
Novel Pathways of Oxidative and Nitrosative Inactivation of the Human MGMT Protein in Colon Cancer and Glioblastoma Cells: Increased Efficacy of Alkylating Agents In Vitro and In Vivo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Chemicals, and Antibodies
2.2. Preparation of Platinated Homoglutathione Disulfide (hGTX)
2.3. Assay of MGMT Activity
2.4. Western Blotting
2.5. Cell Cycle Analysis
2.6. Assay for Interstrand DNA Cross-Linking in Tumor Cells
2.7. Detection of ROS Generation
2.8. Cell Survival Assays
2.9. Tumor Xenografts and Drug Efficacy Studies
2.10. Hematoxylin and Eosin Staining of Tissue Sections
2.11. Statistical Analysis
3. Results
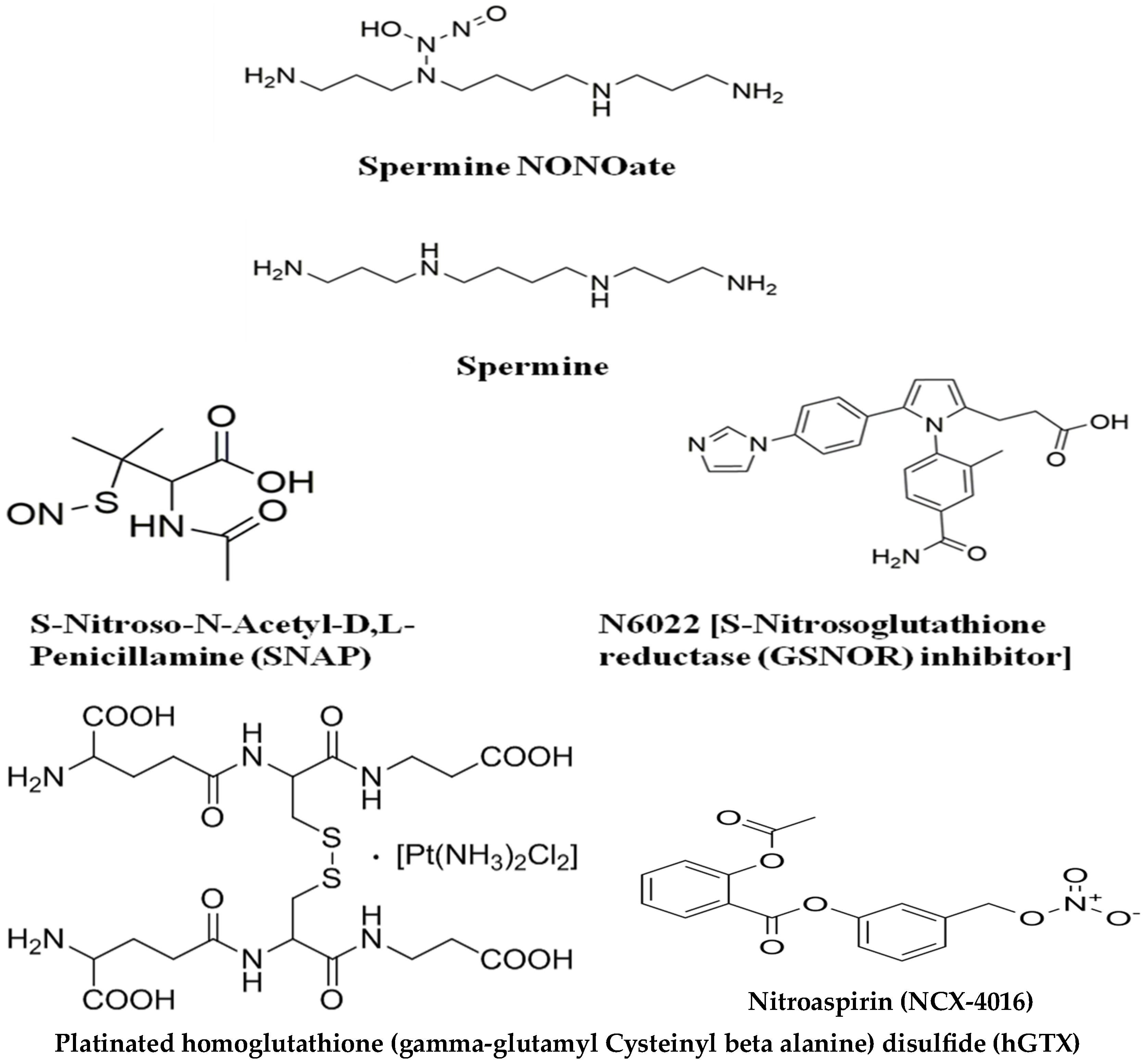
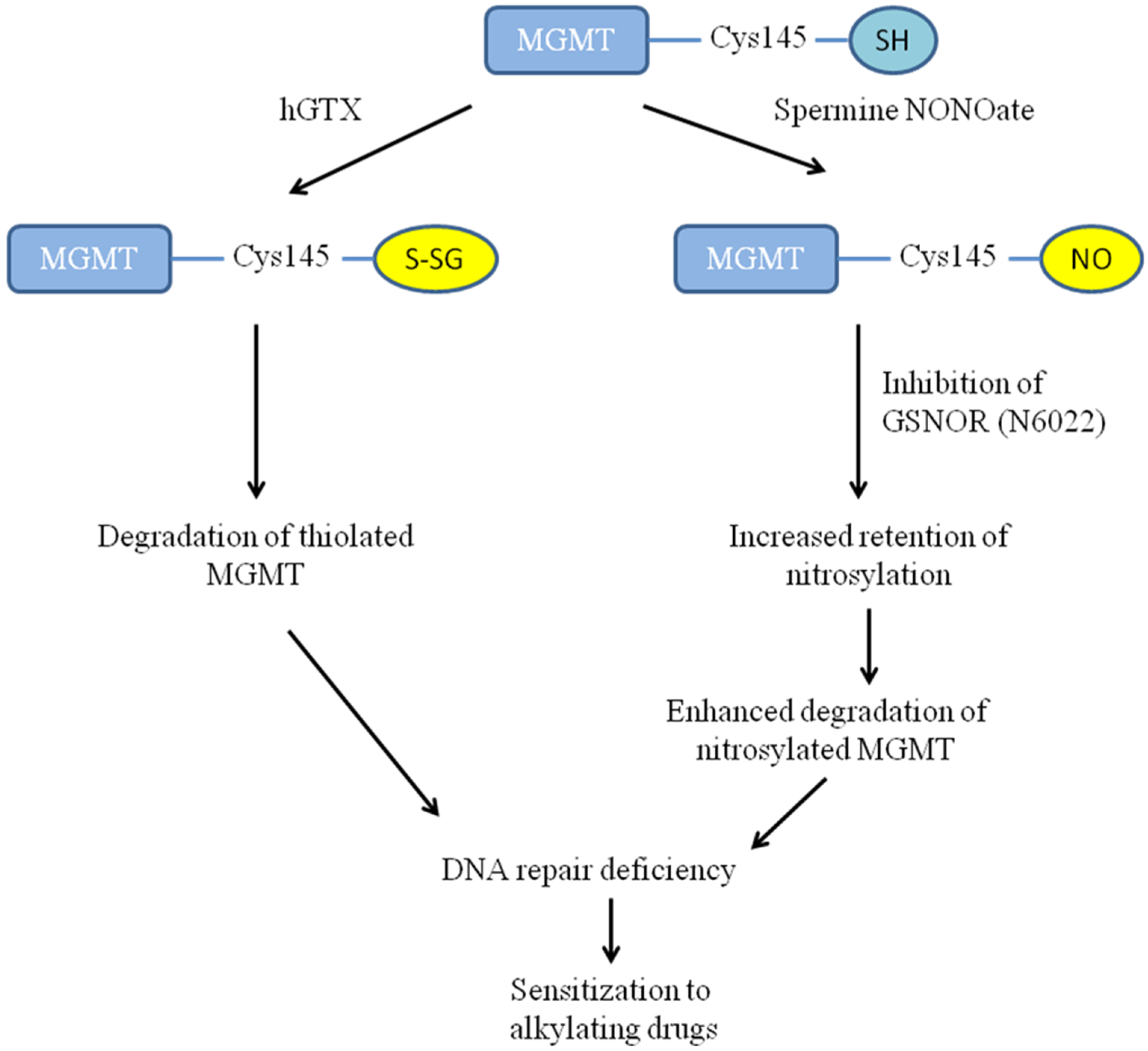
3.1. Overview of Compounds Used for Redox-Driven Inactivation of Human MGMT
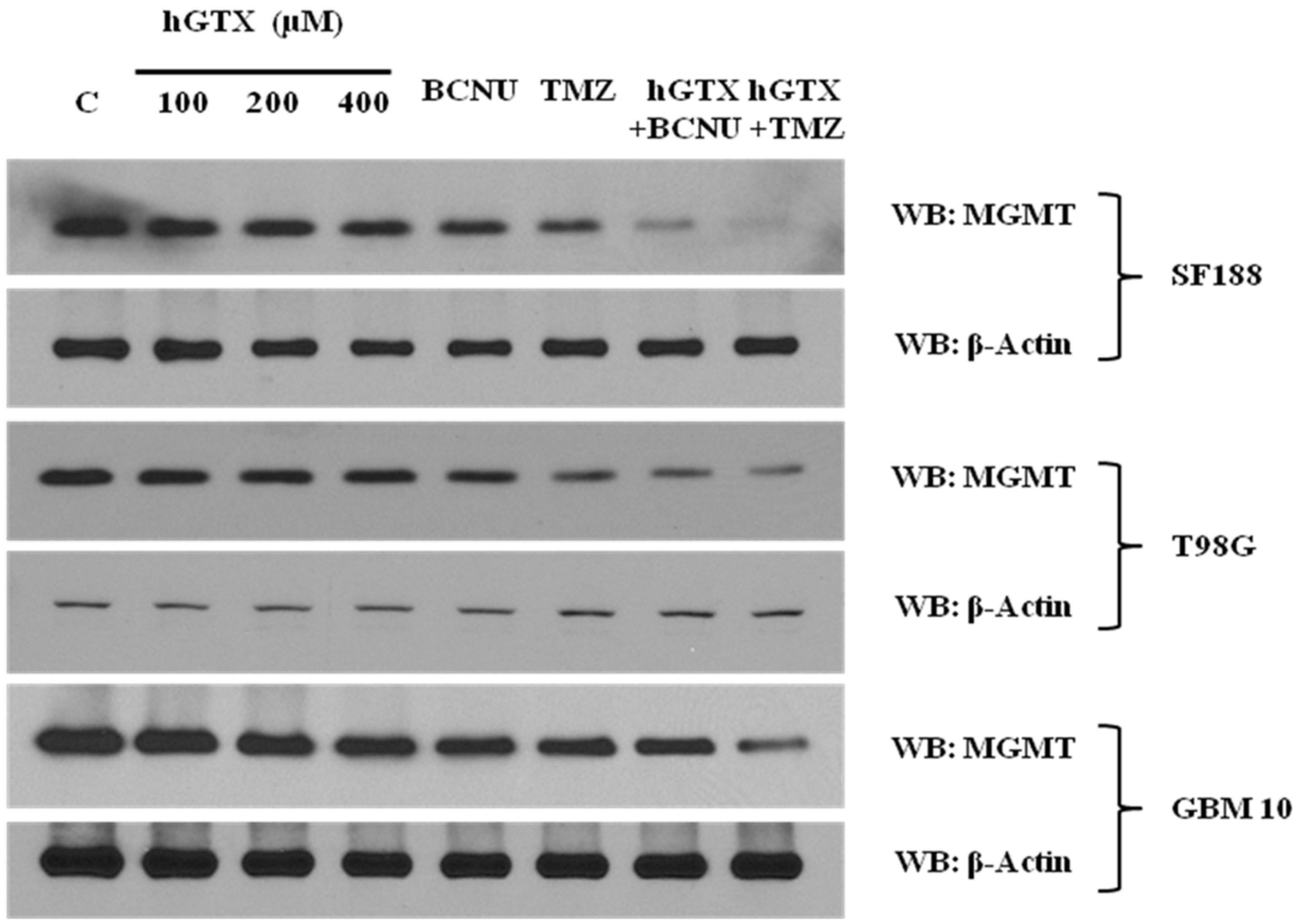
3.2. Treatment with hGTX and Other Nitrosylating Agents Both Alone and in Combination with Alkylating Drugs Resulted in MGMT Inhibition and Loss of MGMT Protein in Glioblastoma Cell Lines
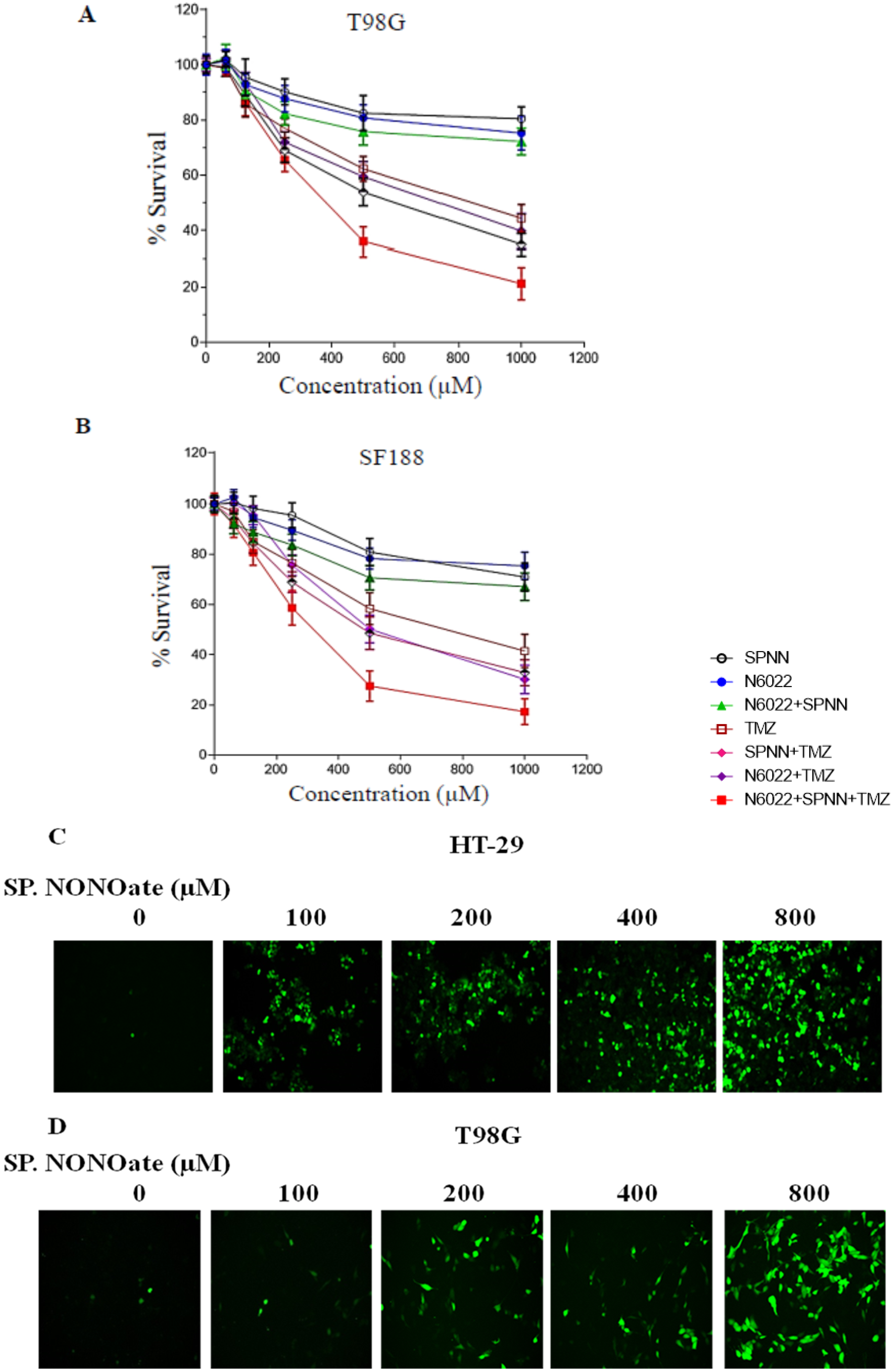
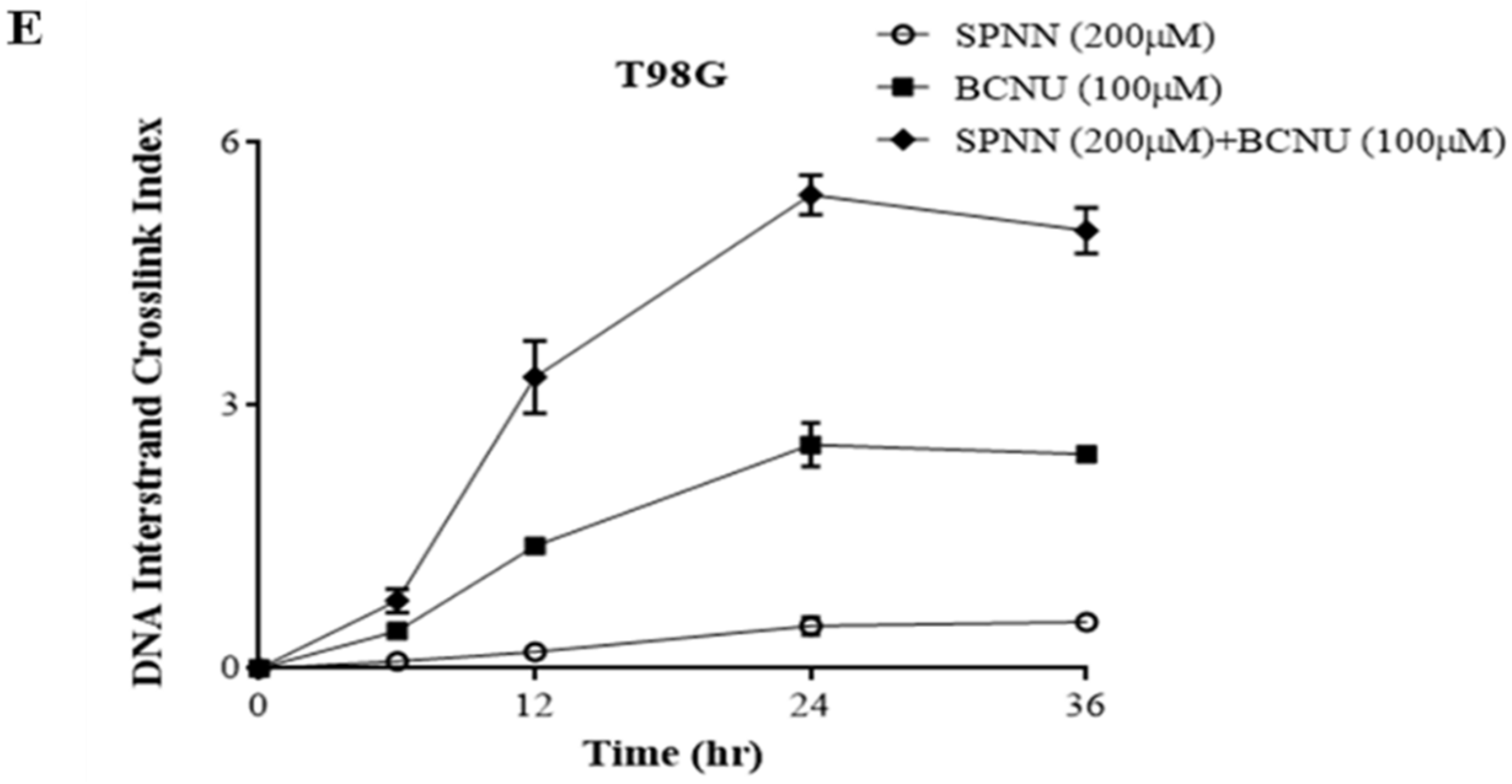
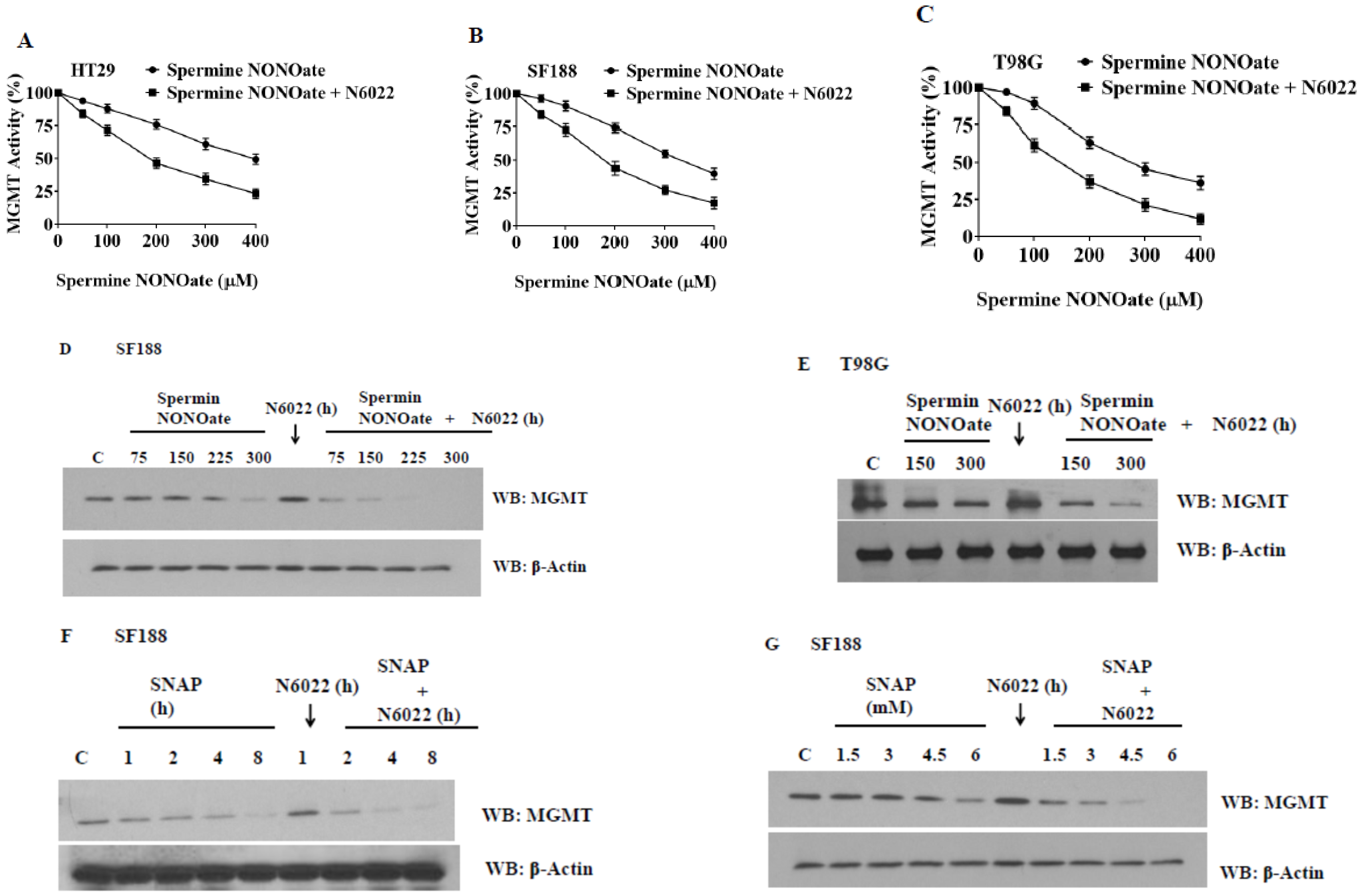
3.3. MGMT Inhibition by Spermine NONOate Markedly Increases the Alkylation-DNA Damage and Greatly Potentiated the Cell-Killing in a Nitrosylation-Dependent Manner
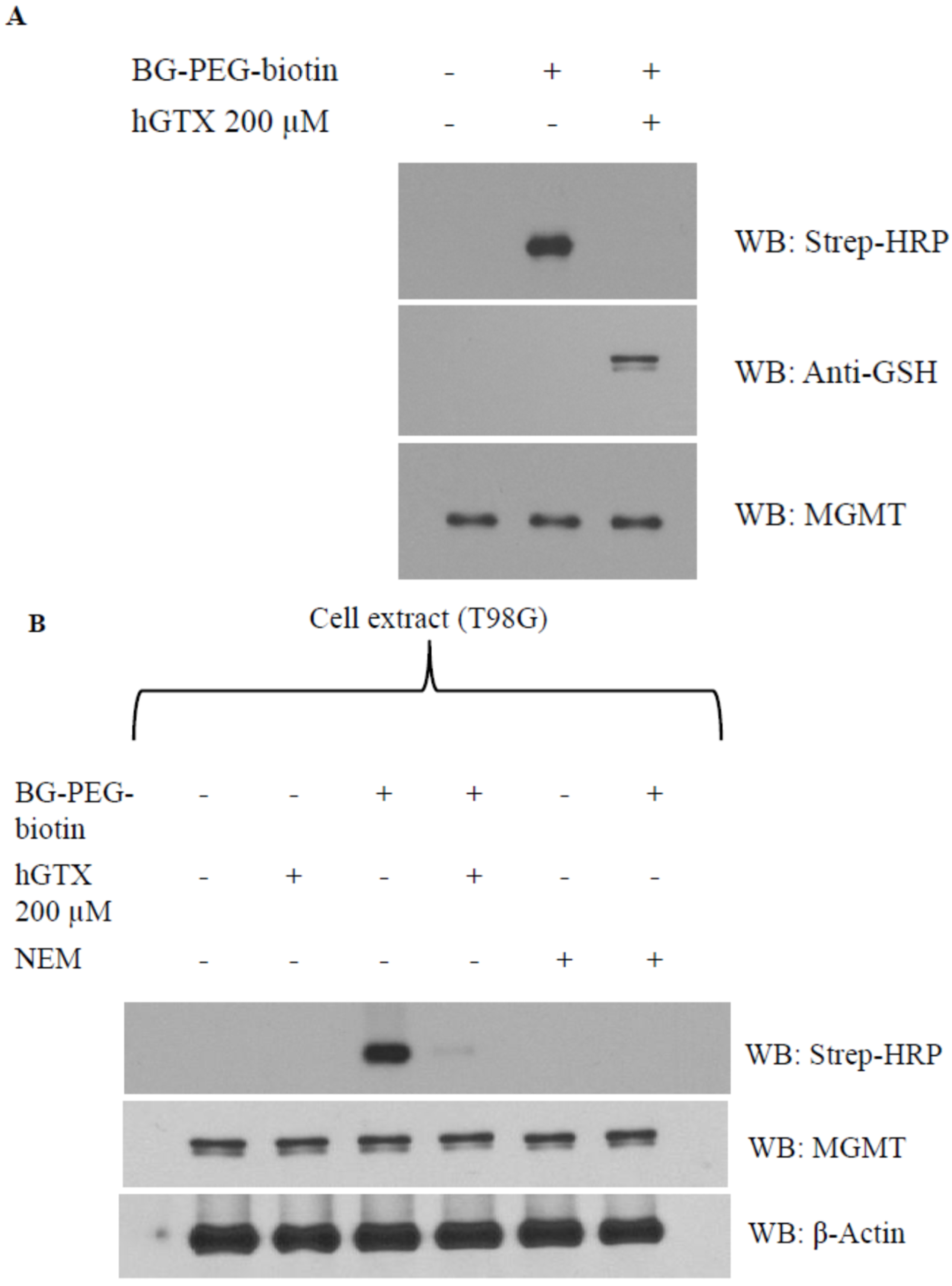
3.4. Evidence That Interaction of hGTX and Spermine NONOate with the Active Site Cysteine of MGMT Leads to DNA Repair Inactivation
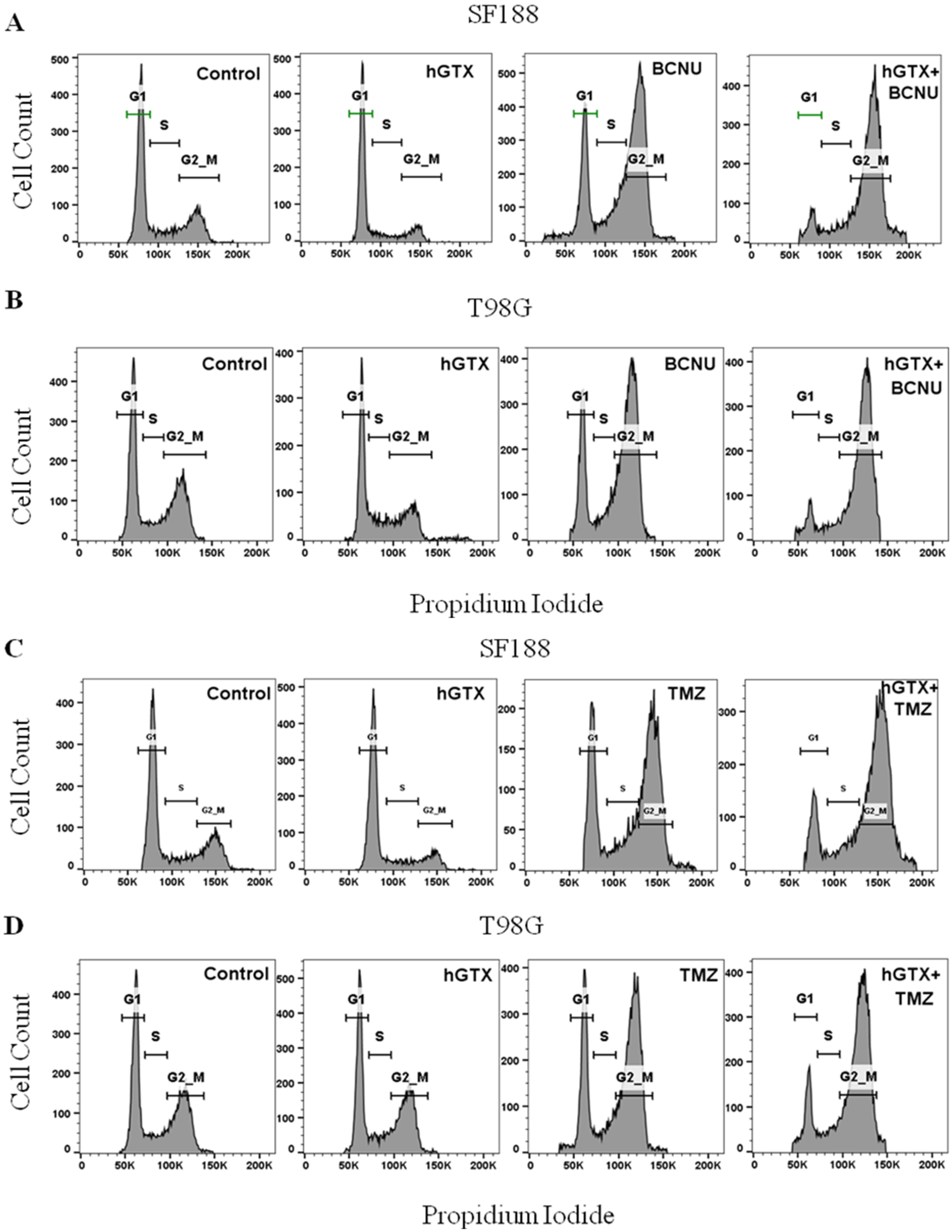
3.5. MGMT Inhibition by hGTX Enhances and Prolongs the BCNU and TMZ-Induced G2/M Cell Cycle Arrest in GBM Cells
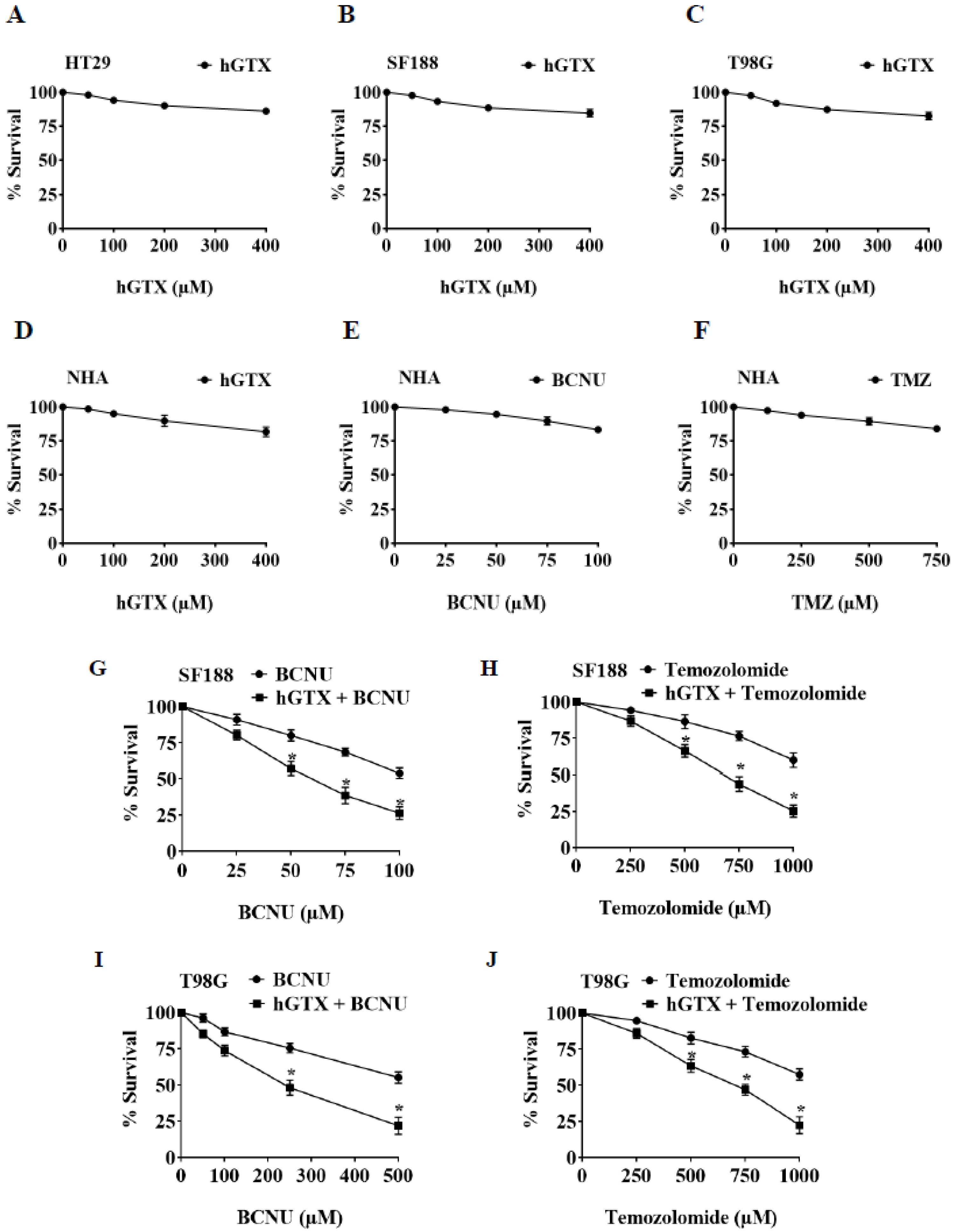
3.6. Brief Pretreatment with hGTX Sensitizes MGMT-Proficient Tumor Cells to Alkylating Agents
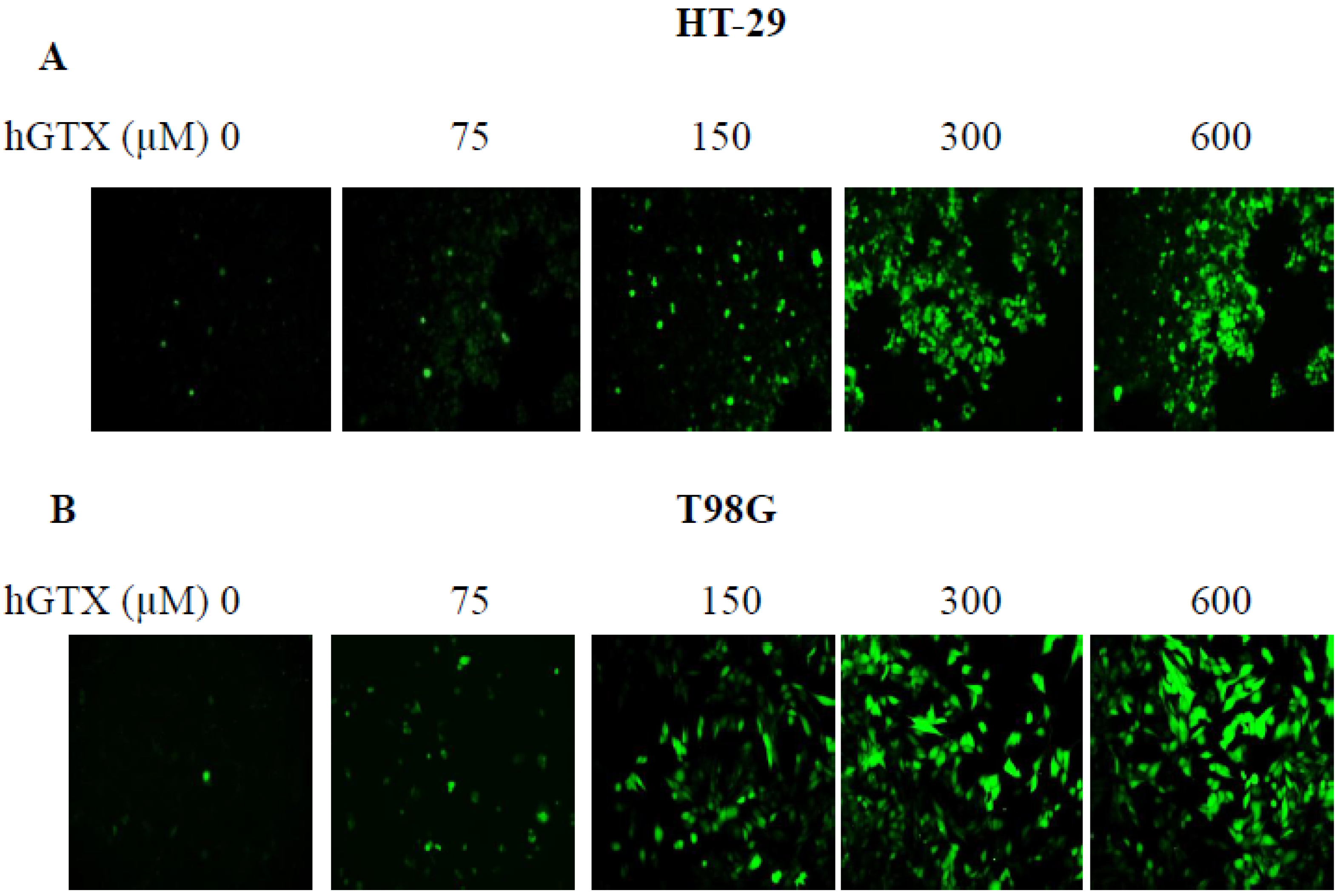
3.7. Treatment with Thiolating and Nitrosylating Agents Promote ROS Generation in MGMT-Proficient Cells
