
Theoretical Investigation of the Enantioselective Complexations between pfDHFR and Cycloguanil Derivatives


Abstract
:1. Introduction
2. Materials and Methods
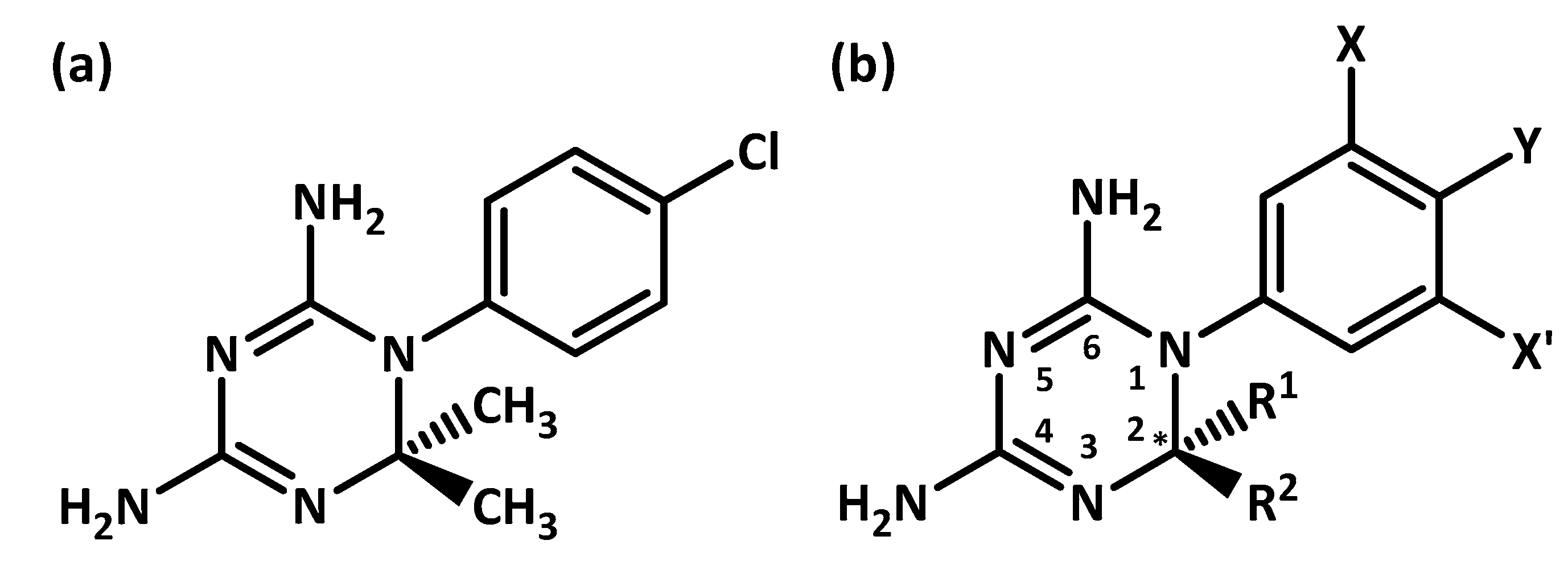
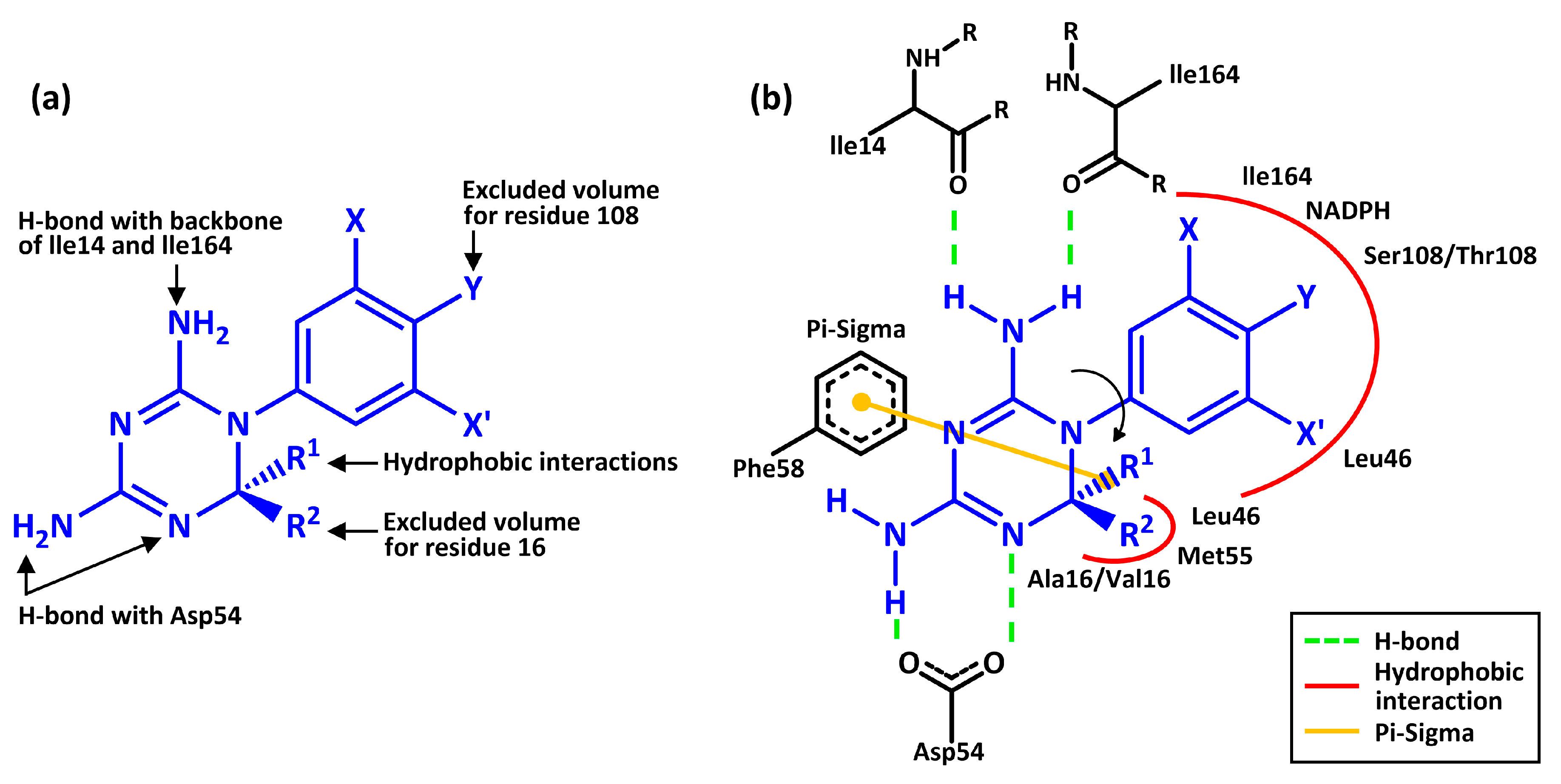
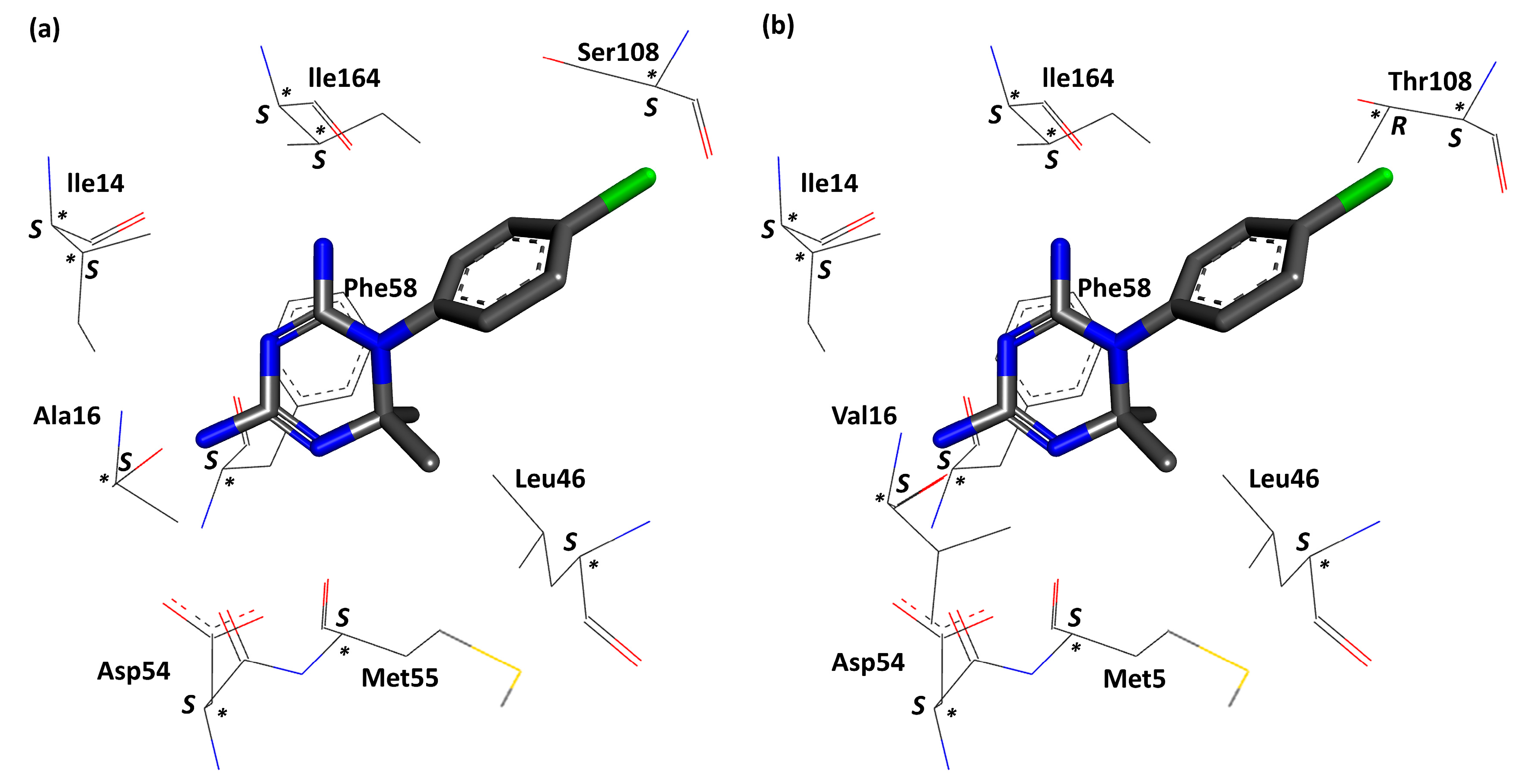
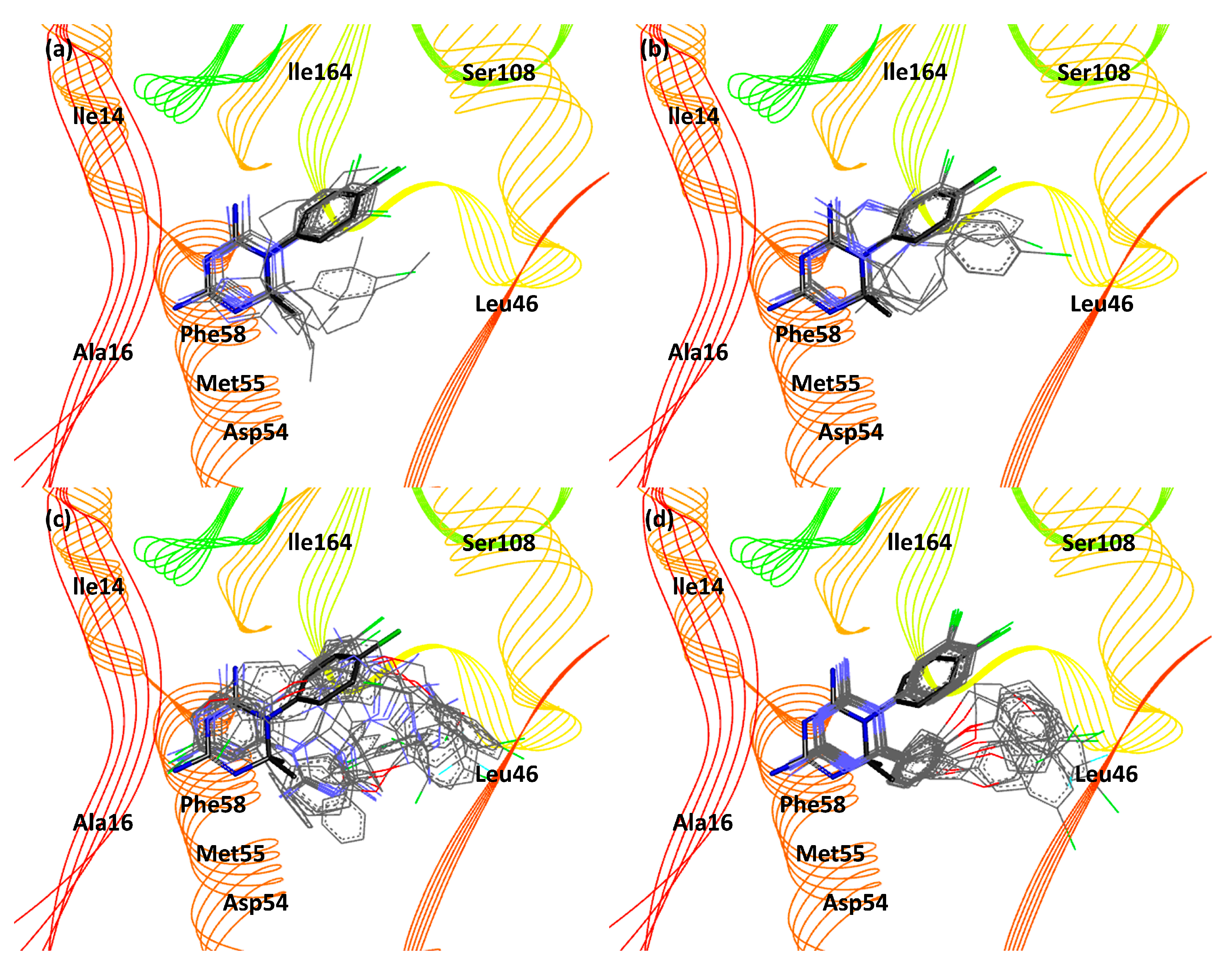
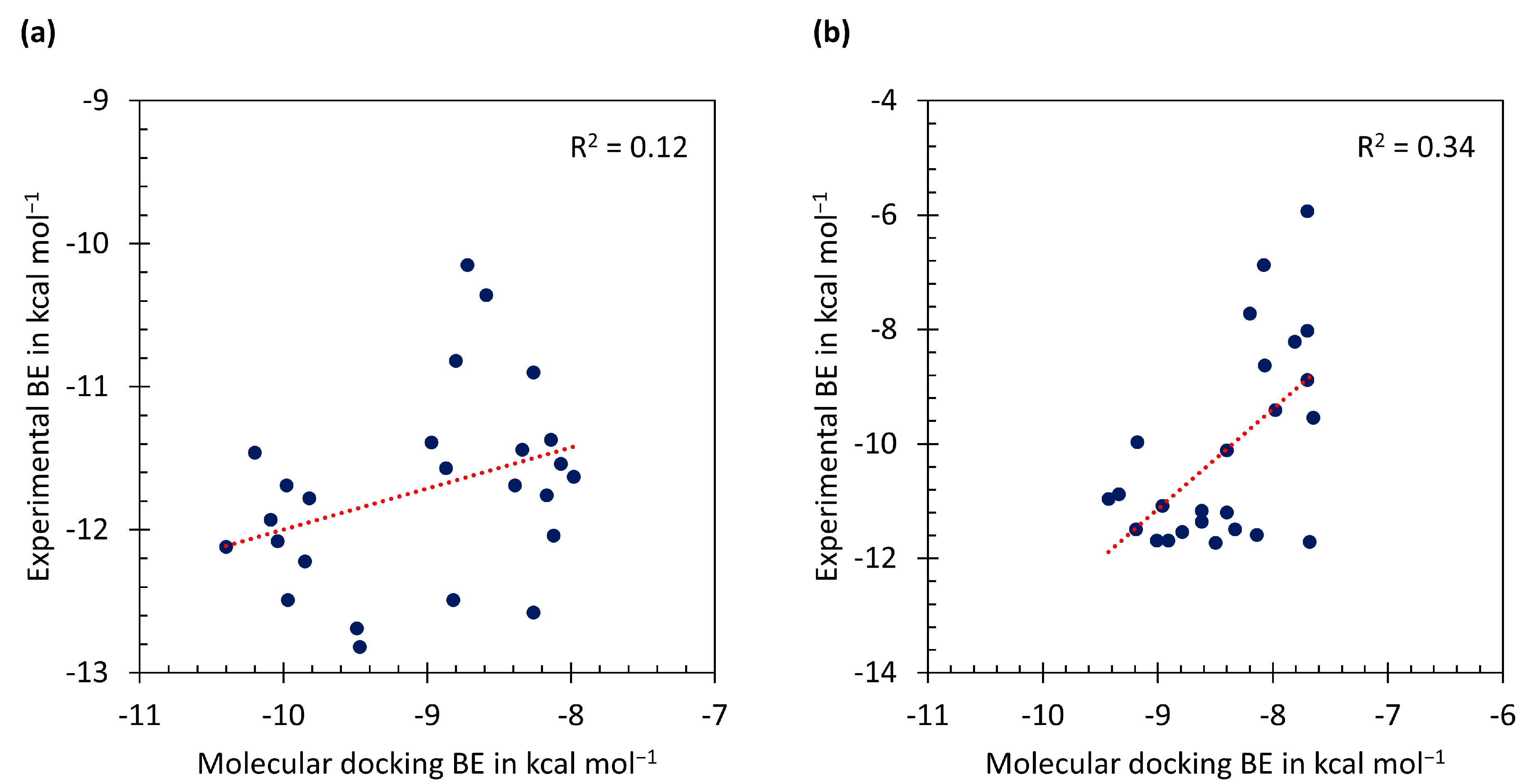
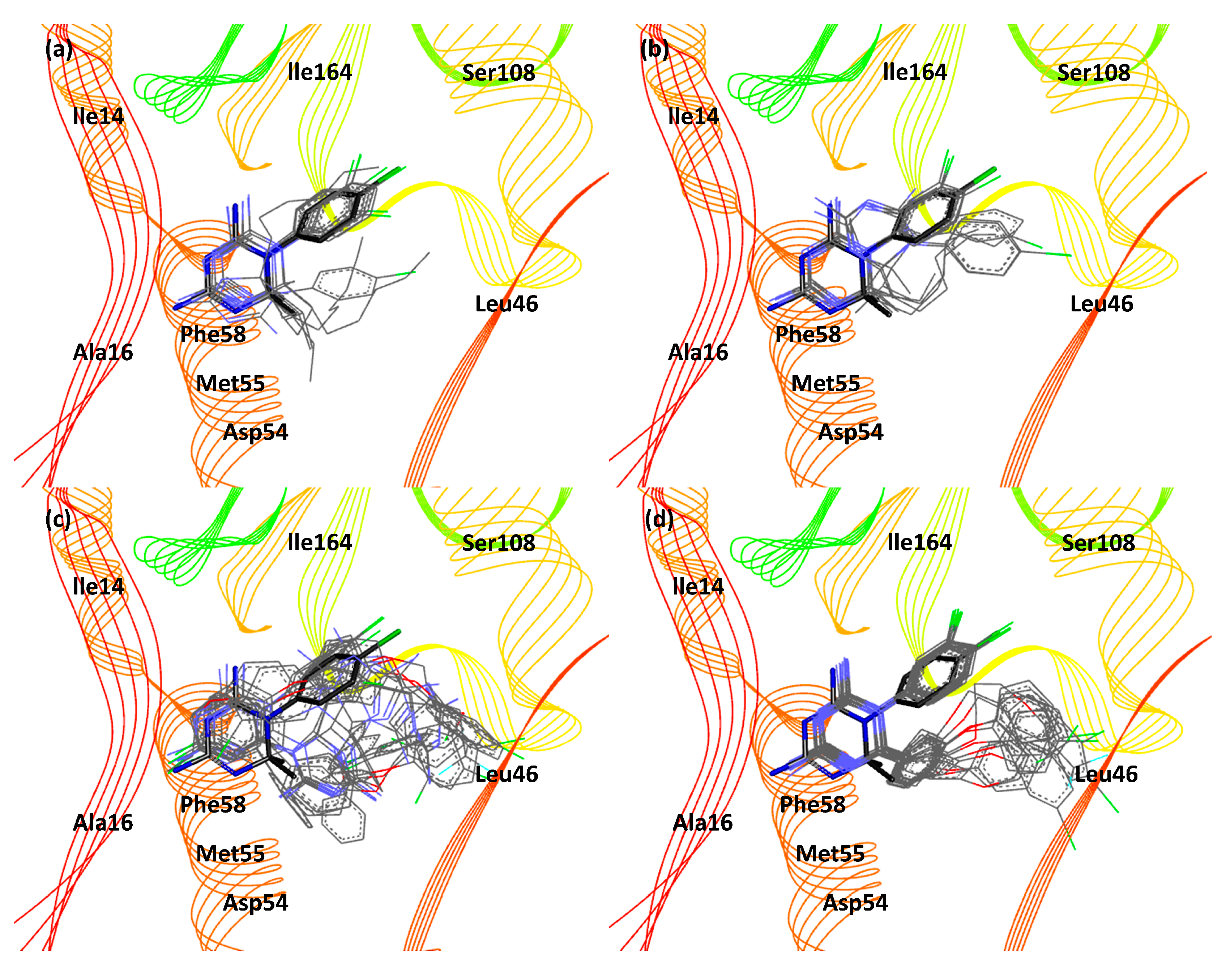
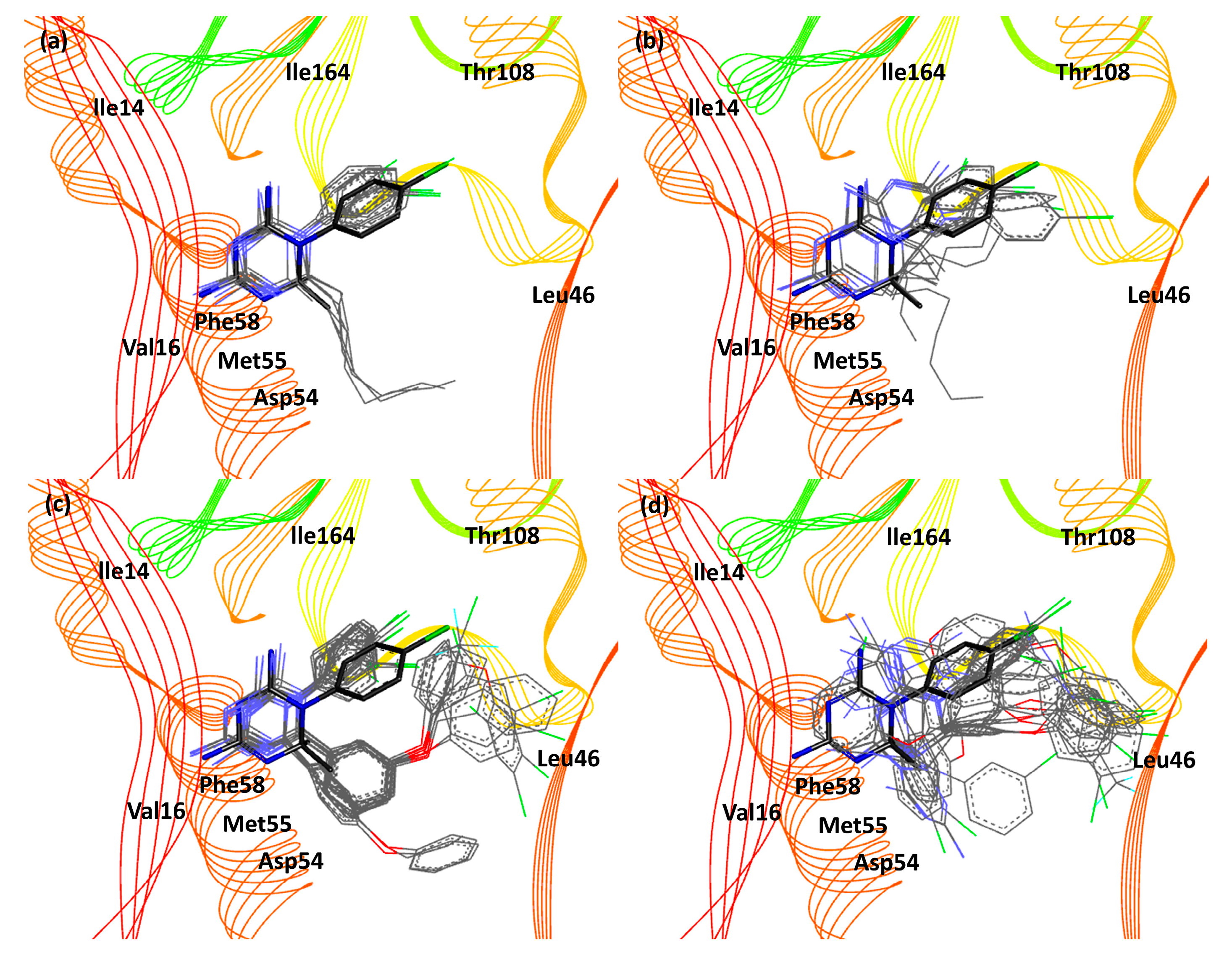
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yuthavong, Y.; Vilaivan, T.; Chareonsethakul, N.; Kamchonwongpaisan, S.; Sirawaraporn, W.; Quarrell, R.; Lowe, G. Development