
Bimodal Release Two-In-One Clonazepam Matrix Lozenge Tablets for Managing Anxiety-Related Disorders: Formulation, Optimization and In Vivo Evaluation



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Clonazepam Solid Dispersion
2.2.1. Preparation of Physical Mixtures (PMs) and Solid Dispersions (SDs) of CLZ
2.2.2. Dissolution Studies
2.3. Optimization of Formulation Excipient Type and Concentration
2.3.1. Optimization of Avicel: Lactose Ratio
2.3.2. Optimization of Gelling Polymer Type and Concentration
2.4. Preparation of Single-Layer Two-in-One Matrix Lozenge Tablets of CLZ
2.5. Characterization of Two-in-One Single-Layer CLZ Lozenge Tablets
2.5.1. Percentage Drug Content
2.5.2. Hardness, Diameter, and Thickness
2.5.3. Friability
2.5.4. Disintegration Test
2.5.5. In Vitro Drug Release
2.5.6. Drug-Release Kinetics
2.5.7. Differential Scanning Calorimetry (DSC)
2.5.8. In Vivo Characterization of Optimized and Commercial Formulations
Ethical Approval
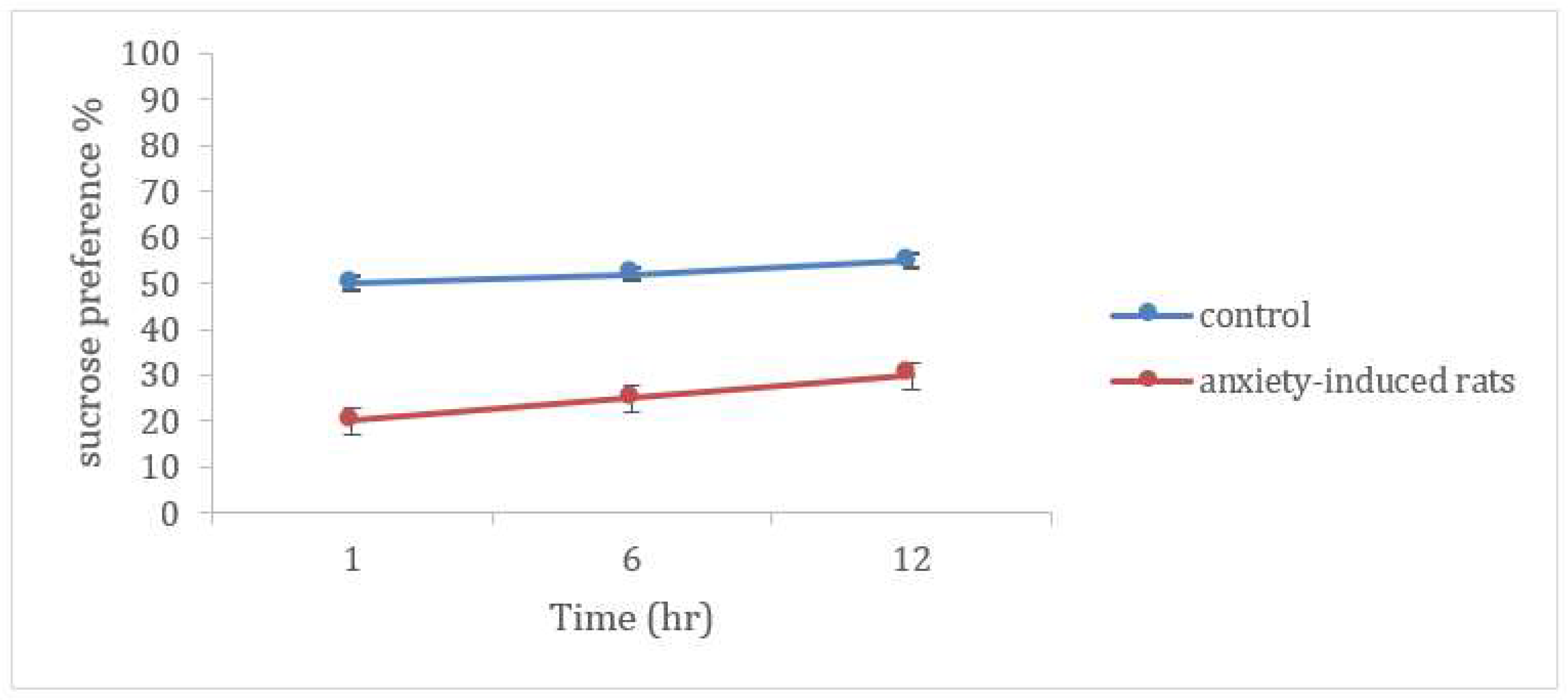
Sucrose Preference Test
Administration and Evaluation of Studied Formulations
- -
- Group1: had free access to water and food for 10 days and served as the negative control. This group was tested under the same conditions and was used to demonstrate the increase in anxiety behavior after induction in the other groups.
- -
- Group2: rats did not receive any treatment and served as positive control.
