Abstract
Our main target was to develop methods for the quality control of the tablet «ramipril» according to the indicators of «Quantitative determination», «Impurities» and «Dissolution». New, precise, accurate and green HPLC methods were developed for the determination of ramipril and its impurities in tablets. The separation was accomplished using a diode array detector at 210 nm with an isocratic and gradient mobile phase consisting of a 0.2 g/L solution of sodium hexanesulfonate (pH 2.7) and the acetonitrile and chromatographic columns Acclaim 120 C18 and Inertsil ODS-3. The developed method was validated in accordance with ICH guidelines. The analysis of impurities was performed within a run duration of less than 25 min, which is about a two times shorter than that of the official Ph. Eur. method. The analysis of ramipril in tablets was performed with a run duration of less than 4.5 min, which is about three times shorter than that of the official USP method. The developed methods were successfully applied for the quality control of the tablet «ramipril» according to the indicators of «Quantitative determination», «Impurities» and «Dissolution». In addition, they proved its superiority over the reported methods in terms of greenness using different assessment tools.
1. Introduction
The quality indicators of medicinal products, which ensure their effectiveness and safety, are established in the registration documentation and the pharmacopoeia. Moreover, the quality of medicinal products is established at the stage of pharmaceutical development, for which a general methodological approach and special approaches are defined in relation to different dosage forms, generic drugs, original drugs, etc. Pharmaceutical enterprises with a large product portfolio are faced with the problem of significant time consumption and the need to involve additional units of equipment for routine drug control. Therefore, quality control methods need constant review and optimization with the involvement of modern technical means. The development and optimization of such analytical methods allow significant reductions in time spent on the analysis and preparation/regeneration of equipment/chromatographic columns, as well as reductions in the cost of the quality control of medicinal products. Taking into account the new approaches to ensuring the quality of API and dosage forms, there is a need to create analytical methods that meet the current requirements of the pharmaceutical regulations of Ukraine, EU countries, the USA, etc., in terms of quality control, as the approaches that were used 10 years ago are now somewhat outdated. To ensure regulatory compliance, there is a need to refine existing methods, and in some cases, to develop new ones.
Hypertension is the most common disease, which is accompanied by high mortality among people of working age and by disabilities from cardiovascular and cerebrovascular diseases [1]. The attention of researchers is focused on the study of methods of the analysis of angiotensin converting enzyme (ACE) inhibitors in medicines and their optimization. Ramipril is a prodrug and nonsulfhydryl ACE inhibitor with antihypertensive activity [2]. Its chemical name is (2S,3aS,6aS)-1-[(2S)-2-[[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino]propanoyl]-3,3a,4,5,6,6a-hexahydro-2H-cyclopenta[b]pyrrole-2-carboxylic acid. Ramipril is sparingly soluble in water and freely soluble in methanol, pKa1 = 3.74 (caboxylic acid); pKa2 = 5.15 (secondary amine), Log P = 2.9 [2,3]. There are a few analytical methods for the simultaneous determination of ramipril with other active substances and for the quantification of ramipril alone [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21]. However, the presented existing analytical methods often have limited application due to having inconsistently sufficient sensitivity, specificity, high time consumption, a long-term and non-compliance with the principles of «green» chemistry. Therefore, the development of new methods and the optimization of existing methods of the quality control of dosage forms of ramipril is an urgent problem that is planned to be worked on during the implementation of this work.
The monograph of ramipril from the European Pharmacopoeia (Ph. Eur.) [3] and US Pharmacopoeia (USP) [22] prescribes the HPLC method for testing the impurities of ramipril as an active substance [3] and of tablets of ramipril [22]. The disadvantages of this method are the following: a long run time (about 50 min) and the consumption of a considerable volume of mobile phase per single run. The monograph of ramipril from the Ph. Eur. [3] prescribes the titrimetric method (alkalimetry) for the assay of ramipril as an active substance. The disadvantage of this method is the impossibility of using dosage forms of ramipril for assay.
The aim of this study was to develop methods for the quality control of tablets of «ramipril» according to the indicators of «Quantitative determination», «Impurities» and «Dissolution». The present study describes a new HPLC method that is intended for routine use in quality control laboratories.
2. Materials and Methods
2.1. Chemicals and Reagents
Ramipril (purity ≥ 98% (HPLC)) was purchased from AARTI Industries Limited (Mumbai, India). Ramipril tablets of 2.5 mg, 5 mg, 10 mg were purchased from a local pharmacy. Standards of the active substance ramipril and its four specified impurities, ramipril impurity A, ramipril impurity B, ramipril impurity C and ramipril impurity D, were supplied by EDQM and USP.
The used chemicals, acetonitrile, sodium hexanesulfonate and phosphoric acid, were gradient grade, purchased from Merck, Darmstadt, Germany. The demineralized water that was used for analyses was an in-house product of Stilman with a conductivity of less than 0.5 µS/cm.