of a lead inhibitor for the A16V + S108T mutant of dihydrofolate reductase from the cycloguanil-resistant strain (T9/94) of Plasmodium falciparum. J. Med. Chem. 2000, 43, 2738–2744. [Google Scholar] [CrossRef] [PubMed]
- Vanichtanankul, J.; Taweechai, S.; Uttamapinant, C.; Chitnumsub, P.; Vilaivan, T.; Yuthavong, Y.; Kamchonwongpaisan, S. Combined spatial limitation around residues 16 and 108 of Plasmodium falciparum dihydrofolate reductase explains resistance to cycloguanil. Antimicrob. Agents Chemother. 2012, 56, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Yuvaniyama, J.; Chitnumsub, P.; Kamchonwongpaisan, S.; Vanichtanankul, J.; Sirawaraporn, W.; Taylor, P.; Walkinshaw, M.D.; Yuthavong, Y. Insights into antifolate resistance from malarial DHFR-TS Structures. Nat. Struct. Mol. Biol. 2003, 10, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Sirawaraporn, W.; Sompornpisut, P.; Vilaivan, T.; Kamchonwongpaisan, S.; Quarrell, R.; Lowe, G.; Thebtaranonth, Y.; Yuthavong, Y. Interaction of pyrimethamine, cycloguanil, WR99210 and their analogues with Plasmodium falciparum dihydrofolate reductase: Structural basis of antifolate resistance. Bioorg. Med. Chem. 2000, 8, 1117–1128. [Google Scholar] [CrossRef]