- -
- Group 3: rats received the marketed tablets of CLZ (Amotril®, containing 2 mg CLZ per tablet, manufactured by Amoun pharmaceuticals, Egypt) as an oral suspension, adjusted to contain 0.25 mg per kg of body weight of rats [24]. The suspension was stabilized with the help of 2% Tween 80 or 1% gum solution, immediately before administration [25].
- -
- Group 4: rats received the optimized formulation containing the same dose as group 3 (0.25 mg/kg) fixed to the buccal cavity, using urethane at a small dose of 2 mg/kg before drug administration only to ensure the retention of test formulations within the buccal cavity for a sufficient period of time.
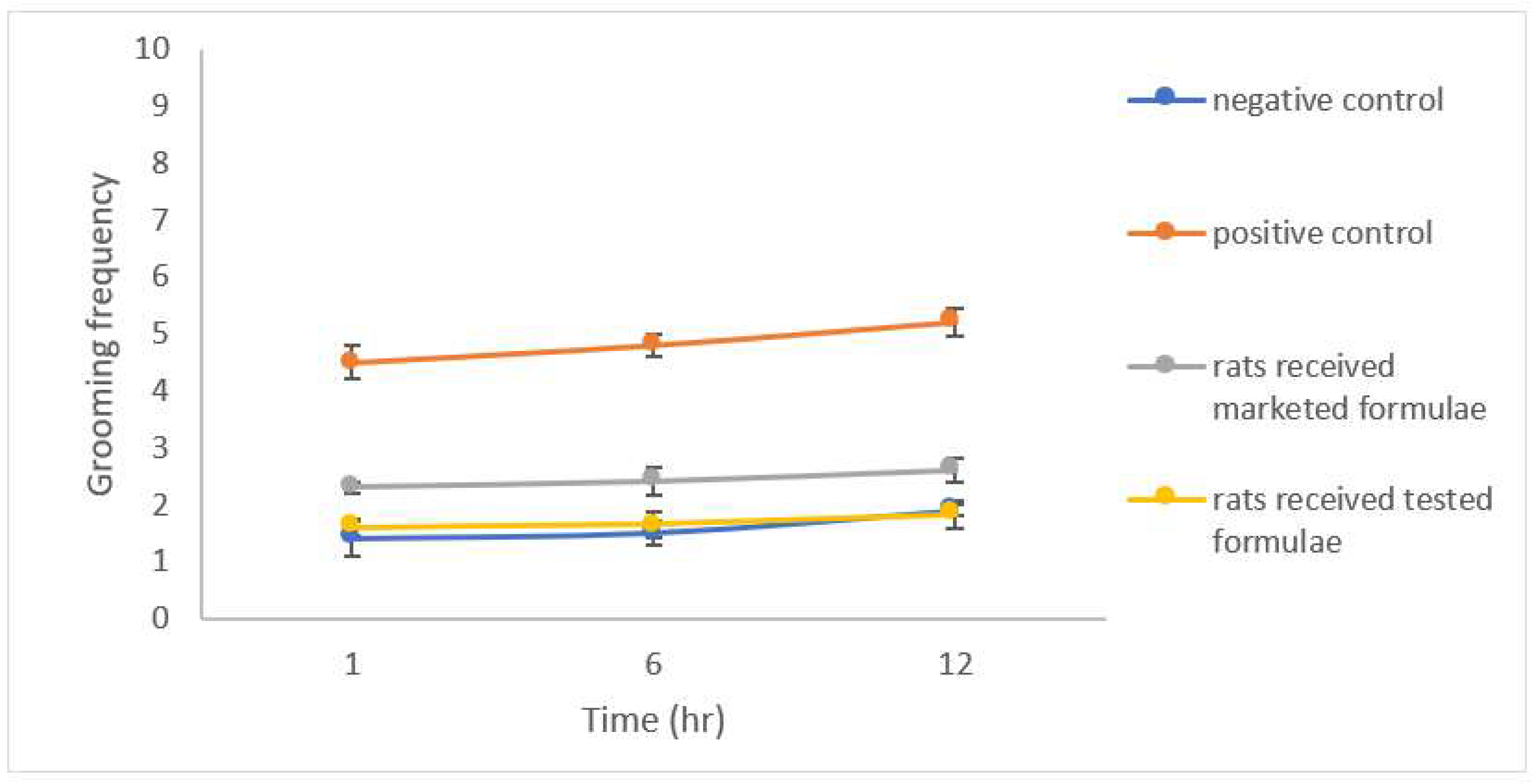
Evaluation of Antianxiety Activity (Behavioral Assessment)
Forced Swim Test
Open Field Test (OFT)
2.5.9. Pharmacokinetic Study
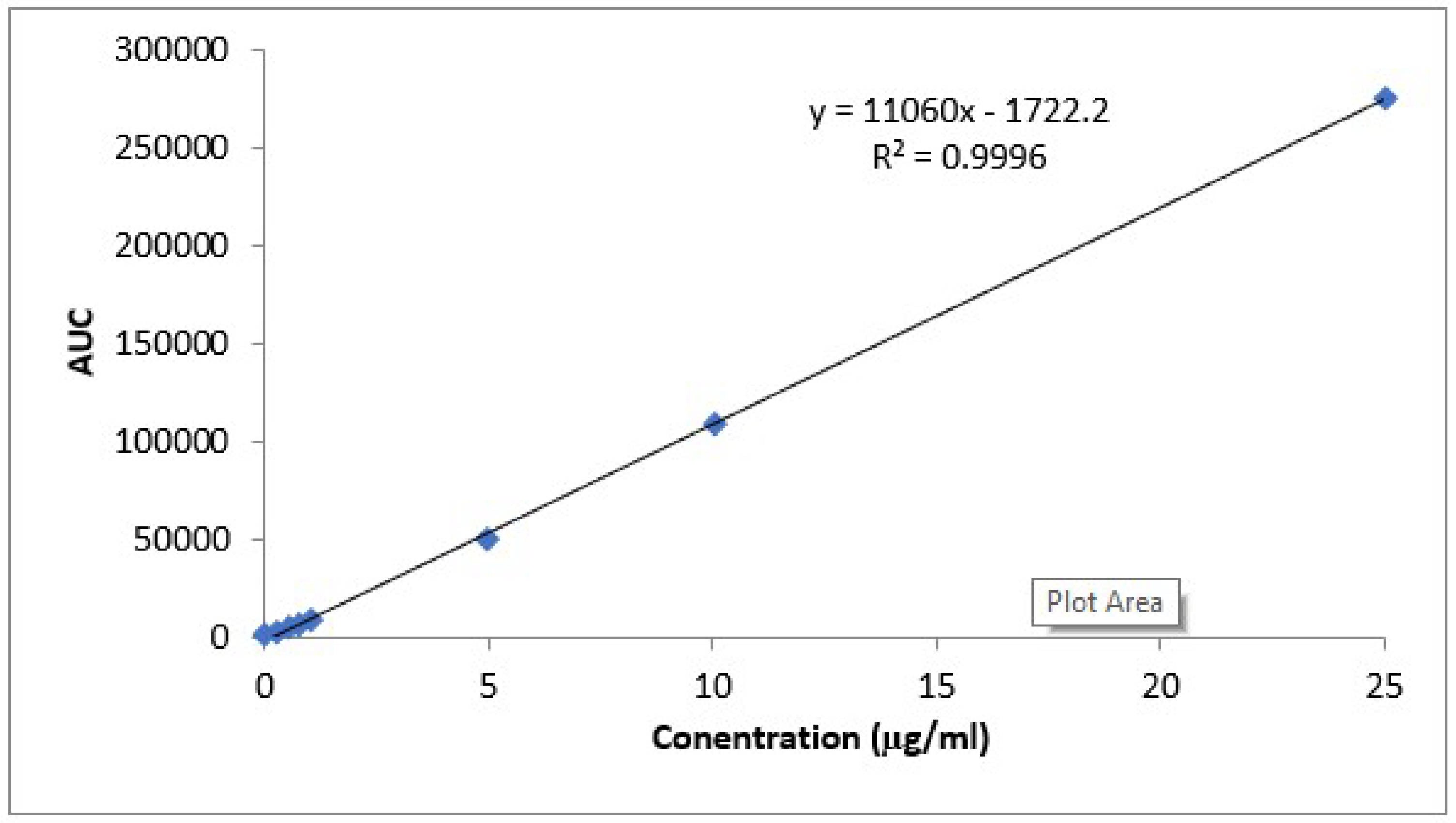
HPLC Analysis Method for CLZ Determination
In Vivo Kinetic Study
Statistical Analysis
3. Results and Discussion
3.1. Dissolution Enhancement of CLZ by Solid Dispersion
3.2. Optimization of Avicel: Lactose Concentration
3.3. Optimization of Polymer Type and Concentration
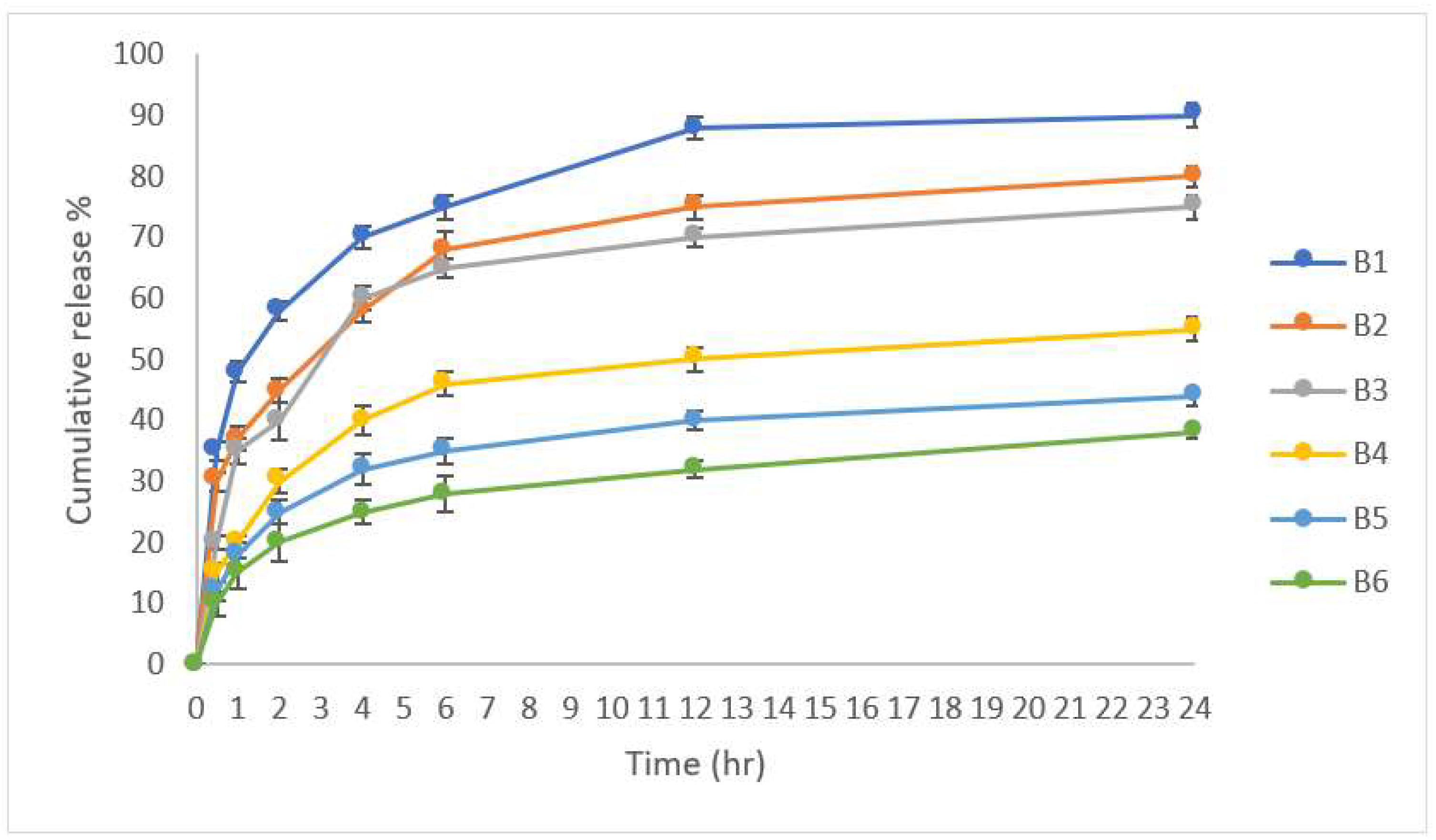
3.4. In Vitro Drug Release
3.5. In Vitro Release Kinetic Study
3.6. Differential Scanning Calorimetry (DSC)
3.7. Evaluation of Anxiety-Related Symptoms
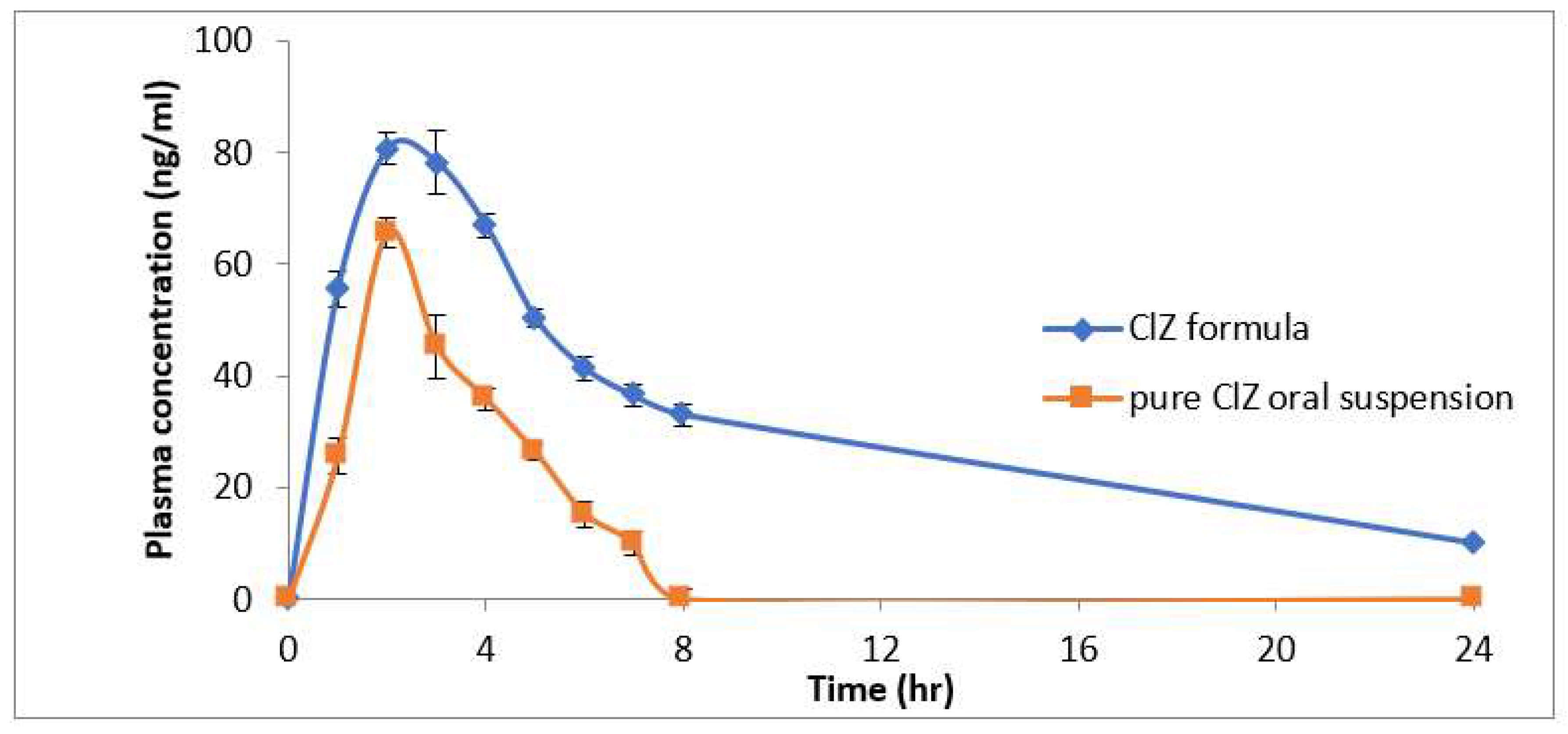
3.8. Pharmacokinetics Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sweetman, S. Martindale: The Complete Drug Reference; Pharmaceutical Press: London, UK, 2009; Volume 36. [Google Scholar]
- Koubeissi, M. Intravenous Clonazepam in Status Epilepticus. Epilepsy Curr. 2016, 16, 89–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirri, M.; Maestrelli, F.; Nerli, G.; Mennini, N.; D’Ambrosio, M.; Luceri, C.; Mura, P.A. Development of a cyclodextrin-based mucoadhesive-thermosensitive in situ gel for clonazepam intranasal delivery. Pharmaceutics 2021, 13, 969. [Google Scholar] [CrossRef] [PubMed]