2.2. Instrumental
The following HPLC columns were used: Inertsil ODS-3 150 mm × 4.6 mm, 3 µm, and Acclaim 120 C18 250 mm × 4.6 mm, 5 µm. The used columns were purchased from GL Sciences, Tokyo, Japan and Thermo Fisher Scientific, Vantaa, Finland.
In this research, the Agilent 1260 Infinity II LC System with a diode array detector controlled by ChemStation Rev. C.01.07 SR1 and Agilent 1200 Infinity with a diode array detector were used (Agilent Technologies, Inc., Santa Clara, CA, USA).
The following additional instrumental equipment was used: analytical balance Mettler Toledo XPE-205 (Mettler-Toledo International Inc., Greifensee, Switzerland), pH meter Mettler Toledo Seven Easy (Mettler-Toledo International Inc., Greifensee, Switzerland), dissolution tester Erweka DT 820 (ERWEKA GmbH, Langen, Germany), US bath Elmasonic P (Elma Schmidbauer GmbH, Singen, Germany) and IKA orbital shaker HS260 (IKA Werke GmbH & Co. KG, Staufen im Breisgau, Germany). The regenerated cellulose (RC) 0.45 µm syringe filters were purchased from Agilent Technologies.
2.3. Sample Preparation and Chromatographic Conditions
2.3.1. Sample Preparation and Chromatographic Conditions for Determination of Impurities of Ramipril in Tablets
For mobile phase A, the pH of a solution of 0.2 g/L of sodium hexanesulfonate was adjusted to 2.7 with phosphoric acid R (for example, 200 mg of sodium hexanesulfonate R is dissolved in 1000 mL of water, and the pH is adjusted to 2.7 with phosphoric acid).
Mobile phase B—Acetonitrile.
Solvent (SLV)—mobile phase A: mobile phase B (1:1 (v/v)).
Preparation of test solution. A solution was prepared with a concentration of 0.5 mg/mL of ramipril in a SLV.
Preparation of reference solution (a). A solution was prepared containing 2 mg/mL CRS ramipril impurity A, 2 mg/mL CRS ramipril impurity B, 2 mg/mL CRS ramipril impurity C, 2 mg/mL CRS ramipril impurity D and 2 mg/mL CRS ramipril in a SLV.
Preparation of reference solution (b). A solution was prepared with a concentration of 2.5 μg/mL of ramipril in a SLV.
Chromatography was carried out on a liquid chromatograph with a spectrophotometric detector under the following conditions: a 150 mm × 4.6 mm octadecylsilyl column, 3 µm (Inertsil ODS-3); a flow rate of 1.5 mL/min; detection at 210 nm; a column temperature of (45 ± 1) °C; injection with 20 μL; and elution mode: gradient according to the following program (Table 1):

Table 1.
Gradient mode for the HPLC method for analysis of ramipril impurities in tablets.
The chromatographic system was considered suitable if the following requirements were met:
-Reference solution (a)
The separation factor between the peaks of ramipril of impurity A and ramipril should be at least 1.5.
The separation factor between the peaks of ramipril of impurity B and ramipril should be at least 1.5.
The calculation does not include peaks with a relative retention time of up to 0.2 (placebo and mobile phase peaks) and peaks present in the chromatogram of the SLV. The limit of quantification of unidentified impurities is 0.09%.
2.3.2. Sample Preparation and Chromatographic Conditions for Quantitative Determination of Ramipril in Tablets
Mobile phase A, mobile phase B and the solvent were the same as those in Section 2.3.1.
Preparation of test solution. A solution was prepared with a concentration of 0.1 mg/mL of ramipril in a SLV.
Preparation of reference solution. A solution was prepared with a concentration of 0.1 mg/mL of CRS ramipril in a SLV.
Chromatography was carried out on a liquid chromatograph with a spectrophotometric detector under the following conditions: a 250 mm × 4.6 mm octadecylsilyl column, 5 µm (Acclaim 120 C18); a flow rate of 1.0 mL/min; a detection at 210 nm; a column temperature of (45 ± 1) °C; injection with 3 μL; elution mode: isocratic; and mobile phase—mobile phase A: mobile phase B (1:1 (v/v)). The chromatographic system was considered suitable if the following requirements were met:
-Reference solution (b)
The efficiency of the chromatographic column, calculated from the ramipril peak, should be at least 2000 theoretical plates.
The relative standard deviation calculated for the peak areas of ramipril should not be more than 1.0%.
The content of ramipril in a tablet should be from 2.25 to 2.625 mg (for dosage form 2.5 mg), 4.5 mg to 5.25 mg (for dosage form 5 mg) and 9.0 mg to 10.5 mg (dosage form 10 mg) based on the average weight of one tablet.
2.3.3. Sample Preparation and Chromatographic Conditions for Quantitative Determination of Ramipril in «Dissolution Test»
The dissolution medium was a 0.1 M solution of hydrochloric acid, the volume of the dissolution medium was 500 mL, the rotation speed was 50 rpm, and the dissolution time was 30 min. Apparatus 2 (Paddle) was used.