3.8. Evidence That GSNOR Inhibition Prolongs the MGMT Deficient State
3.9. Potentiation of Antitumor Efficacy of Alkylating Agents by Thiolating and Nitrosylating Agents in Xenograft Settings
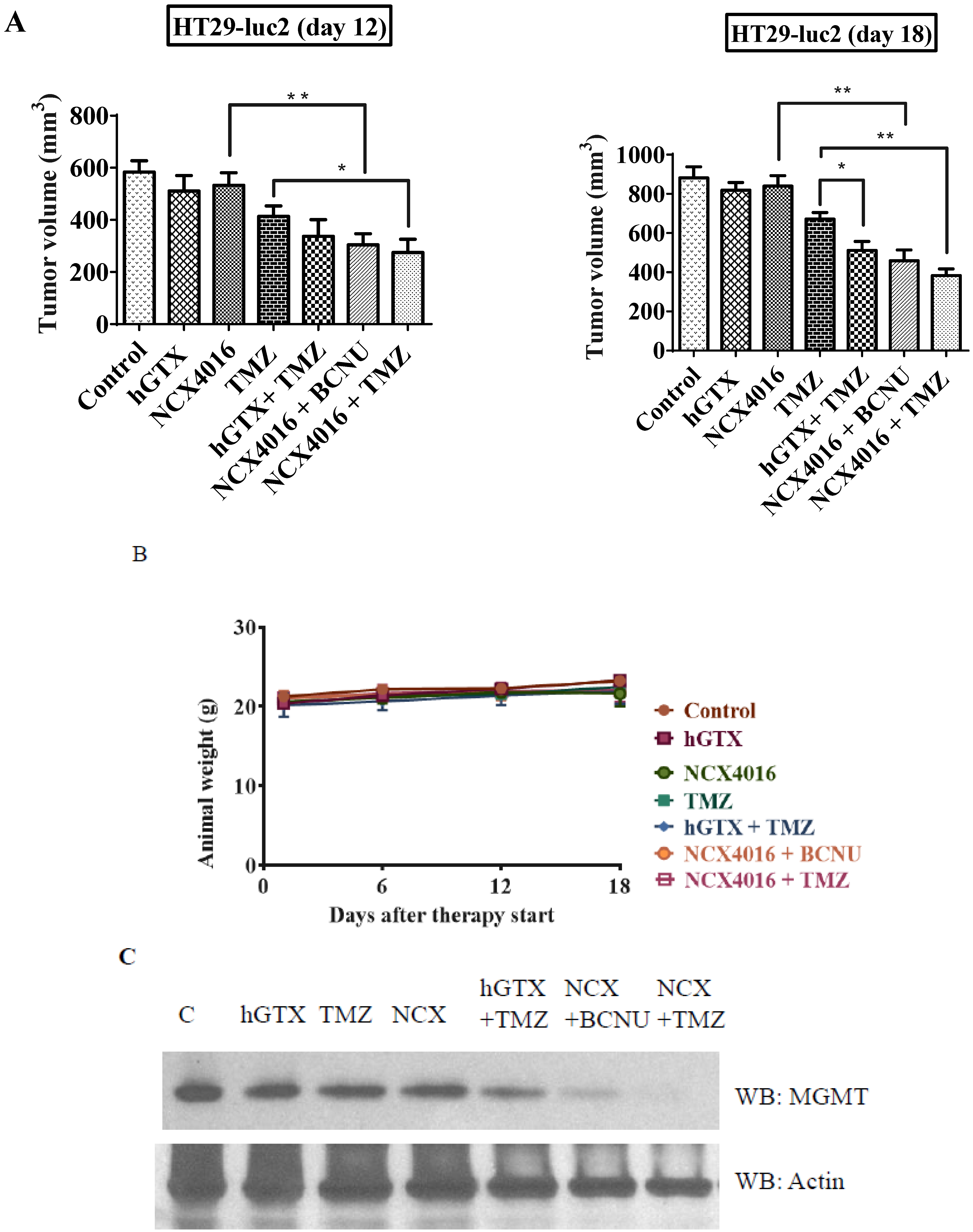

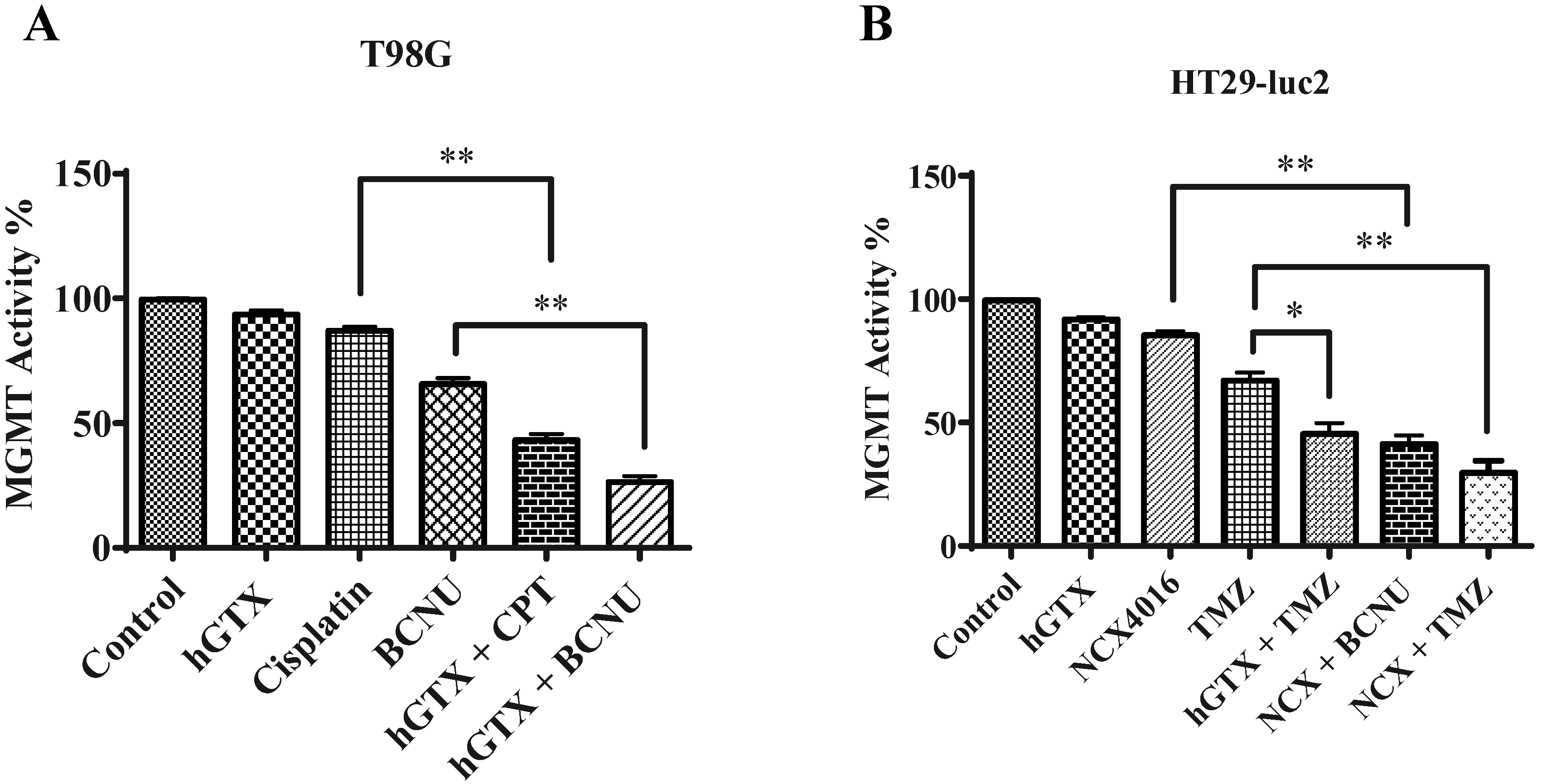


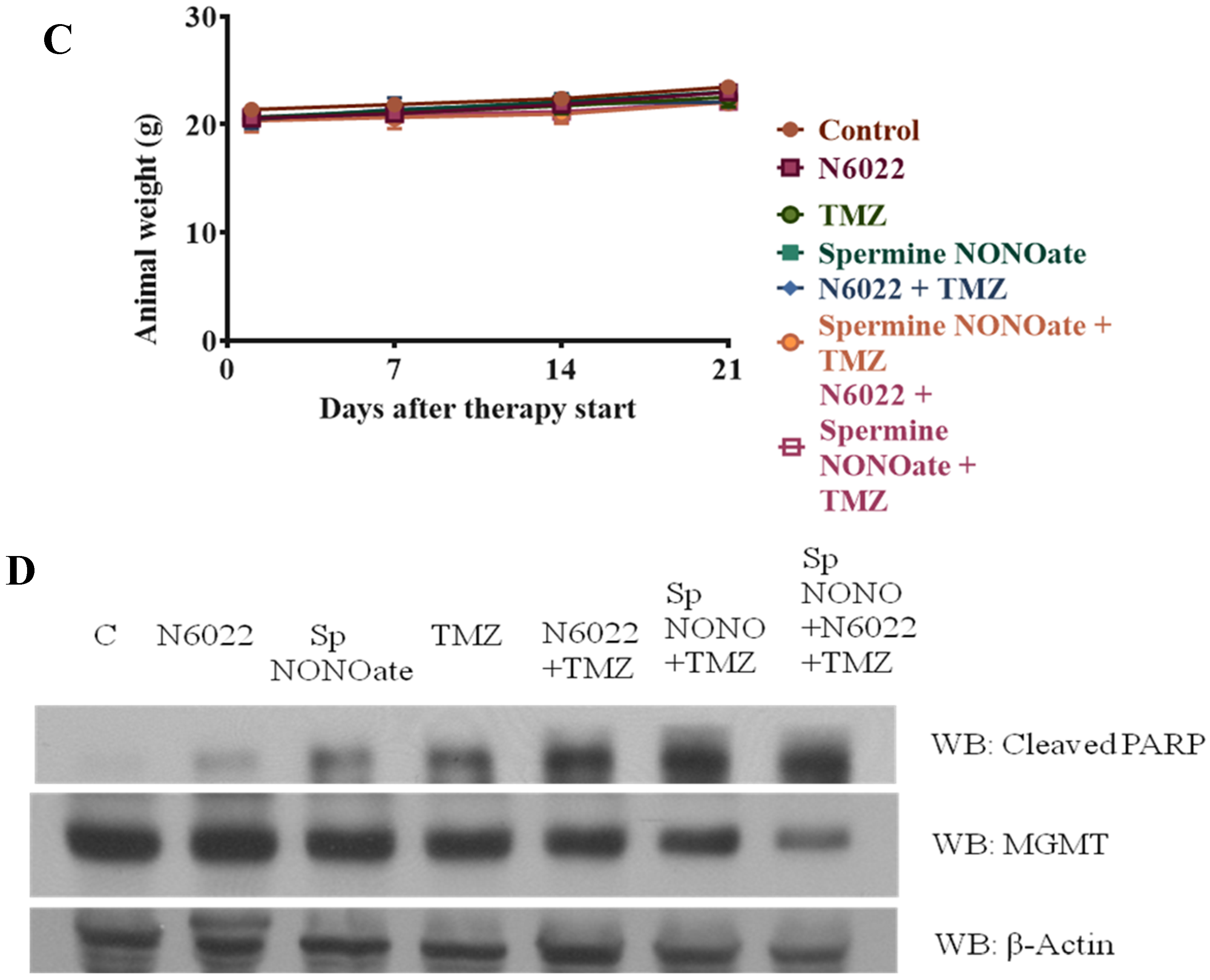
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pegg, A.E. Repair of O6-alkylguanine by alkyltransferases. Mutat. Res. 2000, 462, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Mishina, Y.; Duguid, E.M.; He, C. Direct reversal of DNA alkylation damage. Chem. Rev. 2006, 106, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Margison, G.P.; Santibáñez Koref, M.F.; Povey, A.C. Mechanisms of carcinogenicity/chemotherapy by O6-methylguanine. Mutagenesis 2002, 17, 483–487. [Google Scholar] [CrossRef]
- Srivenugopal, K.S.; Yuan, X.H.; Friedman, H.S.; Ali-Osman, F. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry 1996, 35, 1328–1334. [Google Scholar] [CrossRef]
- Gerson, S.L. Clinical relevance of MGMT in the treatment of cancer. J. Clin. Oncol. 2002, 20, 2388–2399. [Google Scholar] [CrossRef]
- Kaina, B.; Margison, G.P.; Christmann, M. Targeting O6-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell. Mol. Life Sci. 2010, 67, 3663–3681. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.A.; Njoku, M.C.; Dolan, M.E. Inactivation of O6-alkylguanine DNA alkyltransferase as a means to enhance chemotherapy. Cancer Treat. Rev. 2006, 32, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Kreklau, E.L.; Kurpad, C.; Williams, D.A.; Erickson, L.C. Prolonged inhibition of O(6)-methylguanine DNA methyltransferase in human tumor cells by O(6)-benzylguanine in vitro and in vivo. J. Pharmacol. Exp. Ther. 1999, 291, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Chinnasamy, D.; Fairbairn, L.J.; Neuenfeldt, J.; Treisman, J.S.; Hanson, J.P., Jr.; Margison, G.P.; Chinnasamy, N. Lentivirus-mediated expression of mutant MGMTP140K protects human CD34+ cells against