- Kamchonwongpaisan, S.; Quarrell, R.; Charoensetakul, N.; Ponsinet, R.; Vilaivan, T.; Vanichtanankul, J.; Tarnchompoo, B.; Sirawaraporn, W.; Lowe, G.; Yuthavong, Y. Inhibitors of multiple mutants of Plasmodium falciparum dihydrofolate reductase and their antimalarial activities. J. Med. Chem. 2004, 47, 673–680. [Google Scholar] [CrossRef] [PubMed]
- McConathy, J.; Owens, M.J. Stereochemistry in drug action. Prim. Care Companion J. Clin. Psychiatry. 2003, 5, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, B.S. Exploiting the power of stereochemistry in drugs: An overview of racemic and enantiopure drugs. J. Mod. Med. Chem. 2013, 1, 10–36. [Google Scholar] [CrossRef]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar] [PubMed]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- RCSB PDB-3UM8: Wild-Type Plasmodium falciparum DHFR-TS Complexed with Cycloguanil and NADPH Structure Summary Page. Available online: https://www.rcsb.org/pdb/explore.do?structureId=3UM8 (accessed on 30 May 2017).
- RCSB PDB-3UM6: Double Mutant (A16V + S108T) Plasmodium falciparum DHFR-TS (T9/94) Complexed with Cycloguanil, NADPH and dUMP Structure Summary Page. Available online: https://www.rcsb.org/pdb/explore.do?structureId=3UM6 (accessed on 30 May 2017).