Preparation of test solution. One tablet was placed in the vessel of the dissolution apparatus. After 30 min, 25 mL of the solution was taken from the center of the vessel of the dissolution apparatus and filtered through a membrane filter with a pore size of 0.45 μm, discarding the first portions of the filtrate (dilution coefficient: ).
Preparation of reference solution. A solution was prepared with a concentration of 5 μg/mL (for dosage form 2.5 mg/tablet) or 10 μg/mL (for dosage form 5 mg/tablet) or 20 μg/mL (for dosage form 10 mg/tablet) of CRS ramipril in a 0.1 M solution of hydrochloric acid.
Chromatography was carried out on a liquid chromatograph with a spectrophotometric detector under the conditions described in Section 2.3.2.
The amount of ramipril that went into the solution in 30 min should be at least 80% (Q) of the content of the active substance in one tablet.
2.4. Validation of HPLC Method
All validation was carried out in accordance with the requirements of the ICH Validation of Analytical Procedures: Text and Methodology, Q2 (R1) [23].
2.4.1. Validation of HPLC Method for the Determination of Impurities of Ramipril in Tablets
To study the specificity, the absence of the interference of the peaks of ramipril and its specified impurities with the peaks of the SLV and components of the drug was checked.
To study the linearity, 9 solutions containing the following range of concentrations of analytes in the SLV were prepared:
Ramipril impurity A: 0.29–6.84 μg/mL (equivalent to an impurity content of 0.06–1.37%).
Ramipril impurity B: 0.26–6.33 μg/mL (equivalent to an impurity content of 0.05–1.27%).
Ramipril admixture C: 0.30–7.23 μg/mL (equivalent to an admixture content of 0.06–1.45%).
Ramipril impurity D: 2.53–60.60 μg/mL (equivalent to the impurity content of 0.51–12.12%).
Ramipril: 0.25–30.29 μg/mL (equivalent to an impurity content of 0.05–6.06%).
To check the accuracy, 9 model solutions were prepared (3 concentration levels, 3 solutions for each level) containing a known addition of impurities to the test solution, along with 3 parallel test solutions without the additive. Solutions were prepared for each of the 3 dosages.
Precision was studied at the level of repeatability and intermediate precision. Under the conditions of repeatability, 6 parallels of the tested solution were prepared for each of the 3 dosages. Under the conditions of intermediate precision, 12 parallels of the tested solution were prepared for each of the 3 dosages.
2.4.2. Validation of HPLC Method for the Quantitative Determination of Ramipril in Tablets
To study the specificity, the absence of the interference of the peaks of ramipril with the peaks of the SLV and components of the drug was checked.
To study linearity, 7 solutions were prepared containing ramipril at a concentration of 0.066–1.137 mg/mL (corresponding to 66–137% of the nominal concentration of ramipril in the test solution)
To check the accuracy, 9 model solutions were prepared (3 concentration levels, 3 solutions for each level). Solutions were prepared for each of the 3 dosages.
Precision was studied at the level of repeatability. Under conditions of repeatability, 6 parallels of the tested solution were prepared for each of the 3 dosages.
2.4.3. Validation of HPLC Method for the Quantitative Determination of Ramipril in «Dissolution Test»
To study the specificity, the absence of theinterference of the peaks of ramipril with the peaks of the solvent, dissolution media and components of the drug was checked.
To study linearity, 7 solutions were prepared containing ramipril at a concentration of 2.5–25.4 μg/mL (corresponding to a range from 51% of the nominal concentration of ramipril in the test solution for 2.5 mg/tablet to 127% of the nominal concentration of ramipril in the test solution for 10 mg/tablet)
To check the accuracy, 9 model solutions were prepared (3 concentration levels, 3 solutions for each level). Solutions were prepared for each of the 3 dosages.
Precision was studied at the level of repeatability. Under conditions of repeatability, 6 parallels of the tested solution were prepared for each of the 3 dosages.
3. Results
3.1. HPLC Method Development
The first step in the development of methods for the quality control of «ramipril» tablets is the creation of an HPLC method that is suitable for routine use in quality control laboratories according to «Quantitative determination», «Impurities» and «Dissolution».
3.1.1. HPLC Method Development for the Determination of Impurities of Ramipril in Tablets
A precise and accurate green HPLC method was developed for the determination of impurities of ramipril in pharmaceutical dosage forms. A gradient mobile phase consisting of a 0.2 g/L solution of sodium hexanesulfonate (pH 2.7), acetonitrile, at a flow rate of 1.5 mL/min and ambient temperature was used for the analysis on an Inertsil ODS-3 (150 × 4.6 mm, 3 µm) column (Figure 1). The total run time was about 25 min. The injection volume was 20 µL. The Inertsil ODS-3 (4.6 × 150 mm, 3 µm) and Acclaim 120 C18 (250 × 4.6 mm, 5 µm) columns were used for the separation. Experimental studies revealed that the first column was the best one to obtain a good resolution and separation of peaks. The Inertsil ODS-3 (150 × 4.6 mm, 3 µm) column is an ideal modified octadecyl endcapped column with an active surface of 450 m2/gram particles, 100 Å pores and 15% carbon load. The best detection wavelength was 210 nm, which demonstrated the highest sensitivity along with a reasonable response. The effect of the flow rate was investigated using 0.5–1.5 mL/min values. A flow rate of 1.5 mL/min was found to be the best for achieving good separation in a reasonable time.