the combined toxicity of O6-benzylguanine and 1,3-bis(2-chloroethyl)-nitrosourea or temozolomide. Hum. Gene Ther. 2004, 15, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Wiest, L.; Mummert, C.; Stine, L.; Moschel, R.C.; Dolan, M.E. Use of antibodies to human O6-alkylguanine-DNA alkyltransferase to study the content of this protein in cells treated with O6-benzylguanine or N-methyl-N_-nitro-Nnitrosoguanidine. Carcinogenesis 1991, 12, 1679–1683. [Google Scholar] [CrossRef]
- Kanugula, S.; Goodtzova, K.; Pegg, A.E. Probing of conformational changes in human O6-alkylguanine-DNA alkyltransferase protein in its alkylated and DNA bound states by limited proteolysis. Biochem. J. 1998, 329, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Xu-Welliver, M.; Pegg, A.E. Degradation of alkylated form of the DNA repair protein, O6-alkylguanine-DNA alkyltransferase. Carcinogenesis 2002, 23, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Velu, C.S.; Bailey, N.I.; Srivenugopal, K.S. Phosphorylated MGMT in human tumors is insensitive to O6-benzylguanine: Identification of the conserved Tyr114 as a phosphorylation site. Proc. Am. Assoc. Cancer Res. 2005, 46, 1281. [Google Scholar]
- Velu, C.S.; Niture, S.K.; Bailey, N.I.; Srivenugopal, K.S. Posttranslational regulation of human MGMT by sumoylation in brain tumor cells. Proc. Am. Assoc. Cancer Res. 2004, 64, 137. [Google Scholar]
- Srivenugopal, K.S.; Rawat, A.; Niture, S.K.; Paranjpe, A.; Velu, C.; Venugopal, S.N.; Madala, H.R.; Basak, D.; Punganuru, S.R. Posttranslational Regulation of O(6)-Methylguanine-DNA Methyltransferase (MGMT) and New Opportunities for Treatment of Brain Cancers. Mini Rev. Med. Chem. 2016, 16, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Srivenugopal, K.S.; Mullapudi, S.R.S.; Shou, J.; Ali-Osman, F. Mg2+ and ATP-dependent degradation of recombinant O6-methylguanine-DNA methyltransferase protein in human tumor cell extracts. Proc. Am. Assoc. Cancer Res. 2001, 42, 572. [Google Scholar]