- Vyas, T.K.; Babbar, A.; Sharma, R.; Singh, S.; Misra, A. Intranasal mucoadhesive microemulsions of clonazepam: Preliminary studies on brain targeting. J. Pharm. Sci. 2006, 95, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Commission, B.P. British Pharmacopoeia 2017; Stationery Office: London, UK, 2017. [Google Scholar]
- Vårdal, L.; Øiestad, E.L.; Gjelstad, A.; Pedersen-Bjergaard, S. Electromembrane extraction of substances with weakly basic properties: A fundamental study with benzodiazepines. Bioanalysis 2018, 10, 769–781. [Google Scholar] [CrossRef]
- Minhaz, M.; Rahman, M.; Ahsan, M.; Chowdhury, M. Enhancement of solubility and dissolution properties of clonazepam by solid dispersions. Int. J. Pharm. Life Sci. 2012, 3, 1510–1518. [Google Scholar]
- Seshadri, V.C.; Manohari, P.J.; Kunchithapatham, J. Formulation and characterization of mouth dissolving tablets containing benzodiazepine. Indo Am. J. Pharm Res. 2013, 3, 1457–1474. [Google Scholar]
- Shojaei, A.H. Buccal mucosa as a route for systemic drug delivery: A review. J. Pharm. Pharm. Sci. 1998, 1, 15–30. [Google Scholar]
- Malonne, H.; Sonet, B.; Streel, B.; Lebrun, S.; De Niet, S.; Sereno, A.; Vanderbist, F. Pharmacokinetic evaluation of a new oral sustained release dosage form of tramadol. Br. J. Clin. Pharmacol. 2004, 57, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Chowdary, Y.A.; Raparla, R.; Madhuri, M. Formulation and evaluation of multilayered tablets of pioglitazone hydrochloride and metformin hydrochloride. J. Pharm. 2014, 2014, 848243. [Google Scholar] [CrossRef]
- Morovati, A.; Ghaffari, A. Single Layer Extended Release Two-in-One Guaifenesin Matrix Tablet: Formulation Method, Optimization, Release Kinetics Evaluation and Its Comparison with Mucinex® Using Box-Behnken Design. Iran. J. Pharm. Res. IJPR 2017, 16, 1349. [Google Scholar]
- Rashed, S.S.; Tushar, R.R.; Ahmed, F.; Vabna, N.J.; Jahan, L.; Billah, M.M. Comparative in vitro Dissolution Study of Clonazepam Tablets of Bangladesh by UV-Visible Spectrophotometry. ACTA Pharm. Sci. 2021, 59, 641–653. [Google Scholar] [CrossRef]
- ICHHT Guideline. ICH guidelines for Validation of analytical procedures: Text and methodology Q2 (R1). In Proceedings of the International Conference on Harmonization, Geneva, Switzerland, 2–4 March 2005; pp. 11–12. [Google Scholar]
