Alzheimer’s Disease (1998–2018). Bioorg. Med. Chem. 2019, 27, 895–930. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands to Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Jana, A.; Bhattacharjee, A.; Das, S.S.; Srivastava, A.; Choudhury, A.; Bhattacharjee, R.; De, S.; Perveen, A.; Iqbal, D.; Gupta, P.K.; et al. Molecular Insights into Therapeutic Potentials of Hybrid Compounds Targeting Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 3512–3528. [Google Scholar] [CrossRef]
- Liu, L.; Lauro, B.M.; He, A.; Lee, H.; Bhattarai, S.; Wolfe, M.S.; Bennett, D.A.; Karch, C.M.; Young-Pearse, T.; Dominantly Inherited Alzheimer Network (DIAN). Identification of the Aβ37/42 Peptide Ratio in CSF as an Improved Aβ Biomarker for Alzheimer’s Disease. Alzheimers Dement. 2023, 19, 79–96. [Google Scholar] [CrossRef]
- Petit, D.; Fernández, S.G.; Zoltowska, K.M.; Enzlein, T.; Ryan, N.S.; O’Connor, A.; Szaruga, M.; Hill, E.; Vandenberghe, R.; Fox, N.C.; et al. Aβ Profiles Generated by Alzheimer’s Disease Causing PSEN1 Variants Determine the Pathogenicity of the Mutation and Predict Age at Disease Onset. Mol. Psychiatry 2022, 27, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.E.; Carrieri, C.; Dela Cruz, F.; Fullerton, T.; Hajos-Korcsok, E.; He, P.; Kantaridis, C.; Leurent, C.; Liu, R.; Mancuso, J.; et al. Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF -06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies. Clin. Pharmacol. Ther. 2020, 107, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Sturchio, A.; Dwivedi, A.K.; Malm, T.; Wood, M.J.A.; Cilia, R.; Sharma, J.S.; Hill, E.J.; Schneider, L.S.; Graff-Radford, N.R.; Mori, H.; et al. High Soluble Amyloid-Β42 Predicts Normal Cognition in Amyloid-Positive Individuals with Alzheimer’s Disease-Causing Mutations. J. Alzheimers Dis. 2022, 90, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Tudorache, I.F.; Trusca, V.G.; Gafencu, A.V. Apolipoprotein