- Velu, C.S.; Niture, S.K.; Bailey, N.I.; Srivenugopal, K.S. MGMT protein turnover in human tumors is mediated by the ubiquitinproteasome pathway: Phosphorylation-dependent ub-conjugation by the Skp2-SCF complex. Proc. Am. Assoc. Cancer Res. 2005, 65, 1283. [Google Scholar]
- Guengerich, F.P.; Fang, Q.; Liu, L.; Hachey, D.L.; Pegg, A.E. O6-alkylguanine-DNA alkyltransferase: Low pKa and high reactivity of cysteine 145. Biochemistry 2003, 42, 10965–10970. [Google Scholar] [CrossRef]
- Niture, S.K.; Velu, C.S.; Bailey, N.; Srivenugopal, K.S. Human MGMT is a prime target for inactivation by oxidative stress, mediated by glutathionylation and oxidation of the active site cysteine145. Proc. Am. Assoc. Cancer Res. 2004, 64, 355. [Google Scholar]
- Liu, L.; Xu-Welliver, M.; Kanugula, S.; Pegg, A.E. Inactivation and degradation of O(6)-alkylguanine-DNA alkyltransferase after reaction with nitric oxide. Cancer Res. 2002, 62, 3037–3043. [Google Scholar]
- Green, L.S.; Chun, L.E.; Patton, A.K.; Sun, X.; Rosenthal, G.J.; Richards, J.P. Mechanism of inhibition for N6022, a first-in-class drug targeting S-nitrosoglutathione reductase. Biochemistry 2012, 51, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.F.; Iturbe-Ormaetxe, I.; Matamoros, M.A.; Rubio, M.C.; Clemente, M.R.; Brewin, N.J.; Becana, M. Glutathione and homoglutathione synthetases of legume nodules. Cloning, expression, and subcellular localization. Plant Physiol. 2000, 124, 1381–1392. [Google Scholar] [CrossRef]
- Srivenugopal, K.S.; Mullapudi, S.R.; Shou, J.; Hazra, T.K.; Ali-Osman, F. Protein phosphorylation is a regulatory mechanism for O6-alkylguanine-DNA alkyltransferase in human brain tumor cells. Cancer Res. 2000, 60, 282–287. [Google Scholar]
- Myrnes, B.; Norstrand, K.; Giercksky, K.E.; Sjunneskog, C.; Krokan, H. A simplified assay for O6-methylguanine-DNA methyltransferase activity and its application to human neoplastic and nonneoplastic tissues. Carcinogenesis 1984, 5, 1061–1064. [Google Scholar] [CrossRef]