- Bolla, S.; Segji, S.; Nallipogu, V.; Kumar, V.; Chetty, C. Formulation and evaluation of orodispersible tablets of clonazepam using natural superdisintegrants. J. Pharm. Biol. Sci. 2014, 9, 47–52. [Google Scholar] [CrossRef]
- Sharaf, Y.A.; El Deeb, S.; Ibrahim, A.E.; Al-Harrasi, A.; Sayed, R.A. Two Green Micellar HPLC and Mathematically Assisted UV Spectroscopic Methods for the Simultaneous Determination of Molnupiravir and Favipiravir as a Novel Combined COVID-19 Antiviral Regimen. Molecules 2022, 27, 2330. [Google Scholar] [CrossRef] [PubMed]
- Al Ashmawy, A.Z.G.; Eissa, N.G.; El Nahas, H.M.; Balata, G.F. Fast disintegrating tablet of Doxazosin Mesylate nanosuspension: Preparation and characterization. J. Drug Deliv. Sci. Technol. 2021, 61, 102210. [Google Scholar] [CrossRef]
- Abd El-Hay, S.S.; Elhenawee, M.; Maged, K.; Ibrahim, A.E. Cost-effective, green HPLC determination of losartan, valsartan and their nitrosodiethylamine impurity: Application to pharmaceutical dosage forms. R. Soc. Open Sci. 2022, 9, 220250. [Google Scholar] [CrossRef]
- Ashraf, Z.; Khurram, S.; Maqbool, U.; Khan, K.R.; Ajaz, U.; Fatima, S.F.; Naz, S.; Muneer, F.; Irfan, U.; Ayub, M. Assay of clopidogrel hydrogen sulphate in tablet form from different manufacturing sources by using UV spectroscopy. W. J. Pharm. Res. 2015, 4, 377–381. [Google Scholar]
- Ofori-Kwakye, K.; Osei-Yeboah, F.; Kipo, S.L. Formulation and quality evaluation of two conventional release tablet formulations. Int. J. Pharm. Sci. Rev. Res. 2010, 4, 94–99. [Google Scholar]
- Mohire, N.; Yadav, A. Novel approach to formulate β-cyclodextrin complexed mouth dissolving tablet of metronidazole and its in-vitro evaluation. J. Pharm. Res. 2010, 3, 662–667. [Google Scholar]
- Sharma, D.; Singh, M.; Kumar, D.; Singh, G. Formulation development and evaluation of fast disintegrating tablet of cetirizine hydrochloride: A novel drug delivery for pediatrics and geriatrics. J. Pharm. 2014, 2014, 808167. [Google Scholar] [CrossRef]