E—A Multifunctional Protein with Implications in Various Pathologies as a Result of Its Structural Features. Comput. Struct. Biotechnol. J. 2017, 15, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Klickstein, J.; Cannavo, C.; Jackson, R.; Muzikansky, A.; Gandhi, S.; Urick, D.; Sargent, T.; Wrobleski, L.; Roe, A.D.; et al. Opposing Roles of Apolipoprotein E in Aging and Neurodegeneration. Life Sci. Alliance 2019, 2, e201900325. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2023 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E. Protective Effect of Apolipoprotein E Type 2 Allele for Late Onset Alzheimer Disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Liu, P.; Agrawal, A.; Yip, O.; Blumenfeld, J.; Traglia, M.; Kim, M.J.; Koutsodendris, N.; Rao, A.; Grone, B.; et al. The APOE-R136S Mutation Protects against APOE4-Driven Tau Pathology, Neurodegeneration and Neuroinflammation. Nat. Neurosci. 2023, 26, 2104–2121. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Barber, R.C.; Philips, N.; Johnson, L.A.; Hall, J.R.; Subasinghe, K.; Petersen, M.; Toga, A.W.; Yaffe, K.; Rissman, R.A.; et al. The Link between APOE4 Presence and Neuropsychological Test Performance among Mexican Americans and Non-Hispanic Whites of the Multiethnic Health & Aging Brain Study—Health Disparities Cohort. Dement. Geriatr. Cogn. Disord. 2022, 51, 26–31. [Google Scholar] [CrossRef]
- Kerchner, G.A.; Berdnik, D.; Shen, J.C.; Bernstein, J.D.; Fenesy, M.C.; Deutsch, G.K.; Wyss-Coray, T.; Rutt, B.K. APOE Ε4 Worsens Hippocampal CA1 Apical Neuropil Atrophy and Episodic Memory. Neurology 2014, 82, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N.; Malek-Ahmadi, M.; Dugger, B.N.; Lee, K.; Sue, L.I.; Serrano, G.; Walker, D.G.; Davis, K.; Jacobson, S.A.; Beach, T.G. The Influence of Apolipoprotein E Genotype on Regional Pathology in Alzheimer’s Disease. BMC Neurol. 2013, 13, 44. [Google Scholar] [CrossRef]