- Punganuru, S.R.; Madala, H.R.; Mikelis, C.M.; Dixit, A.; Arutla, V.; Srivenugopal, K.S. Conception, synthesis, and characterization of a rofecoxib-combretastatin hybrid drug with potent cyclooxygenase-2 (COX-2) inhibiting and microtubule disrupting activities in colon cancer cell culture and xenograft models. Oncotarget 2018, 9, 26109–26129. [Google Scholar] [CrossRef] [PubMed]
- Ali-Osman, F.; Rairkar, A.; Young, P. Formation and repair of 1,3-bis-(2-chloroethyl)-1-nitrosourea and cisplatin induced total genomic DNA interstrand crosslinks in human glioma cells. Cancer Biochem. Biophys. 1995, 14, 231–241. [Google Scholar]
- Madala, H.R.; Punganuru, S.R.; Ali-Osman, F.; Zhang, R.; Srivenugopal, K.S. Brain- and brain tumor-penetrating disulfiram nanoparticles: Sequence of cytotoxic events and efficacy in human glioma cell lines and intracranial xenografts. Oncotarget 2018, 9, 3459–3482. [Google Scholar] [CrossRef]
- Punganuru, S.R.; Madala, H.R.; Arutla, V.; Zhang, R.; Srivenugopal, K.S. Characterization of a highly specific NQO1-activated near-infrared fluorescent probe and its application for in vivo tumor imaging. Sci. Rep. 2019, 9, 8577. [Google Scholar] [CrossRef]
- Townsend, D.M.; He, L.; Hutchens, S.; Garrett, T.E.; Pazoles, C.J.; Tew, K.D. NOV-002, a Glutathione disulfide mimetic, as a modulator of cellular redox balance. Cancer Res. 2008, 68, 2870–2877. [Google Scholar] [CrossRef]
- Sara, J.; Lin, H.; Garrett, T.; Tew, K.D.; Townsend, D.M. Protective effects of a glutathione disulfide mimetic (NOV-002) against cisplatin induced kidney toxicity. Biomed. Pharmacother. 2010, 64, 73–76. [Google Scholar]
- Gumireddy, K.; Li, A.; Cao, L.; Yan, J.; Liu, L.; Xu, X.; Pazoles, C.; Huang, Q. NOV-002, A glutathione disulfide mimetic, suppresses tumor cell invasion and metastasis. J. Carcinog. Mutagen. 2013, 2013, S7-002. [Google Scholar]