- Accelrys. Discovery Studio Modeling Environment Release 4.0; Accelrys Inc.: San Diego, CA, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comp. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Yuthavong, Y. Basis for antifolate action and resistance in malaria. Microbes Infect. 2002, 4, 175–182. [Google Scholar] [CrossRef]

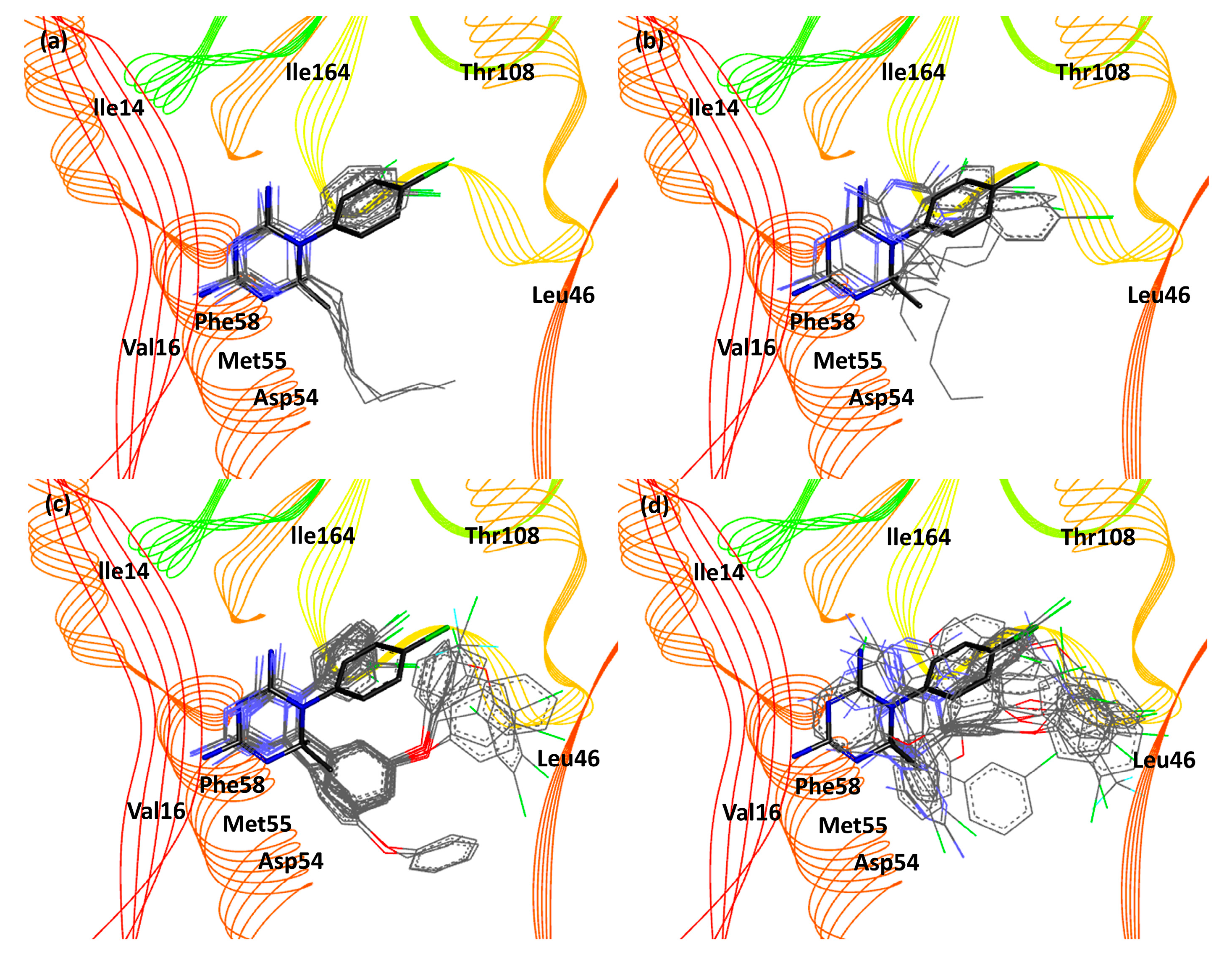

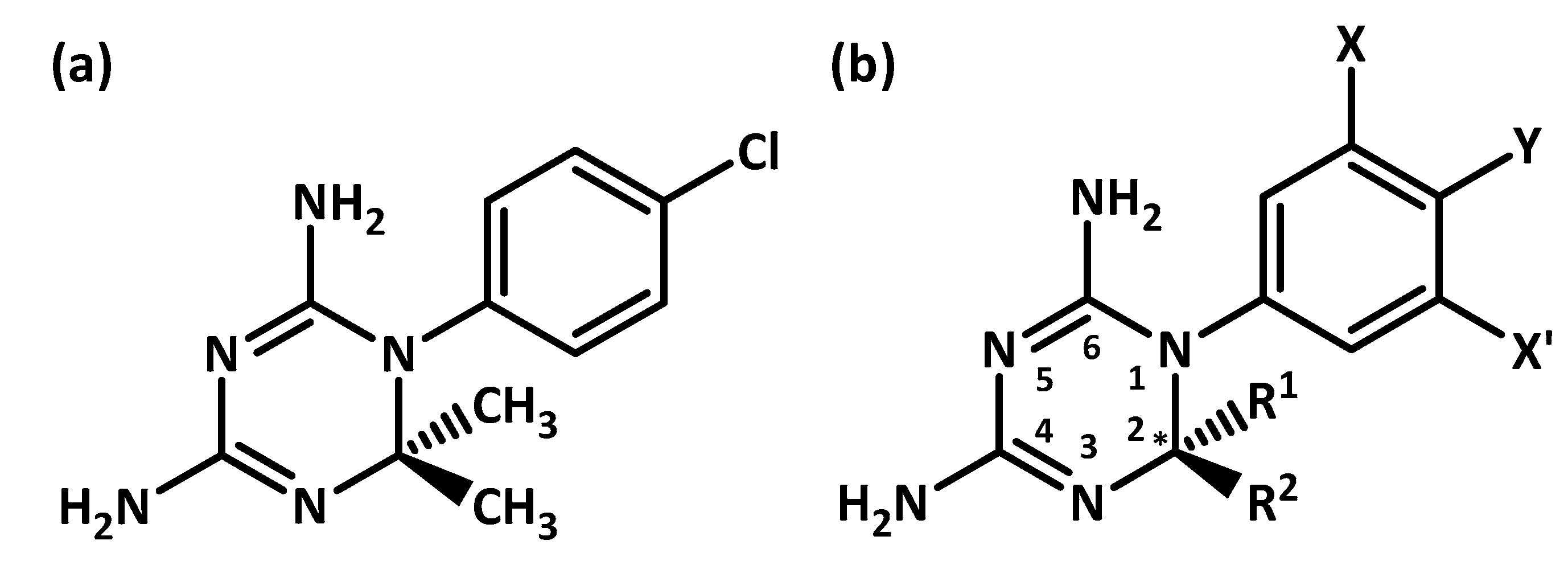{kind=link}
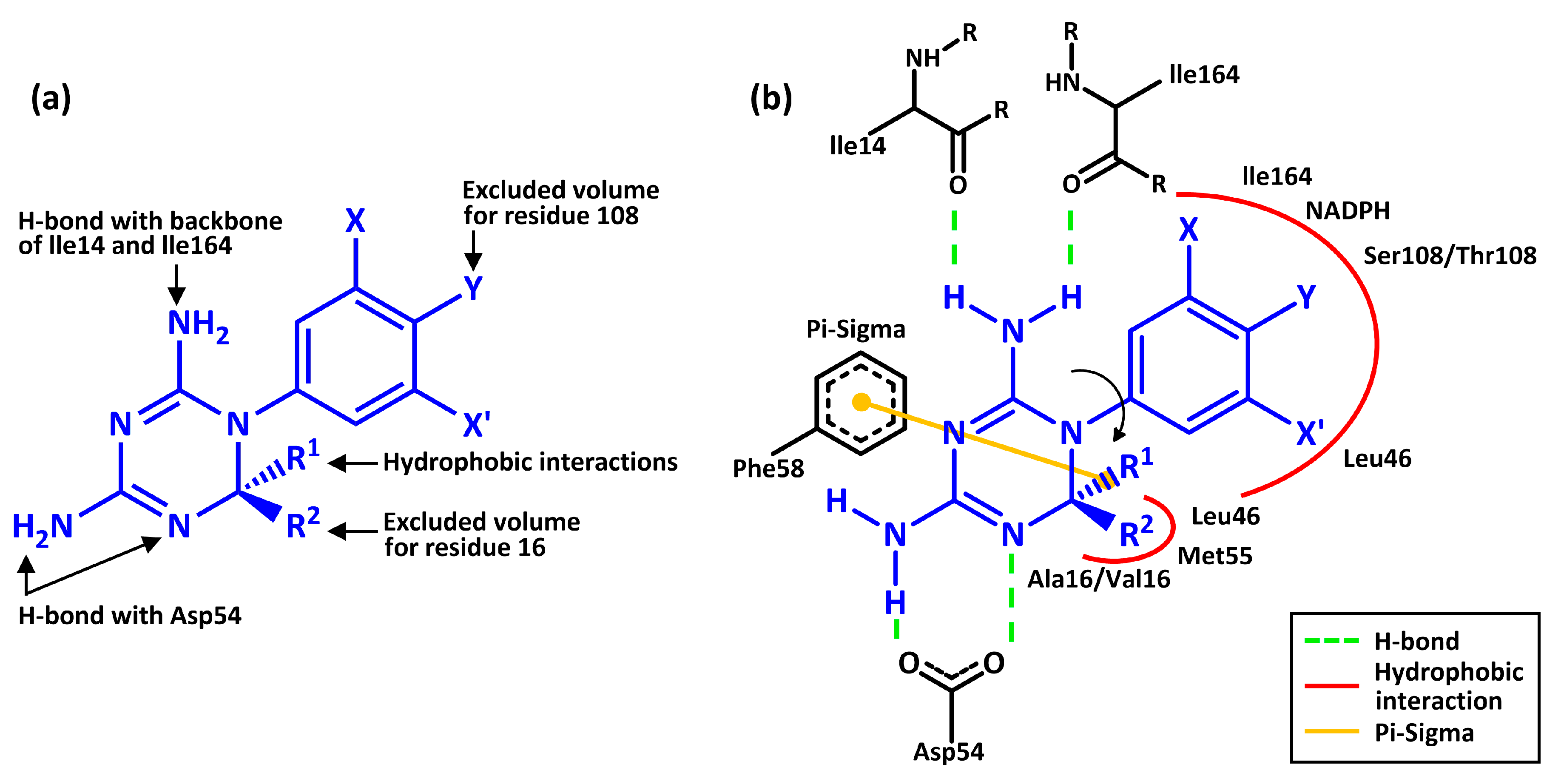{kind=link}
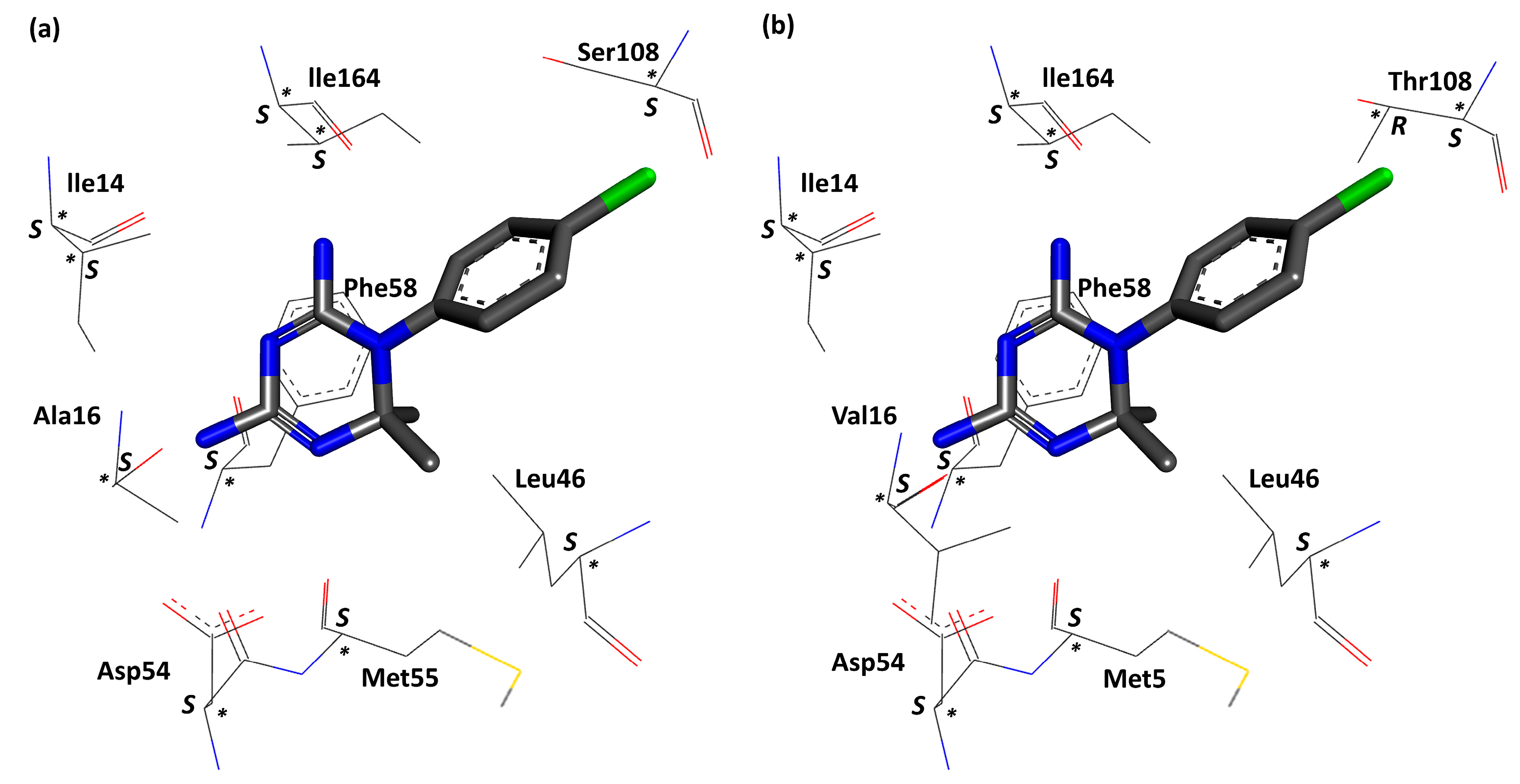{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3UM8 | 3UM6 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | X | Y | R1 | R2 | R | S | Exp. | R | S | Exp. |
| Cyc | H | Cl | Me | Me | −8.12 −7.98 | −12.04 | −7.70 −7.70 | −8.02 | ||
| 23 | Cl | H | Me | Me | −11.63 | −8.88 | ||||
| 24 | H | Cl | Me | nPr | −8.07 | −6.85 b | −11.54 | −8.08 | −7.09 b | −6.87 |
| 25 | Cl | H | Me | iPr | −8.59 | −7.30 b | −10.36 | −8.20 a | −7.41 b | −7.72 |
| 26 | H | Cl | Me | iPr | −8.72 | −7.12 b | −10.15 | −7.70 | −7.37 b | −5.93 |
| 27 | Cl | H | Me | nPr | −8.14 | −8.01 | −11.37 | −8.07 | −7.57 a | −8.63 |
| 28 | H | Cl | Me | nHex | −7.75 b | −8.26 | −12.58 | −7.81 | −7.79 | −8.21 |
| 29 | Cl | H | Me | nHex | −7.85 | −8.17 | −11.76 | −7.65 a | −7.62 a | −9.54 |