- Shinohara, M.; Sato, N. The Roles of Apolipoprotein E, Lipids, and Glucose in the Pathogenesis of Alzheimer’s Disease. In Diabetes Mellitus; Nakabeppu, Y., Ninomiya, T., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1128, pp. 85–101. [Google Scholar] [CrossRef]
- Morgen, K.; Frölich, L.; Tost, H.; Plichta, M.M.; Kölsch, H.; Rakebrandt, F.; Rienhoff, O.; Jessen, F.; Peters, O.; Jahn, H.; et al. APOE-Dependent Phenotypes in Subjects with Mild Cognitive Impairment Converting to Alzheimer’s Disease. J. Alzheimers Dis. 2013, 37, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Sluimer, J.D.; Vrenken, H.; Blankenstein, M.A.; Fox, N.C.; Scheltens, P.; Barkhof, F.; Van Der Flier, W.M. Whole-Brain Atrophy Rate in Alzheimer Disease: Identifying Fast Progressors. Neurology 2008, 70 Pt 2, 1836–1841. [Google Scholar] [CrossRef]
- Van Der Flier, W.M.; Pijnenburg, Y.A.; Fox, N.C.; Scheltens, P. Early-Onset versus Late-Onset Alzheimer’s Disease: The Case of the Missing APOE Ɛ4 Allele. Lancet Neurol. 2011, 10, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Van Der Vlies, A.E.; Koedam, E.L.G.E.; Pijnenburg, Y.A.L.; Twisk, J.W.R.; Scheltens, P.; Van Der Flier, W.M. Most Rapid Cognitive Decline in APOE Ε4 Negative Alzheimer’s Disease with Early Onset. Psychol. Med. 2009, 39, 1907–1911. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Betensky, R.A.; Hyman, B.T. APOE Ε2 Is Associated with Milder Clinical and Pathological A Lzheimer Disease. Ann. Neurol. 2015, 77, 917–929. [Google Scholar] [CrossRef]
- William Rebeck, G.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in Sporadic Alzheimer’s Disease: Allelic Variation and Receptor Interactions. Neuron 1993, 11, 575–580. [Google Scholar] [CrossRef]