- Montero, A.J.; Diaz-Montero, C.M.; Deutsch, Y.E.; Hurley, J.; Koniaris, L.G.; Rumboldt, T.; Yasir, S.; Jorda, M.; Garret-Mayer, E.; Avisar, E.; et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Res. Treat. 2012, 132, 215–223. [Google Scholar] [CrossRef]
- Miller, M.R.; Megson, L. Recent developments in nitric oxide donor drugs. Br. J. Pharmacol. 2007, 151, 305–321. [Google Scholar] [CrossRef]
- Corazzi, T.; Leone, M.; Maucci, R.; Corazzi, L.; Gresele, P. Direct and irreversible inhibition of cyclooxygenase-1 by nitroaspirin (NCX 4016). J Pharmacol Exp Ther. 2005, 315, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, A.; Srivenugopal, K.S. Discovery and characterization of nitroaspirin (NCX-4016) as a powerful and clinically relevant inhibitor of human MGMT for increasing the efficacy of alkylating agents. Cancer Res. 2012, 72 (Suppl. S8), 4685. [Google Scholar] [CrossRef]
- Srivenugopal, K.S.; Paranjpe, A. The NO-releasing aspirin inactivates and degrades human MGMT more efficiently than O6-benzylguanine and greatly sensitizes brain tumor cells to alkylating agents. Cancer Res. 2013, 73 (Suppl. S8), 4473. [Google Scholar] [CrossRef]
- Paranjpe, A.; Srivenugopal, K.S. Degradation of NF-κB, p53 and other regulatory redox-sensitive proteins by thiol-conjugating and -nitrosylating drugs in human tumor cells. Carcinogenesis 2013, 34, 990–1000. [Google Scholar] [CrossRef]
- Barnett, S.D.; Buxton, I.L.O. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Donze, J.R.; Liu, L. Inactivated MGMT by O6-benzylguanine is associated with prolonged G2/M arrest in cancer cells treated with BCNU. Oncogene 2005, 24, 2175–2183. [Google Scholar] [CrossRef]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 2000, 267, 4928–4944. [Google Scholar] [CrossRef]
- Ghezzi, P.; Bonetto, V.; Fratelli, M. Thiol-disulfide balance: From the concept of oxidative stress to that of redox regulation. Antioxid. Redox Signal 2005, 7, 964–972. [Google Scholar] [CrossRef]