| 30 | H | Cl | H | Me | −8.34 | −7.76 | −11.44 | −7.98 | −7.53 | −9.41 |
| 31 | Cl | H | H | Me | −8.26 a | −7.83 | −10.90 | −8.40 a | −7.48 | −10.11 |
| 32 | H | Cl | H | C6H5 | −8.97 | −8.39 | −11.39 | −9.18 | −7.21 b | −9.97 |
| 33 | Cl | H | H | C6H5 | −8.80 a | −8.67 | −10.82 | −9.34 a | −7.19 | −10.88 |
| 34 | H | Cl | H | 4-C6H5OC6H5 | −8.57 b | −9.47 | −12.82 | −9.19 | −9.04 b | −11.49 |
| 35 | Cl | H | H | 4-C6H5OC6H5 | −8.47 b | −9.97 | −12.49 | −9.01 a | −7.22 b | −11.69 |
| 36 | H | Cl | H | 3-C6H5OC6H5 | −8.60 | −9.49 | −12.69 | −8.91 | −6.81 | −11.69 |
| 37 | Cl | H | H | 3-C6H5OC6H5 | −8.73 b | −9.85 | −12.22 | −8.50 | −8.55 b | −11.73 |
| 38 | H | Cl | H | 3-C6H5CH2OC6H4 | −8.12 | −8.82 | −12.49 | −8.40 | −6.74 | −11.20 |
| 39 | Cl | H | H | 3-C6H5CH2OC6H4 | −9.31 b | −9.82 | −11.78 | −8.14 | −7.01 | −11.59 |
| 40 | H | Cl | H | 3-(4-ClC6H4O)C6H4 | −8.82 | −10.04 | −12.08 | −8.96 | −7.27 | −11.08 |
| 41 | Cl | H | H | 3-(4-ClC6H4O)C6H4 | −9.25 b | −10.40 | −12.12 | −8.79 | −7.39 | −11.54 |
| 42 | Cl | H | H | nC7H15 | −8.39 a | −8.03 | −11.69 | −8.33 a | −7.59 | −11.49 |
| 43 | Cl | H | H | 4-PrOC6H4 | −7.43 b | −8.87 | −11.57 | −9.43 a | −8.05 b | −10.96 |
| 44 | Cl | H | H | 3-(3,5-Cl2C6H3O)C6H4 | −8.90 b | −10.09 | −11.93 | −8.62 | −6.93 b | −11.36 |