- Jones, P.B.; Adams, K.W.; Rozkalne, A.; Spires-Jones, T.L.; Hshieh, T.T.; Hashimoto, T.; Von Armin, C.A.F.; Mielke, M.; Bacskai, B.J.; Hyman, B.T. Apolipoprotein E: Isoform Specific Differences in Tertiary Structure and Interaction with Amyloid-β in Human Alzheimer Brain. PLoS ONE 2011, 6, e14586. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE Influences Amyloid-β (Aβ) Clearance despite Minimal apoE/Aβ Association in Physiological Conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Strickland, M.R.; Holtzman, D.M. Dr. Jekyll and Mr. Hyde: ApoE Explains Opposing Effects of Neuronal LRP1. J. Clin. Investig. 2019, 129, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.A.; Tai, L.M.; LaDu, M.J.; Rebeck, G.W. Human APOE4 Increases Microglia Reactivity at Aβ Plaques in a Mouse Model of Aβ Deposition. J. Neuroinflamm. 2014, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Atagi, Y.; Liu, C.-C.; Painter, M.M.; Chen, X.-F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-J.; Wang, M.; Li, R.-Y.; Wei, T.; Yang, H.-C.; Yin, Y.-S.; Mi, Y.-X.; Qin, Q.; Tang, Y. TREM2 and Microglia Contribute to the Synaptic Plasticity: From Physiology to Pathology. Mol. Neurobiol. 2023, 60, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Keene, C.D.; Cudaback, E.; Li, X.; Montine, K.S.; Montine, T.J. Apolipoprotein E Isoforms and Regulation of the Innate Immune Response in Brain of Patients with Alzheimer’s Disease. Curr. Opin. Neurobiol. 2011, 21, 920–928. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, S.M.; Kirschner, A.W.T.; Lindner, K.; Rust, R.; Budny, V.; Wolski, W.E.; Gavin, A.-C.; Nitsch, R.M.; Tackenberg, C. APOE2, E3, and E4 Differentially Modulate Cellular Homeostasis, Cholesterol Metabolism, and Inflammatory Response in Isogenic iPSC-Derived Astrocytes. Stem Cell Rep. 2022, 17, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, L.-M.; Wu, J. Cross-Talk between Apolipoprotein E and Cytokines. Mediators Inflamm. 2011, 2011, 949072. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Shinohara, M.; Yamazaki, A.; Ren, Y.; Asmann, Y.W.; Kanekiyo, T.; Bu, G. ApoE (Apolipoprotein E) in Brain Pericytes Regulates Endothelial Function in an Isoform-Dependent Manner by Modulating Basement Membrane Components. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 128–144. [Google Scholar] [CrossRef]