- Thomas, J.A.; Poland, B.; Honzatko, R. Protein sulfhydryls and their role in the antioxidant function of protein S-thiolation. Arch. Biochem. Biophys. 1995, 319, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-glutathionylation: From redox regulation of protein functions to human diseases. J. Cell Mol. Med. 2004, 8, 201–212. [Google Scholar] [CrossRef]
- Laval, F.; Wink, D.A. Inhibition by nitric oxide of the repair protein, O6-methylguanine-DNA methyltransferase. Carcinogenesis 1994, 5, 443–447. [Google Scholar] [CrossRef]
- Liu, L.; Xu-Welliver, M.; Pegg, A.E. Inactivation of DNA repair protein O6-alkylguanine-DNA alkyltransferase by reaction with nitric oxide. FASEB J. 2001, 15, A519. [Google Scholar]
- Kurimoto, M.; Endo, S.; Hirashima, Y.; Hamada, H.; Ogiichi, T.; Takaku, A. Growth inhibition and radiosensitization of cultured glioma cells by nitric oxide generating agents. J. Neurooncol. 1999, 42, 35–44. [Google Scholar] [CrossRef]
- Safdar, S.; Payne, C.A.; Tu, N.H.; Taite, L.J. Targeted nitric oxide delivery preferentially induces glioma cell chemosensitivity via Altered p53 and O6-methylguanine-DNA Methyltransferase activity. Biotechnol. Bioeng. 2012, 110, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Kogias, E.; Osterberg, N.; Baumer, B.; Psarras, N.; Koentges, C.; Papazoglou, A.; Saavedra, J.E.; Keefer, L.K.; Weyerbrock, A. Growth-inhibitory and chemosensitizing effects of the glutathione-S-transferase-π-activated nitric oxide donor PABA/NO in malignant gliomas. Int. J. Cancer 2012, 130, 1184–1194. [Google Scholar] [CrossRef]
- Wei, W.; Li, B.; Hanes, M.A.; Kakar, S.; Chen, X.; Liu, L. S-nitrosylation from GSNOR Deficiency Impairs DNA Repair and Promotes Hepatocarcinogenesis. Sci. Transl. Med. 2010, 2, 19ra13. [Google Scholar] [CrossRef]
- American Brain Tumor Association: Brain Tumor Statistics. Available online: http://www.abta.org/about-us/news/brain-tumor-statistics/ (accessed on 26 December 2024).