| 45 | Cl | H | H | 3-[2,4,5-Cl3C6H2O(CH2)3O]C6H4 | −9.18 b | −10.20 | −11.46 | −7.68 | −3.49 b | −11.71 |
| 46 | Cl | H | H | 3-(3-CF3C6H4O)C6H4 | −8.20 b | −9.98 | −11.69 | −8.62 | −6.74 | −11.17 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulatee, S.; Toochinda, P.; Suksangpanomrung, A.; Lawtrakul, L. Theoretical Investigation of the Enantioselective Complexations between pfDHFR and Cycloguanil Derivatives. Sci. Pharm. 2017, 85, 37. https://doi.org/10.3390/scipharm85040037
Kulatee S, Toochinda P, Suksangpanomrung A, Lawtrakul L. Theoretical Investigation of the Enantioselective Complexations between pfDHFR and Cycloguanil Derivatives. Scientia Pharmaceutica. 2017; 85(4):37. https://doi.org/10.3390/scipharm85040037
Chicago/Turabian StyleKulatee, Suriyawut, Pisanu Toochinda, Anotai Suksangpanomrung, and Luckhana Lawtrakul. 2017. "Theoretical Investigation of the Enantioselective Complexations between pfDHFR and Cycloguanil Derivatives" Scientia Pharmaceutica 85, no. 4: 37. https://doi.org/10.3390/scipharm85040037
APA StyleKulatee, S., Toochinda, P., Suksangpanomrung, A., & Lawtrakul, L. (2017). Theoretical Investigation of the Enantioselective Complexations between pfDHFR and Cycloguanil Derivatives. Scientia Pharmaceutica, 85(4), 37. https://doi.org/10.3390/scipharm85040037