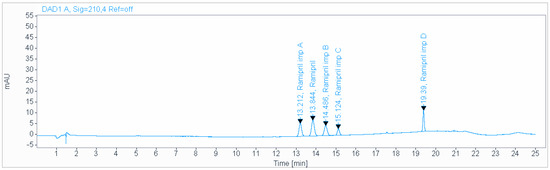
Figure 1.
Chromatogram obtained using gradient mobile phase consisting of 0.2 g/L solution of sodium hexanesulfonate (pH 2.7), acetonitrile, at a flow rate of 1.5 mL/min and ambient temperature, used for the analysis on an Inertsil ODS-3 (4.6 × 150 mm, 3 µm) column.
The usage of ion pairing reagents in HPLC is a known approach in HPLC method development, which allow for the separation of ionic and highly polar substances on reversed-phase HPLC columns [24]. Given the previous experience of our scientific group, we decided to apply sodium hexanesulfonate. The use of gradient elution made it possible to separate ramipril impurities. The results of peak identification with calculated LOQ are given in Table 2.
Table 2.
Identification of peaks (reference solution (a)).
Rationing at the time of release: impurities A, B, C: no more than 0.5% of each; impurity D: no more than 0.5%; any impurity: no more than 0.2%; and the amount of impurity: no more than 1.0%.
Rationing during the shelf life: impurities A, B, C: no more than 0.5% of each; impurities D, E: no more than 5.0%; any impurity: no more than 0.5%; and the amount of impurity: no more than 5.0%.
3.1.2. HPLC Method Development for the Quantitative Determination of Ramipril in Tablets
A precise and accurate green HPLC method was developed for the determination of ramipril in pharmaceutical dosage forms. An isocratic mobile phase consisting of a 0.2 g/L solution of sodium hexanesulfonate (pH 2.7), acetonitrile (50:50 v/v), at a flow rate of 1.0 mL/min and ambient temperature was used for the analysis on an Acclaim 120 C18 (250 × 4.6 mm, 5 µm) column (Figure 2). The retention time was 4.15 min. The injection volume was 3 µL. The Acclaim 120 C18 (250 × 4.6 mm, 5 µm) or equivalent and Inertsil ODS-3 (4.6 × 150 mm, 3 µm) columns were used for the separation. Experimental studies revealed that the first column was the best one to obtain a good retentive time. The best detection wavelength was 210 nm, which demonstrated the highest sensitivity along with a reasonable response. It did not interfere with the SLV peak. The effect of the flow rate was investigated using 0.5–1.5 mL/min values. A flow rate of 1 mL/min was found to be the best for achieving good separation in a reasonable time.
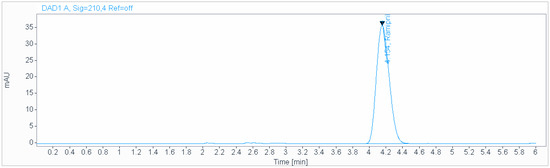
Figure 2.
Chromatogram obtained using isocratic mobile phase consisting of 0.2 g/L solution of sodium hexanesulfonate (pH 2.7), acetonitrile (50:50 v/v), at a flow rate of 1.0 mL/min and ambient temperature used for the analysis on an Acclaim 120 C18 (250 × 4.6 mm, 5 µm) column.
Different proportions of the mobile phase components were tested (Figure 3). The obtained results showed the best results using a mobile phase composed of a 0.2 g/L solution of sodium hexanesulfonate (pH 2.7), acetonitrile (50:50 v/v). As the acetonitrile percentage increased, the analyte peak interfered with the SLV peak, whereas as the acetonitrile percentage decreased, tailing increased, and a low number of theoretical plates was observed.
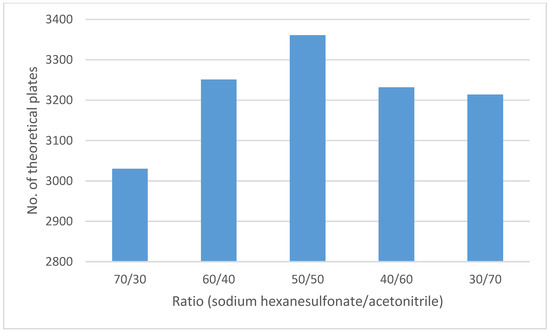
Figure 3.
Optimization of chromatographic conditions of the mobile phase composition.
3.1.3. HPLC Method Development for the Quantitative Determination of Ramipril in «Dissolution Test»
A precise and accurate green HPLC method was developed for the determination of ramipril in a «Dissolution test». We used the same chromatographic conditions as those used for the quantitative determination of ramipril in dosage forms.