- He, L.W.; Zeng, L.; Tian, N.; Li, Y.; He, T.; Tan, D.M.; Zhang, Q.; Tan, Y. Optimization of food deprivation and sucrose preference test in SD rat model undergoing chronic unpredictable mild stress. Anim. Models Exp. Med. 2020, 3, 69–78. [Google Scholar] [CrossRef]
- Nin, M.S.; Couto-Pereira, N.S.; Souza, M.F.; Azeredo, L.A.; Ferri, M.K.; Dalprá, W.L.; Gomez, R.; Barros, H.M. Anxiolytic effect of clonazepam in female rats: Grooming microstructure and elevated plus maze tests. Eur. J. Pharmacol. 2012, 684, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, R.; Barros, H.M. Clonazepam increases in vivo striatal extracellular glucose in diabetic rats after glucose overload. Pharmacol. Biochem. Behav. 2003, 76, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Crusio, W.E.; Gerlai, R.T. Handbook of Molecular-Genetic Techniques for Brain and Behavior Research; Elsevier: Amsterdam, The Netherland, 1999. [Google Scholar]
- Prut, L.; Belzung, C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur. J. Pharmacol. 2003, 463, 3–33. [Google Scholar] [CrossRef]
- Mura, P.; Bragagni, M.; Mennini, N.; Cirri, M.; Maestrelli, F. Development of liposomal and microemulsion formulations for transdermal delivery of clonazepam: Effect of randomly methylated β-cyclodextrin. Int. J. Pharm. 2014, 475, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Sakata, O.; Onishi, H.; Machida, Y. Clonazepam oral droplets for the treatment of acute epileptic seizures. Drug Dev. Ind. Pharm. 2008, 34, 1376–1380. [Google Scholar] [CrossRef] [PubMed]
- Al-Tahan, F.; Löscher, W.; Frey, H. Pharmacokinetics of clonazepam in the dog. Arch. Int. Pharmacodyn. Thérapie 1984, 268, 180–193. [Google Scholar]
- Gombas, A.; Szabó-Révész, P.; Kata, M.; Regdon, G.; Erős, I. Quantitative determination of crystallinity of α-lactose monohydrate by DSC. J. Therm. Anal. Calorim. 2002, 68, 503–510. [Google Scholar] [CrossRef]
- Wójcik-Pastuszka, D.; Krzak, J.; Macikowski, B.; Berkowski, R.; Osiński, B.; Musiał, W. Evaluation of the release kinetics of a pharmacologically active substance from model intra-articular implants replacing the cruciate ligaments of the knee. Materials 2019, 12, 1202. [Google Scholar] [CrossRef] [Green Version]