- Jackson, R.J.; Meltzer, J.C.; Nguyen, H.; Commins, C.; Bennett, R.E.; Hudry, E.; Hyman, B.T. APOE4 Derived from Astrocytes Leads to Blood–Brain Barrier Impairment. Brain 2022, 145, 3582–3593. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E Controls Cerebrovascular Integrity via Cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Liu, C.; Yamazaki, Y.; Heckman, M.G.; Martens, Y.A.; Jia, L.; Yamazaki, A.; Diehl, N.N.; Zhao, J.; Zhao, N.; DeTure, M.; et al. Tau and Apolipoprotein E Modulate Cerebrovascular Tight Junction Integrity Independent of Cerebral Amyloid Angiopathy in Alzheimer’s Disease. Alzheimers Dement. 2020, 16, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 Leads to Blood–Brain Barrier Dysfunction Predicting Cognitive Decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood–Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Zhao, J.; Fu, Y.; Inoue, Y.; Ren, Y.; Chen, Y.; Doss, S.V.; Shue, F.; Jeevaratnam, S.; Bastea, L.; et al. Peripheral apoE4 Enhances Alzheimer’s Pathology and Impairs Cognition by Compromising Cerebrovascular Function. Nat. Neurosci. 2022, 25, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood–Brain Barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.E.; Esparza, T.J.; Lewis, H.A.; Kim, E.; Mac Donald, C.L.; Sullivan, P.M.; Brody, D.L. Human Apolipoprotein E4 Worsens Acute Axonal Pathology but Not Amyloid-β Immunoreactivity After Traumatic Brain Injury in 3×TG-AD Mice. J. Neuropathol. Exp. Neurol. 