- Williamson, J.M.; Meister, A. Stimulation of hepatic glutathione formation by administration of L-2-oxothiazolidine-4-carboxylate, a 5-oxo-L-prolinase substrate. Proc. Natl. Acad. Sci. USA 1981, 78, 936–939. [Google Scholar] [CrossRef]
- Williamson, J.M.; Boettcher, B.; Meister, A. Intracellular cysteine delivery system that protects against toxicity by promoting glutathione synthesis. Proc. Natl. Acad. Sci. USA 1982, 79, 6246–6249. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Crawford, J.M.; Holden, S.A.; Lin, Y.; Cathcart, K.N.; Luchette, C.A.; Flatow, J. Glutathione monoethyl ester can selectively protect liver from high dose BCNU or cyclophosphamide. Cancer 1988, 62, 1275–1281. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basak, D.; Mostofa, A.; Madala, H.R.; Srivenugopal, K.S. Novel Pathways of Oxidative and Nitrosative Inactivation of the Human MGMT Protein in Colon Cancer and Glioblastoma Cells: Increased Efficacy of Alkylating Agents In Vitro and In Vivo. Diseases 2025, 13, 32. https://doi.org/10.3390/diseases13020032
Basak D, Mostofa A, Madala HR, Srivenugopal KS. Novel Pathways of Oxidative and Nitrosative Inactivation of the Human MGMT Protein in Colon Cancer and Glioblastoma Cells: Increased Efficacy of Alkylating Agents In Vitro and In Vivo. Diseases. 2025; 13(2):32. https://doi.org/10.3390/diseases13020032
Chicago/Turabian StyleBasak, Debasish, Agm Mostofa, Hanumantha Rao Madala, and Kalkunte S. Srivenugopal. 2025. "Novel Pathways of Oxidative and Nitrosative Inactivation of the Human MGMT Protein in Colon Cancer and Glioblastoma Cells: Increased Efficacy of Alkylating Agents In Vitro and In Vivo" Diseases 13, no. 2: 32. https://doi.org/10.3390/diseases13020032
APA StyleBasak, D., Mostofa, A., Madala, H. R., & Srivenugopal, K. S. (2025). Novel Pathways of Oxidative and Nitrosative Inactivation of the Human MGMT Protein in Colon Cancer and Glioblastoma Cells: Increased Efficacy of Alkylating Agents In Vitro and In Vivo. Diseases, 13(2), 32. https://doi.org/10.3390/diseases13020032