- Patra, C.N.; Kumar, A.B.; Pandit, H.K.; Singh, S.P.; MEDURI, V.D. Design and evaluation of sustained release bi-layer tablets of propranolol hydrochloride. Acta Pharm. 2007, 57, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Barakat, N.S.; Elbagory, I.M.; Almurshedi, A.S. Controlled-release carbamazepine granules and tablets comprising lipophilic and hydrophilic matrix components. AAPS PharmSciTech 2008, 9, 1054–1062. [Google Scholar] [CrossRef] [Green Version]
- Merchant, H.A.; Shoaib, H.M.; Tazeen, J.; Yousuf, R.I. Once-daily tablet formulation and in vitro release evaluation of cefpodoxime using hydroxypropyl methylcellulose: A technical note. AAPS PharmSciTech 2006, 7, E178–E183. [Google Scholar] [CrossRef] [PubMed]
- Roni, M.A.; Islam, M.S.; Kibria, G.; Sadat, S.M.A.; Rony, R.; Rahman, H.; Jalil, R.U. Effects of poloxamer and HPMC on the dissolution of clonazepam-polyethylene glycol solid dispersions and tablets. Ind. J. Pharm. Educ. Res. 2011, 45, 139. [Google Scholar]
- O’Neil, M.J. The Merck Index: An Encyclopedia of Chemicals, Drugs and Biologicals; Royal Society of Chemistry: London, UK, 2013; Volume 15. [Google Scholar]
- Awasthi, R.; Kulkarni, G.T.; Ramana, M.V.; Pinto, T.d.J.A.; Kikuchi, I.S.; Ghisleni, D.D.M.; de Souza Braga, M.; De Bank, P.; Dua, K. Dual crosslinked pectin–alginate network as sustained release hydrophilic matrix for repaglinide. Int. J. Biol. Macromol. 2017, 97, 721–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, R. Floating in-situ raft forming liquid gastroretentive drug delivery system containing poorly water soluble drug. Bull. Pharm. Sci. 2021, 44, 301–312. [Google Scholar] [CrossRef]
- Larhrib, H.; Martin, G.P.; Prime, D.; Marriott, C. Characterisation and deposition studies of engineered lactose crystals with potential for use as a carrier for aerosolised salbutamol sulfate from dry powder inhalers. Eur. J. Pharm. Sci. 2003, 19, 211–221. [Google Scholar] [CrossRef]