2013, 72, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Tesseur, I.; Kekonius, L.; Wyss-Coray, T.; Fish, J.D.; Masliah, E.; Hopkins, P.C.; Scearce-Levie, K.; et al. Carboxyl-Terminal-Truncated Apolipoprotein E4 Causes Alzheimer’s Disease-like Neurodegeneration and Behavioral Deficits in Transgenic Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10966–10971. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease Neuroimaging Initiative; Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; et al. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.-C.; Van Ingelgom, A.J.; Linares, C.; Kurti, A.; Knight, J.A.; Heckman, M.G.; Diehl, N.N.; Shinohara, M.; Martens, Y.A.; et al. APOE Ε2 Is Associated with Increased Tau Pathology in Primary Tauopathy. Nat. Commun. 2018, 9, 4388. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 Is a Master Regulator of Tau Uptake and Spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Xian, X.; Pohlkamp, T.; Durakoglugil, M.S.; Wong, C.H.; Beck, J.K.; Lane-Donovan, C.; Plattner, F.; Herz, J. Reversal of ApoE4-Induced Recycling Block as a Novel Prevention Approach for Alzheimer’s Disease. eLife 2018, 7, e40048. [Google Scholar] [CrossRef]
- De Luca, V.; Orfei, M.D.; Gaudenzi, S.; Caltagirone, C.; Spalletta, G. Inverse Effect of the APOE Epsilon4 Allele in Late- and Early-Onset Alzheimer’s Disease. Eur. Arch. Psychiatry Clin. Neurosci. 2016, 266, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Valdez-Gaxiola, C.A.; Maciel-Cruz, E.J.; Hernández-Peña, R.; Dumois-Petersen, S.; Rosales-Leycegui, F.; Gallegos-Arreola, M.P.; Moreno-Ortiz, J.M.; Figuera, L.E. Potential Modifying Effect of the APOEε4 Allele on Age of Onset and Clinical Manifestations in Patients with Early-Onset Alzheimer’s Disease with and without a Pathogenic Variant in PSEN1 in a Sample of the Mexican Population. Int. J. Mol. Sci. 2023, 24, 15687. [Google Scholar] [CrossRef] [PubMed]