An isocratic mobile phase consisting of a 0.2 g/L solution of sodium hexanesulfonate (pH 2.7), acetonitrile (50:50 v/v), at a flow rate of 1.0 mL/min and ambient temperature was used for the analysis on an Acclaim 120 C18 (250 × 4.6 mm, 5 µm) column (Figure 4). The injection volume was 3 µL.
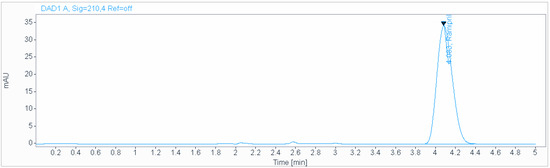
Figure 4.
Chromatogram obtained in «Dissolution test» using isocratic mobile phase consisting of 0.2 g/L solution of sodium hexanesulfonate (pH 2.7), acetonitrile (50:50 v/v), at a flow rate of 1.0 mL/min and ambient temperature used for the analysis on an Acclaim 120 C18 (250 × 4.6 mm, 5 µm) column.
3.2. Method Validation
The procedure was validated in compliance with the standards in accordance with the International Conference on Harmonization (ICH) [23].
3.2.1. Validation of HPLC Method for the Determination of Impurities of Ramipril in Tablets
To confirm the efficiency of the method, the following parameters were studied: specificity; linearity in the range of application; accuracy; precision; limit of detection (LOD); and limit of quantification (LOQ) [23]. The formulas that were used for the calculations are given in Appendix A. The summary of the validation results is given in Appendix B, Table A1.
The separation of impurities and other components in the spiking sample was appropriate. No interference was obtained among the ramipril peak, the impurity peaks and the matrix peaks.
The linear regression parameters were calculated in accordance with the recommendations of the ICH Validation of Analytical Procedures: Text and Methodology, Q2 (R1) [23]. Excellent linearity of the method for the analysis of impurities of ramipril was confirmed by the RSD of the obtained response factors, which was lower than 7.0% (2.2%, 2.0%, 2.3%, 1.8%), and the obtained correlation coefficient was almost ideal (Table 3, Appendix B, Table A1).
Table 3.
Results of the calculation of the linear regression parameters and accuracy and precision of the method according to the calibration curve.
Recoveries from the spiked samples were close to 100% (93.9–105.4% for impurity A, 96.4–105.1% for impurity B, 95.1–105.7% for impurity C and 96.0–105.7% for impurity D). The RSD of the obtained results was lower than 4.0%. Excellent precision for the method for the analysis of ramipril was confirmed by the RSD of the detected content between the samples that were prepared by two analysts, which was lower than 15.0% (3.1%, 3.7%, 2.4%), and the difference between the content obtained in the results by the two analysts was lower than 20.0% (3.1%, 1.9%, 2.0%). The obtained results demonstrate good accuracy and precision for the proposed method (Table 4 and Table 5).
Table 4.
Calculations for the parameter “accuracy”.
Table 5.
Calculations for the parameter “precision”.
The LOD of the method for the determination of ramipril impurity A was determined to be ~0.030%, that for impurity B was ~0.037%, that for impurity C was ~0.070%, and that for impurity D was ~0.030% (acceptability criteria was LOD ≤ 0.15%). The LOQ of the method for the determination of ramipril impurity A was determined to be ~0.092%, that for impurity B was ~0.107%, that for impurity C was ~0.213%, and that for impurity D was ~0.091% (acceptability criteria was LOQ ≤ 0.25%).
The calculation of the conversion factor for impurity A was 1.0, that for impurity B was 1.1, that for impurity C was 2.5, and that for impurity D was 1.2 (acceptability criteria—0.8 ≤ k ≤ 1.2). To calculate the content of impurity C, the peak area of impurity C was multiplied by the correction factor (2.5). To calculate the content of other specified impurities, the correction factor may not be applied.
3.2.2. Validation of HPLC Method for the Quantitative Determination of Ramipril in Tablets
To confirm the efficiency of the method, the following parameters were studied: specificity; linearity in the range of application; accuracy; and precision [23]. The formulas that were used for the calculations are given in Appendix A. The summary of the validation results is given in Appendix C, Table A2.
The linear regression parameters were calculated in accordance with the recommendations of the ICH Validation of Analytical Procedures: Text and Methodology, Q2 (R1) [23]. A high determination coefficient was obtained for the regression analysis in the concentration ranges of 0.066–0.137 mg/mL. Excellent linearity of the method for the analysis of ramipril was confirmed by the RSD of the obtained response factors, which was lower than 3% (0.42%), and the obtained correlation coefficient was almost ideal (0.9999) (Table 6, Appendix C, Table A2).
Table 6.
Results of the calculation of the linear regression parameters and accuracy and precision of the method according to the calibration curve.