- Kaialy, W.; Martin, G.P.; Ticehurst, M.D.; Royall, P.; Mohammad, M.A.; Murphy, J.; Nokhodchi, A. Characterisation and deposition studies of recrystallised lactose from binary mixtures of ethanol/butanol for improved drug delivery from dry powder inhalers. AAPS J. 2011, 13, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.M.; Mohammed, E.J. Preparation and In-Vitro Evaluation of Clopidogrel Bisulfate Liquisolid Compact. Iraqi J. Pharm. Sci. 2018, 135–149. [Google Scholar] [CrossRef] [Green Version]
- Barros, H.; Tannhauser, S.; Tannhauser, M.; Tannhauser, M. The effects of GABAergic drugs on grooming behaviour in the open field. Pharmacol. Toxicol. 1994, 74, 339–344. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition (mg) | Pure Drug | PM1 | PM2 | PM3 | SD1 | SD2 | SD3 | PM4 | PM5 | PM6 | SD4 | SD5 | SD6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clonazepam | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| HPMCk4000 | - | 1 | 2 | 3 | 1 | 2 | 3 | - | - | - | - | - | - |
| PEG6000 | - | - | - | - | - | - | - | 1 | 2 | 3 | 1 | 2 | 3 |
| Ratio % w/w Drug: polymer | 1:0 | 1:1 | 1:2 | 1:3 | 1:1 | 1:2 | 1:3 | 1:1 | 1:2 | 1:3 | 1:1 | 1:2 | 1:3 |
| Composition (mg) | Formulation Code | |||||
|---|---|---|---|---|---|---|
| Ingredients | A1 | A2 | A3 | A4 | A5 | A6 |
| Mixture 1 (1 mg Clonazepam) | ||||||
| Avecil: Lactose ratio % w/w | 0.1:1 | 0.2:1 | 0.4:1 | 0.6:1 | 0.8:1 | 1:1 |
| Mixture 2 (1 mg Clonazepam) | ||||||
| Ingredients | B1 | B2 | B3 | B4 | B5 | B6 |
| Lactose: Avecil ratio % w/w Na Alginate % w/w HPMC % w/w | 1:1 1 - | 1:1 2 - | 1:1 3 - | 1:1 - 1 | 1:1 - 2 | 1:1 - 3 |
| Mg Stearate | 1 | 1 | 1 | 1 | 1 | 1 |
| Total weight (mg) | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 |
| A | ||||
|---|---|---|---|---|
| Formula | Drug Content % | Hardness (Kg/cm2) | Thickness (mm) | Friability % |
| A1 | 98.2 ± 1.5 | 9.0 ± 0.5 | 2.1 ± 0.05 | 0.5 ± 0.01 |
| A2 | 95.1 ± 2.5 | 9.8 ± 0.5 | 2.1 ± 0.02 | 0.6 ± 0.02 |
| A3 | 96.3 ± 1.0 | 9.7 ± 0.5 | 2.2 ± 0.05 | 0.4 ± 0.01 |
| A4 | 98.1 ± 0.5 | 9.0 ± 1.1 | 2.2 ± 0.01 | 0.3 ± 0.01 |
| A5 | 95.5 ± 2.5 | 9.1 ± 1.1 | 2.3 ± 0.05 | 0.6 ± 0.02 |
| A6 | 97.3 ± 1.5 | 10.5 ± 0.1 | 1.9 ± 0.02 | 0.7 ± 0.01 |
| B | ||||
| B1 | 98.5 ± 0.5 | 10.8 ± 0.5 | 2.1 + 0.01 | 0.9 ± 0.01 |
| B2 | 95.0 + 2.1 | 11.8 ± 1.5 | 1.9 ± 0.01 | 15.0 ± 0.5 |
| B3 | 96.5 ± 1.5 | 12.0 ± 0.8 | 2.1 ± 0.02 | 15.0 ± 0.5 |