- Langella, S.; Barksdale, N.G.; Vasquez, D.; Aguillon, D.; Chen, Y.; Su, Y.; Acosta-Baena, N.; Acosta-Uribe, J.; Baena, A.Y.; Garcia-Ospina, G.; et al. Effect of Apolipoprotein Genotype and Educational Attainment on Cognitive Function in Autosomal Dominant Alzheimer’s Disease. Nat. Commun. 2023, 14, 5120. [Google Scholar] [CrossRef]
- Ryan, N.S.; Nicholas, J.M.; Weston, P.S.J.; Liang, Y.; Lashley, T.; Guerreiro, R.; Adamson, G.; Kenny, J.; Beck, J.; Chavez-Gutierrez, L.; et al. Clinical Phenotype and Genetic Associations in Autosomal Dominant Familial Alzheimer’s Disease: A Case Series. Lancet Neurol. 2016, 15, 1326–1335. [Google Scholar] [CrossRef]
- Van Der Vlies, A.E.; Pijnenburg, Y.A.L.; Koene, T.; Klein, M.; Kok, A.; Scheltens, P.; Van Der Flier, W.M. Cognitive Impairment in Alzheimer’s Disease Is Modified by APOE Genotype. Dement. Geriatr. Cogn. Disord. 2007, 24, 98–103. [Google Scholar] [CrossRef]
- Smits, L.L.; Pijnenburg, Y.A.L.; Van Der Vlies, A.E.; Koedam, E.L.G.E.; Bouwman, F.H.; Reuling, I.E.W.; Scheltens, P.; Van Der Flier, W.M. Early Onset APOE E4-Negative Alzheimer’s Disease Patients Show Faster Cognitive Decline on Non-Memory Domains. Eur. Neuropsychopharmacol. 2015, 25, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Marra, C.; Bizzarro, A.; Daniele, A.; De Luca, L.; Ferraccioli, M.; Valenza, A.; Brahe, C.; Tiziano, F.D.; Gainotti, G.; Masullo, C. Apolipoprotein E Ε4 Allele Differently Affects the Patterns of Neuropsychological Presentation in Early- and Late-Onset Alzheimer’s Disease Patients. Dement. Geriatr. Cogn. Disord. 2004, 18, 125–131. [Google Scholar] [CrossRef]
- Lehtovirta, M.; Soininen, H.; Helisalmi, S.; Mannermaa, A.; Helkala, E.-L.; Hartikainen, P.; Hanninen, T.; Ryynanen, M.; Riekkinen, P.J. Clinical and Neuropsychological Characteristics in Familial and Sporadic Alzheimer’s Disease: Relation to Apolipoprotein E Polymorphism. Neurology 1996, 46, 413–419. [Google Scholar] [CrossRef]