Excellent accuracy for the method for the analysis of ramipril was confirmed by the RSD of the found content for each concentration level, which was lower than 3% (1.1%, 1.3%, 2.1%), and the deviation of the mean “found/put” value was lower than 0.51% (0.01%). The obtained results demonstrate good accuracy of the proposed method.
Excellent precision for the method for analysis of the ramipril was confirmed by the RSD of the found content between samples, which was lower than 2% (0.45%, 1.70%, 0.54%), and the confidence interval of the “found/put” values was lower than 1.60% (0.82%) (Appendix C, Table A2). The obtained results demonstrate good accuracy and precision of the proposed HPLC method (Table 7 and Table 8).
Table 7.
Calculations for the parameter “accuracy” for the ramipril tablets.
Table 8.
Calculations for the parameter “precision”.
3.2.3. Validation of HPLC Method for the Quantitative Determination of Ramipril in «Dissolution Test»
To confirm the efficiency of the method, the following parameters were studied: specificity; linearity in the range of application; accuracy; and precision [23]. The formulas that were used for the calculations are given in Appendix A. The summary of validation results is given in Appendix D, Table A3. The linear regression parameters were calculated in accordance with the recommendations of the ICH Validation of Analytical Procedures: Text and Methodology, Q2 (R1) [23]. Excellent linearity of the method for the analysis of ramipril was confirmed by the RSD of the obtained response factors, which was lower than 5% (0.91%), and the obtained correlation coefficient was ideal (1.0000) (Table 9).
Table 9.
Results of the calculation of the linear regression parameters and accuracy and precision of the method according to the calibration curve.
Excellent accuracy for the method for the analysis of ramipril was confirmed by the RSD of the found content for each concentration level, which was lower than 5% (2.5%, 0.6%, 0.6%), and the deviation of the mean “found/put” value was lower than 0.96% (0.65%, 0.75%, 0.28%). The obtained results demonstrate good accuracy for the proposed HPLC method. Excellent precision for the method for the analysis of ramipril was confirmed by the RSD of the found content between samples, which was lower than 4.0% (1.2%, 2.1%, 0.8%), and the confidence interval of the “found/put” values was lower than 3.00% (1.76%, 1.79%, 1.77%) (Appendix D, Table A3). The obtained results demonstrate good accuracy and precision for the proposed HPLC method (Table 10 and Table 11).
Table 10.
Calculations for the parameter “accuracy” for ramipril tablets.
Table 11.
Calculations for the parameter “precision”.
3.3. Greenness Profile of the Developed Methods
Three greenness metrics were applied in the assessment of the environmental impact of the proposed methods: AES [25], AGREE [26] and GAP [27,28,29]. The calculated total penalty points for the proposed HPLC method for the determination of ramipril in tablets were equal to 22, so the obtained score was 78. The calculated total penalty points for the proposed HPLC method for the determination of ramipril impurities in tablets were equal to 24, so the obtained score was 76. As shown in Table 12, the AGREE assessment shows that the developed method is superior to the reported ones because of using biodegradable solvents indicated by the green sectors and its final score (0.75). As shown in Table 13, the AGREE assessment shows that the developed methods are superior to the reported ones because of using biodegradable solvents indicated by the green sectors and its final score (0.75, 0.69). Therefore, the suggested approach has a low ecological impact according to the above-mentioned greenness assessment metrics compared with the reported methods.
Table 12.
Comparison of the greenness profile between the proposed and reported method for determining ramipril in tablets.
Table 13.
Comparison of the greenness profile between the proposed and reported method for determining ramipril impurities in tablets.
4. Discussion
During the development of our method, we focused on choosing a selective, simple and «green» mobile phase and chromatographic column in order to achieve and express reproducible separations. The column Inertsil ODS-3 (150 mm × 4.6 mm, 3 µm) and Acclaim 120 C18 (250 mm × 4.6 mm, 5 µm) achieved the best separation with the highest values for critical pair resolutions, with the shortest run time for analysis.
Taking into consideration the experience of our research group in the development and validation of analytical methods with an API for the purposes of routine pharmaceutical analysis, we chose acetonitrile and sodium hexanesulfonate as mobile phase components, which allowed us to impose the principles of «green» chemistry. All three proposed HPLC methods involved the use of the same mobile phase consisting of 0.2 g/L solution of sodium hexanesulfonate (pH 2.7) and acetonitrile. The HPLC method for the determination of impurities of ramipril in tablets involved the usage of gradient elution, and the HPLC method for the quantitative determination of ramipril in tablets and the «Dissolution test» involved isocratic elution (50:50 v/v). The simplicity of the HPLC methods is convenient for use by chemists, so the methods can be widely used for the purposes of routine pharmaceutical analysis. Additionally, the HPLC methods are rapid (the run time for the determination of impurities is about 25 min, and quantitative determination is 4.5 min) and «green».
We validated all three HPLC methods. The acceptability criteria for all validation characteristics are clearly formulated. The quality control method for the drug “ramipril, with 2.5 mg, 5 mg and 10 mg tablets” according to the indicators of “Impurities”, “Quantitative determination” and “Dissolution test” is presented in Appendix A, Appendix B, Appendix C and Appendix D.