| B4 | 97.5 ± 0.5 | 6.5 ± 0.2 | 2.2 ± 0.01 | 0.5 ± 0.01 |
| B5 | 99.6 + 0.5 | 7.4 ± 2.5 | 1.8 ± 0.2 | 15.0 ± 0.5 |
| B6 | 99.5 ± 1.5 | 9.0 ± 0.5 | 2.0 ± 0.1 | 15.0 ± 0.5 |
| Batch Code | Zero Order | First Order | Higuchi |
|---|---|---|---|
| R2 | R2 | R2 | |
| B1 | 0.6692 | 0.8375 | 0.8519 |
| B2 | 0.7068 | 0.8251 | 0.8798 |
| B3 | 0.6098 | 0.7291 | 0.7995 |
| B4 | 0.6686 | 0.7370 | 0.8499 |
| B5 | 0.6926 | 0.7457 | 0.8679 |
| B6 | 0.7806 | 0.8253 | 0.9254 |
| Pharmacokinetic Parameters | Buccal CLZ Lozenges (0.25 mg/kg) (Group I) | Oral CLZ Suspension (0.25 mg/kg) (Group II) |
|---|---|---|
| Cmax (ng/mL) | 70.8 ± 2.1 | 50.69 ± 2.81 |
| Tmax (h) | 4 ± 0.01 | 2 ± 0.002 |
| AUC 0–t (ng/mL h) | 735.5 ± 10.22 | 168.68 ± 5.98 |
| t1/2 (h) | 5.3 ± 0.3 | 1.4 ± 0.1 |
| Vz/F (mg/kg)/(ng/mL) | 0.0025 ± 0.0002 | 0.0020 ± 0.0001 |
| Cl/F (mg/kg)/(ng/mL)/h | 0.0012 ± 0.0001 | 0.0025 ± 0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomaa, E.; El Deeb, S.; Ibrahim, A.E.; Faisal, M.M. Bimodal Release Two-In-One Clonazepam Matrix Lozenge Tablets for Managing Anxiety-Related Disorders: Formulation, Optimization and In Vivo Evaluation. Sci. Pharm. 2022, 90, 43. https://doi.org/10.3390/scipharm90030043
Gomaa E, El Deeb S, Ibrahim AE, Faisal MM. Bimodal Release Two-In-One Clonazepam Matrix Lozenge Tablets for Managing Anxiety-Related Disorders: Formulation, Optimization and In Vivo Evaluation. Scientia Pharmaceutica. 2022; 90(3):43. https://doi.org/10.3390/scipharm90030043
Chicago/Turabian StyleGomaa, Eman, Sami El Deeb, Adel Ehab Ibrahim, and Mennatullah M. Faisal. 2022. "Bimodal Release Two-In-One Clonazepam Matrix Lozenge Tablets for Managing Anxiety-Related Disorders: Formulation, Optimization and In Vivo Evaluation" Scientia Pharmaceutica 90, no. 3: 43. https://doi.org/10.3390/scipharm90030043
APA StyleGomaa, E., El Deeb, S., Ibrahim, A. E., & Faisal, M. M. (2022). Bimodal Release Two-In-One Clonazepam Matrix Lozenge Tablets for Managing Anxiety-Related Disorders: Formulation, Optimization and In Vivo Evaluation. Scientia Pharmaceutica, 90(3), 43. https://doi.org/10.3390/scipharm90030043