- Almkvist, O.; Johansson, C.; Laffita-Mesa, J.; Thordardottir, S.; Graff, C. APOE Ε4 Influences Cognitive Decline Positively in APP and Negatively in PSEN1 Mutation Carriers with Autosomal-dominant Alzheimer’s Disease. Eur. J. Neurol. 2022, 29, 3580–3589. [Google Scholar] [CrossRef]
- Almkvist, O.; Graff, C. The APOE Ε4 Allele Affects Cognitive Functions Differently in Carriers of APP Mutations Compared to Carriers of PSEN1 Mutations in Autosomal-Dominant Alzheimer’s Disease. Genes 2021, 12, 1954. [Google Scholar] [CrossRef] [PubMed]
- Gharbi-Meliani, A.; Dugravot, A.; Sabia, S.; Regy, M.; Fayosse, A.; Schnitzler, A.; Kivimäki, M.; Singh-Manoux, A.; Dumurgier, J. The Association of APOE Ε4 with Cognitive Function over the Adult Life Course and Incidence of Dementia: 20 Years Follow-up of the Whitehall II Study. Alzheimers Res. Ther. 2021, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Vélez, J.I.; Lopera, F.; Sepulveda-Falla, D.; Patel, H.R.; Johar, A.S.; Chuah, A.; Tobón, C.; Rivera, D.; Villegas, A.; Cai, Y.; et al. APOE*E2 Allele Delays Age of Onset in PSEN1 E280A Alzheimer’s Disease. Mol. Psychiatry 2016, 21, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Raulin, A.-C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.-C. ApoE in Alzheimer’s Disease: Pathophysiology and Therapeutic Strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Korczyn, A.D.; Grinberg, L.T. Is Alzheimer Disease a Disease? Nat. Rev. Neurol. 2024, 20, 245–251. [Google Scholar] [CrossRef]
- Ellouze, I.; Sheffler, J.; Nagpal, R.; Arjmandi, B. Dietary Patterns and Alzheimer’s Disease: An Updated Review Linking Nutrition to Neuroscience. Nutrients 2023, 15, 3204. [Google Scholar] [CrossRef]
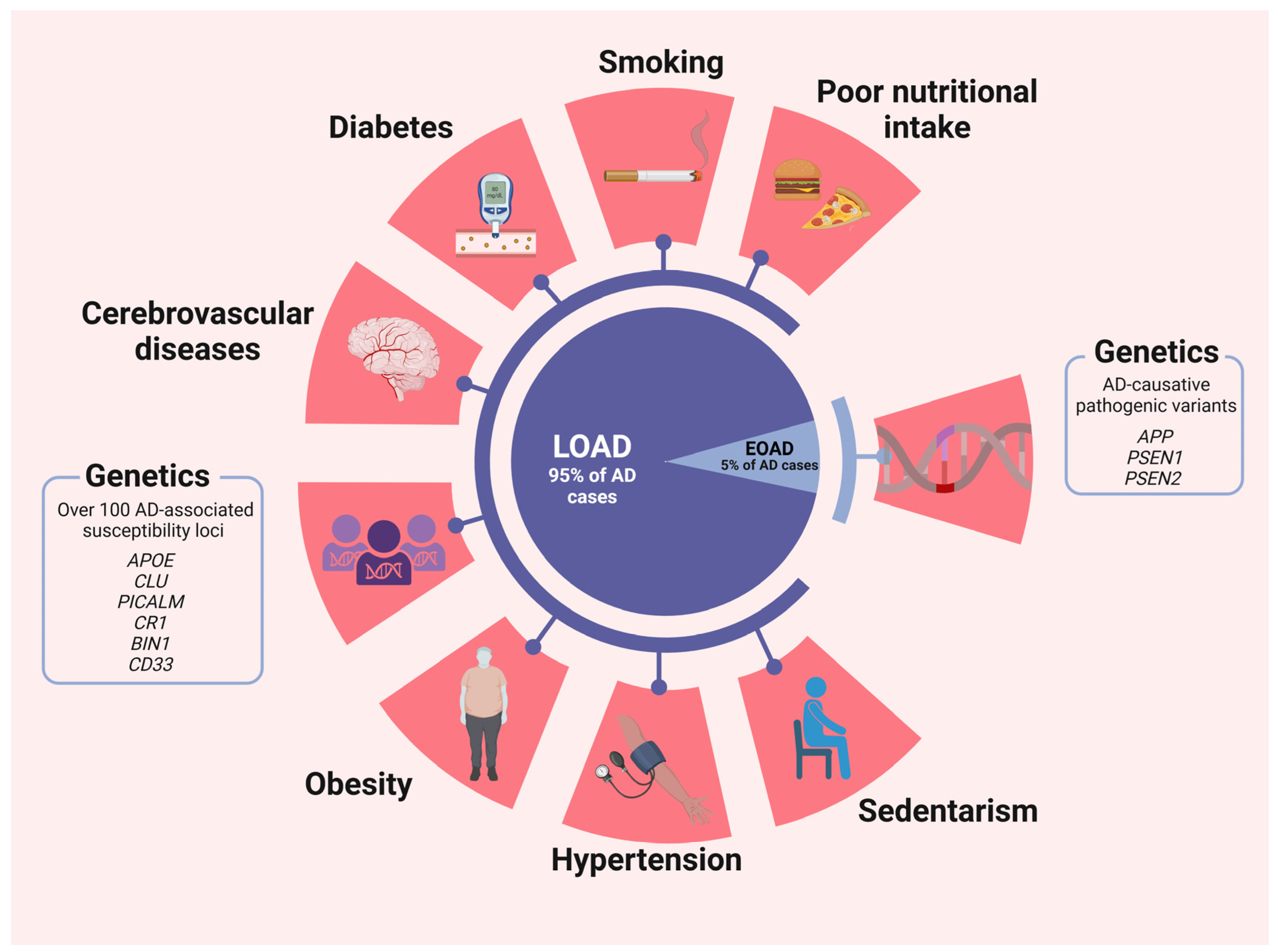
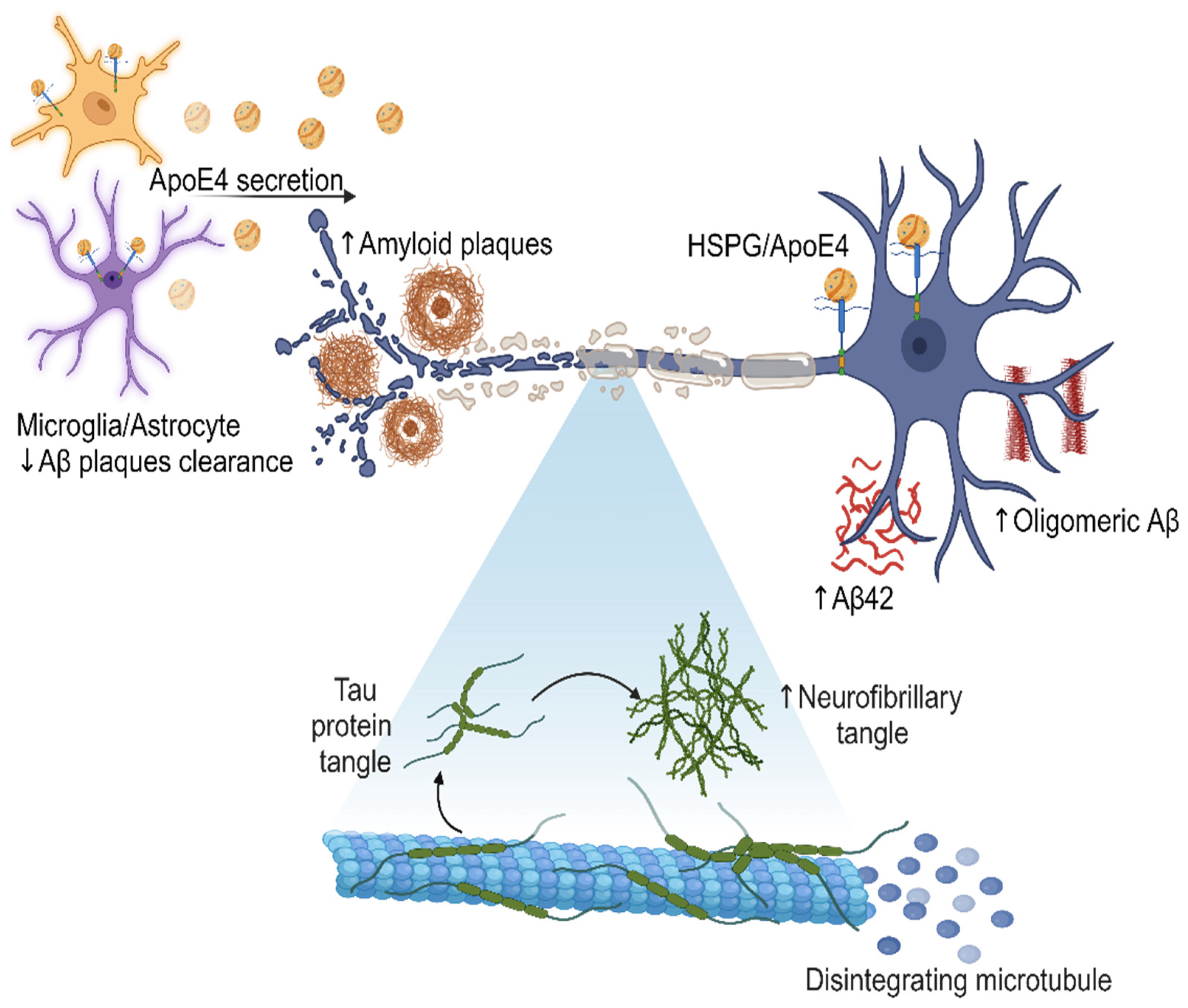
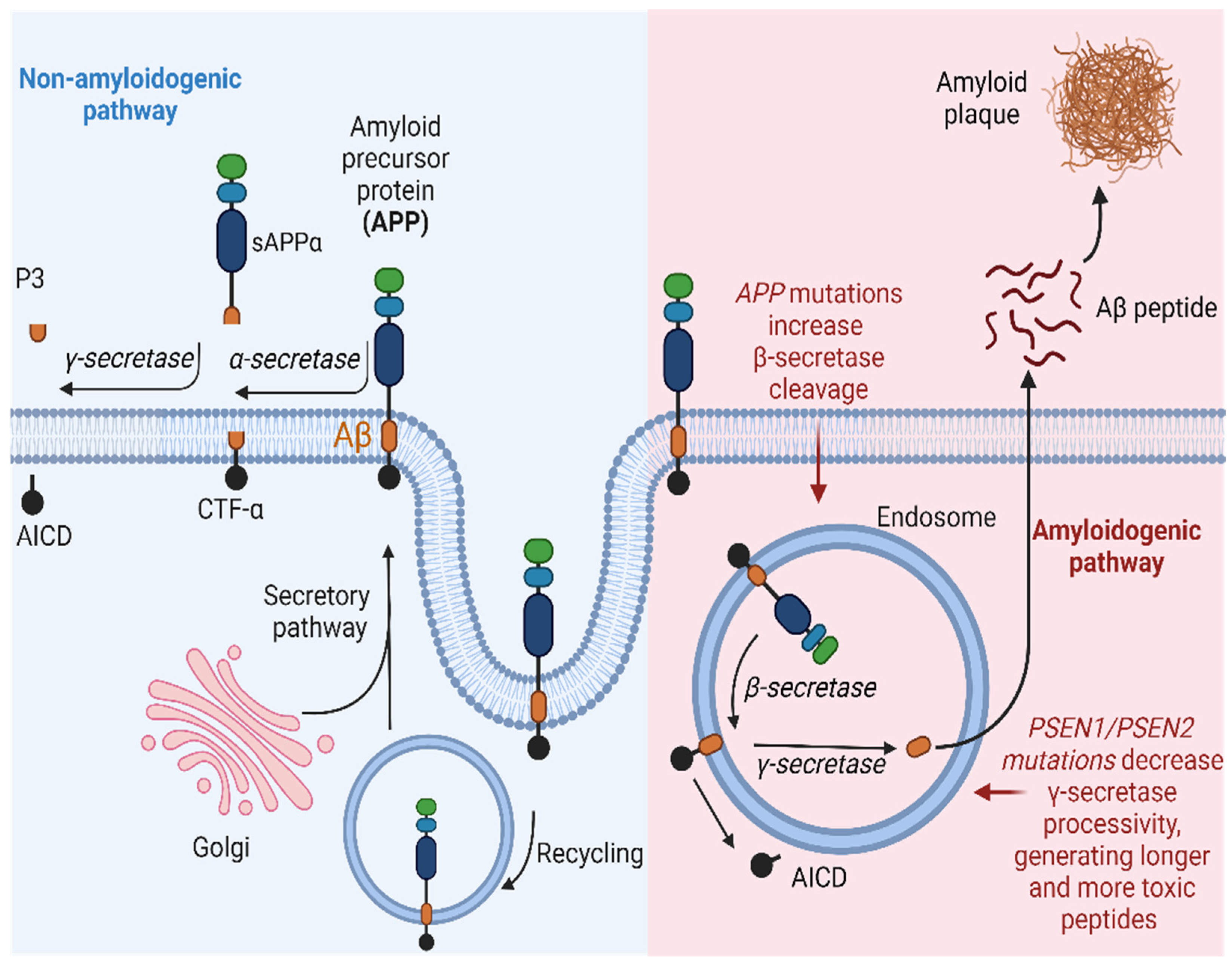
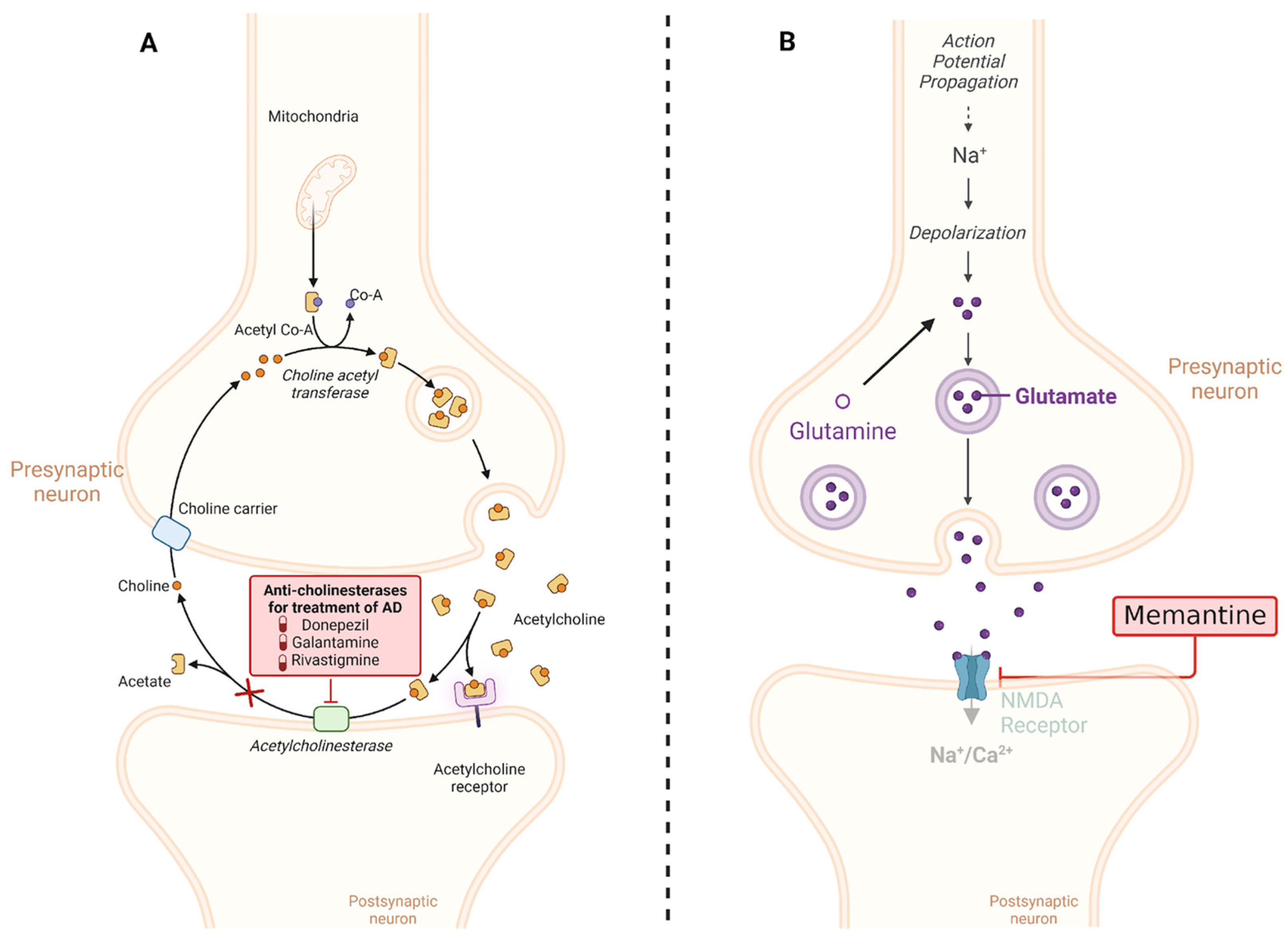
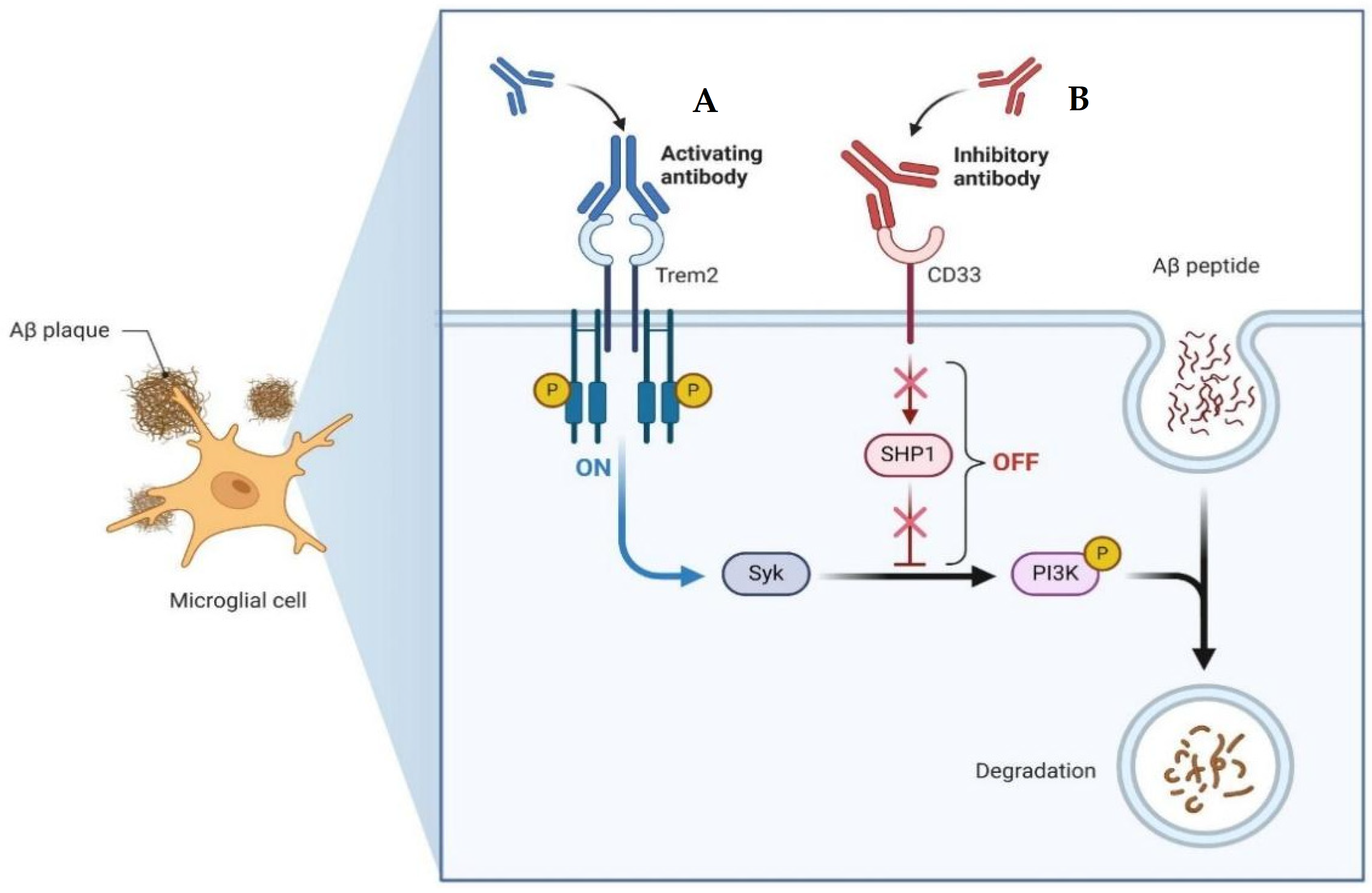
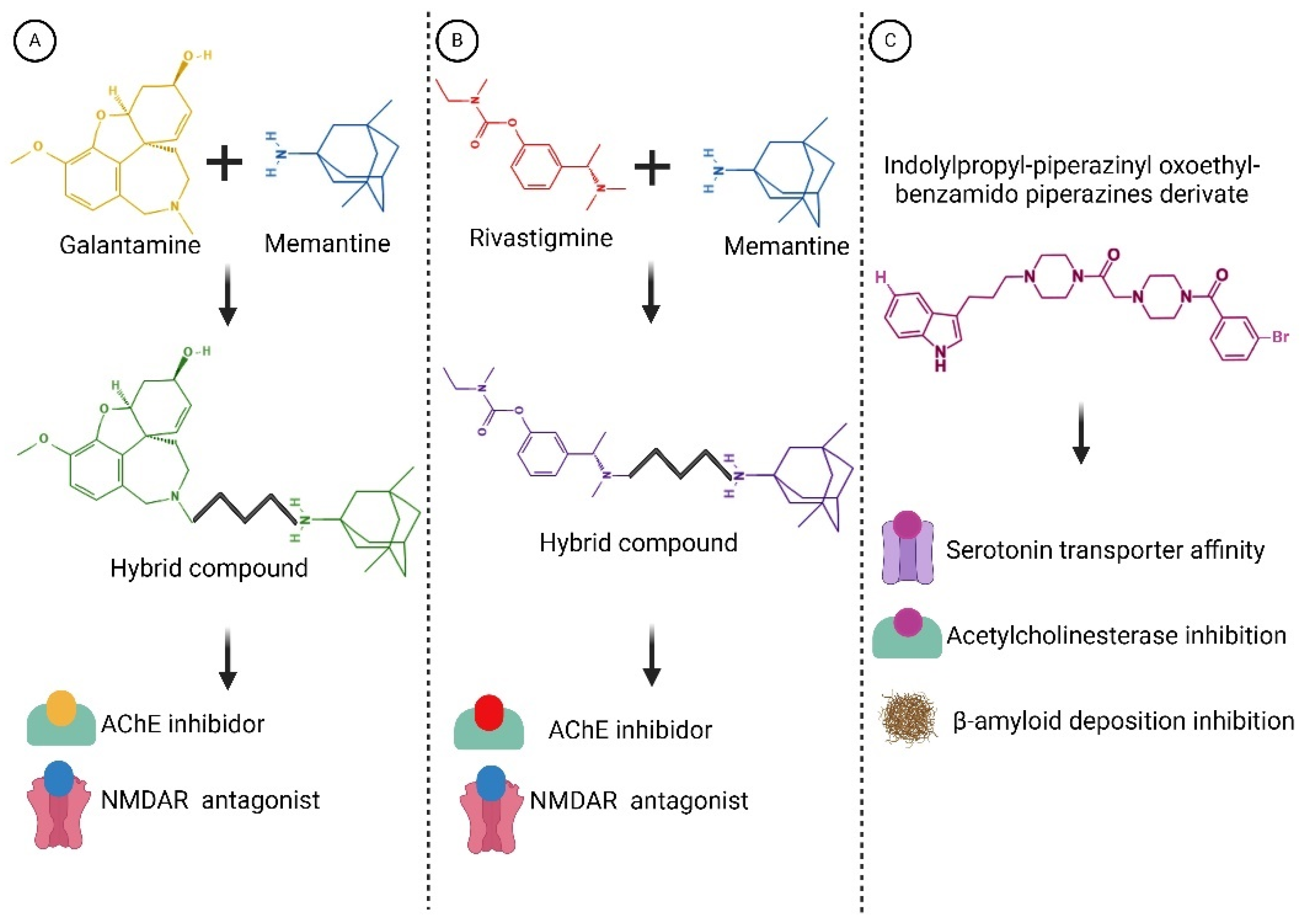
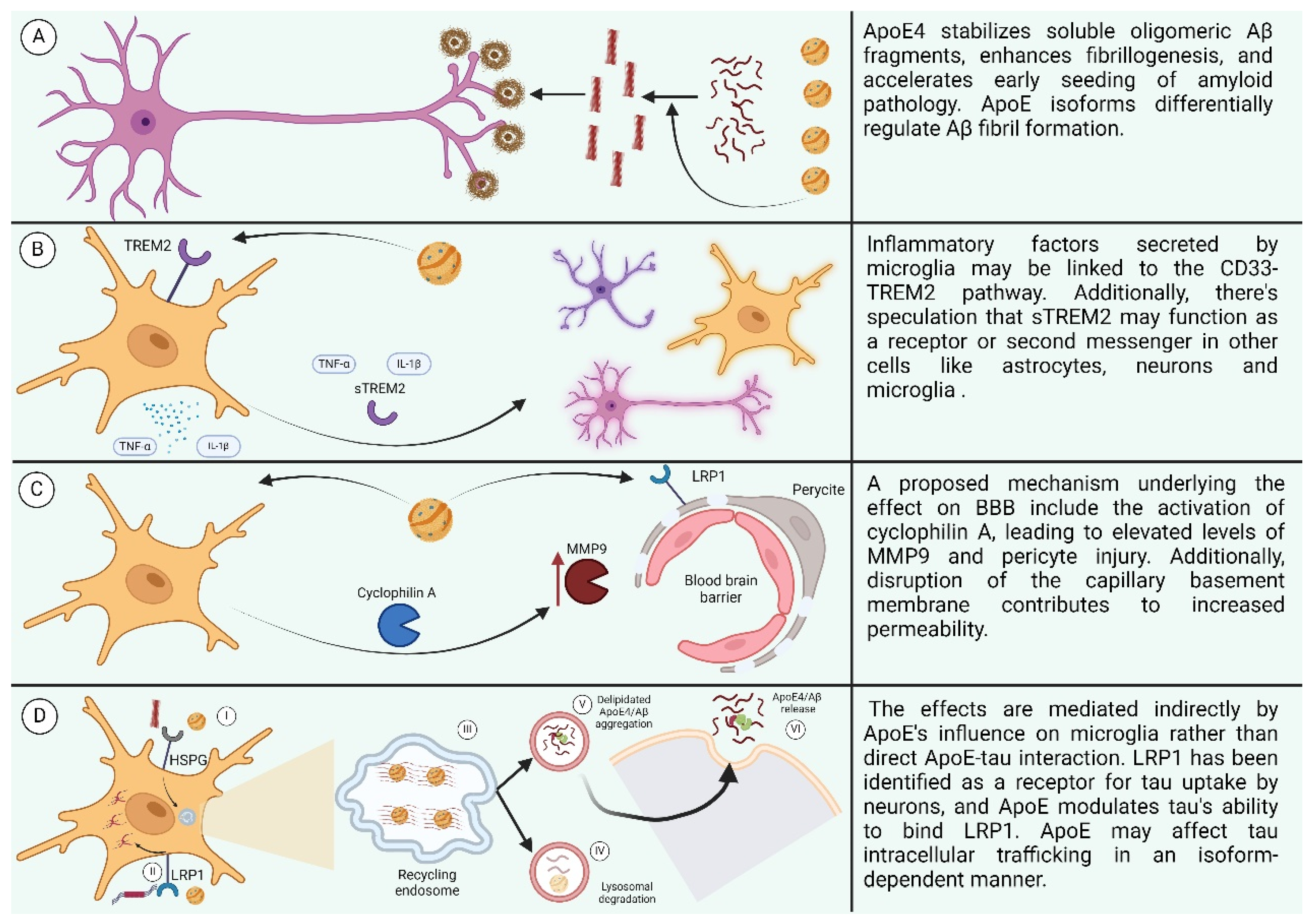
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).