Greenness metrics (AES, AGREE and GAPI) were applied in the assessment of the environmental impact of the proposed methods. The proposed HPLC methods have a low ecological impact according to the above-mentioned greenness assessment metrics compared with the reported methods.
5. Conclusions
Precise and accurate green HPLC methods were developed for the determination of ramipril and its impurities in tablets. They were successfully validated according to ICH guidelines in terms of specificity, linearity in the range of application, accuracy, precision, LOD and LOQ. The AES, AGREE and GAPI assessment tools were used for the evaluation of the greenness degree of our proposed methods. The proposed methods have the ability to separate ramipril and impurities found in tablet dosage forms and tablet excipients. Therefore, the chromatographic methods can be used for the routine analysis of ramipril in dosage forms. In addition, the procedure can be applied to the detection and determination of impurities in tablets and in a “Dissolution test”. Quality control methods for the drug “ramipril, with 2.5 mg, 5 mg and 10 mg tablets” according to the indicators of “Impurities”, “Quantitative determination” and “Dissolution test” are suggested.
Author Contributions
Conceptualization, Y.K., K.T. and L.L.; methodology, Y.K., K.T. and L.L.; investigation, K.T.; resources, Y.K., K.T. and L.L.; data curation, Y.K., K.T. and L.L.; writing—original draft preparation, K.T. and L.L.; writing—review and editing, Y.K., K.T. and L.L.; investigation, Y.K., K.T. and L.L.; supervision, Y.K. and L.L.; project administration, Y.K., K.T. and L.L.; funding acquisition, Y.K., K.T. and L.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are available from the corresponding author upon request.
Acknowledgments
The authors would like to thank all the brave defenders of Ukraine who made the finalization of this article possible.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Formulas for calculations
The difference between the retention times for the peaks of ramipril on the chromatograms of the reference solution and the tested solution was calculated using the following formula:
where
is the retention time of impurity A (B, C or D) of ramipril on the chromatogram of the reference solution;
is the retention time of impurity A (B, C or D) of ramipril on the chromatogram of the test solution.
Concentration values in normalized coordinates were calculated using the following formula:
The value of the response of the device in normalized coordinates was calculated according to the following formula:
The “found/put” ratio was calculated using the following formula:
Coefficient b was calculated using the following formula:
where
m is the number of model solutions.
Coefficient a was calculated using the following formula:
Correlation coefficient r was calculated using the following formula:
The deviation of the average “found/put” value from 100% was calculated according to the following equation:
The maximum permissible deviation of the “found/put” value from 100% was calculated according to the following equation:
The confidence interval of the spread of the “found/put” values was calculated according to the following equation:
where
is the standard deviation of the “found/entered” ratios for all solutions;
is the one-sided Student’s coefficient for a probability of 95% (for degrees of freedom 9 − 1 = 8 is 1.8595).
The concentration of ramipril impurities in the tested solutions with additives was calculated using the following formula (“found”):
where
C0 is the concentration of ramipril impurities in the reference solution;
S1 is the peak area of ramipril impurities in the test solution;
S0 is the peak area of ramipril impurities in the reference solution.
The theoretical concentration of ramipril impurities in the test solutions with additives was calculated according to the following formula (“put”):
where
is the concentration of ramipril impurities in the drug, determined in model solutions P1–P3.
V is an aliquot of the reference solution added to the model solution;
is the concentration of ramipril impurities in the reference solution.
The “found”/“put” ratio (in percent) was calculated using the following formula:
The calculation of the content of ramipril impurity D in ramipril in 2.5 mg, 5 mg and 10 mg tablets was carried out according to the following formula:
The limit of detection (LOD) and limit of quantification (LOQ) were calculated using the signal/noise ratio using the following formula:
where
C is the concentration of ramipril impurities in the model solution;
is the signal/noise ratio from the chromatogram of the model solution.
To establish the need to introduce a conversion factor, the response factors of impurities were calculated according to the following formula:
where
is the average value of the peak area of the impurity (ramipril);
is the concentration of the impurity (ramipril) in the solution, μg/mL.
The conversion factor was calculated according to the following formula:
where
is the response factor for ramipril;
is the response factor for the impurity.
The theoretical concentration of ramipril in the test solutions with additives (for the quantitative determination of ramipril in tablets) was calculated according to the following formula (“put”):
where
C1 is the concentration of ramipril in the drug, determined in accordance with the MCQ;
3.2 is an aliquot of the drug taken to prepare a model solution;
V is an aliquot of the original reference solution added to the model solution;
is the concentration of ramipril in the reference solution.
The theoretical concentration of the ramipril tablet of 2.5 mg in the test solutions with additives (for dissolution test) was calculated according to the following formula (“put”):
The theoretical concentration of the ramipril tablet of 5 mg in the test solutions with additives (for dissolution test) was calculated according to the following formula (“put”):
The theoretical concentration of the ramipril tablet of 10 mg in the test solutions with additives (for dissolution test) was calculated according to the following formula (“put”):
where
C1 is the concentration of ramipril in the drug, determined in accordance with the MCQ;
0.5 (1.0; 2.0) is an aliquot of the drug taken to prepare a model solution;
V is an aliquot of the original reference solution added to the model solution;
is the concentration of ramipril in the reference solution.
Appendix B
Table A1.
Summary of validation results of the quality control method of the drug “ramipril, with 2.5 mg, 5 mg and 10 mg tablets” according to the indicator “Impurities”.
Table A1.
Summary of validation results of the quality control method of the drug “ramipril, with 2.5 mg, 5 mg and 10 mg tablets” according to the indicator “Impurities”.
| Investigation | Parameter | Eligibility Criteria | Evaluation | |
|---|---|---|---|---|
| Specificity | The purity of the peak of ramipril, calculated for the peaks of the tested solution | PFTS ≥ 990 | ≥999.935 | |
| The difference in retensive time of ramipril impurity A on the chromatogram of the reference solution and the test solution | ΔRT ≤ 2.0% | 0.11% | ||
| The difference in retensive time of ramipril impurity B on the chromatogram of the reference solution and the test solution | ΔRT ≤ 2.0% | 0.14% | ||
| The difference in retensive time of ramipril impurity C on the chromatogram of the reference solution and the test solution | ΔRT ≤ 2.0% | 0.17% | ||
| The difference in retensive time of ramipril impurity D on the chromatogram of the reference solution and the test solution | ΔRT ≤ 2.0% | 0.25% | ||
| The resolution between the peak of ramipril and the peak closest to it | Rs ≥ 1.5 | ≥2.21 | ||
| Linearity | Range of application (50% of the specification limit for an unidentified impurity—120% of the specification limit of the sum of impurities D) | 1.25–25.00 μg/mL | 0.25–30.29 μg/mL | |
| Intercept | impurity A | |a| ≤ 5.0 | 0.1 | |
| impurity B | 0.1 | |||
| impurity C | 0.6 | |||
| impurity D | 0.4 | |||
| ramipril | 0.1 | |||
| Correlation coefficient | impurity A | r ≥ 0.990 | 0.9989 | |
| impurity B | 0.9996 | |||
| impurity C | 0.9988 | |||
| impurity D | 0.9999 | |||
| ramipril | 0.9996 | |||
| The response factor of the individual concentration level, in % to the response factor of the target concentration | impurity A | 93.0% ≤ Z ≤ 107.0% | 98.7–105.4% | |
| impurity B | 98.4–103.6% | |||
| impurity C | 98.0–104.8% | |||
| impurity D | 97.2–102.9% | |||
| ramipril | 99.9–104.1% | |||
| Residual standard deviation | impurity A | RSD ≤ 7.0% | 2.2% | |
| impurity B | 2.0% | |||
| impurity C | 2.3% | |||
| impurity D | 1.8% | |||
| ramipril | 1.4% | |||
| Accuracy (calibration curve) | Deviation of the mean “found/put” value | impurity A | δ ≤ 1.60% | 1.52% |
| impurity B | 0.86% | |||
| impurity C | 1.16% | |||
| impurity D | 0.85% | |||
| ramipril | 1.12% | |||
| Accuracy (spiked samples) | Content in samples with an additive (impurity A) | 2.5 mg | 80.0% ≤ Z ≤ 120.0% | 94.2–105.4% |
| 5 mg | 93.9–104.0% | |||
| 10 mg | 95.5–102.7% | |||
| Content in samples with an additive (impurity B) | 2.5 mg | 80.0% ≤ Z ≤ 120.0% | 99.5–105.1% | |
| 5 mg | 99.6–101.3% | |||
| 10 mg | 96.4–104.5% | |||
| Content in samples with an additive (impurity C) | 2.5 mg | 80.0% ≤ Z ≤ 120.0% | 95.7–105.1% | |
| 5 mg | 95.1–102.5% | |||
| 10 mg | 97.9–105.7% | |||
| Content in samples with an additive (impurity D) | 2.5 mg | 80.0% ≤ Z ≤ 120.0% | 96.0–103.1% | |
| 5 mg | 96.0–104.4% | |||
| 10 mg | 97.4–105.7% | |||
| Standard deviation of the found content for each concentration level (impurity A) | 2.5 mg | RSD ≤ 15.0% | 2.4% | |
| 5 mg | 2.4% | |||
| 10 mg | 3.5% | |||
| Standard deviation of the found content for each concentration level (impurity B) | 2.5 mg | RSD ≤ 15.0% | 2.9% | |
| 5 mg | 1.2% | |||
| 10 mg | 2.3% | |||
| Standard deviation of the found content for each concentration level (impurity C) | 2.5 mg | RSD ≤ 15.0% | 2.5% | |
| 5 mg | 1.3% | |||
| 10 mg | 4.0% | |||
| Standard deviation of the found content for each concentration level (impurity D) | 2.5 mg | RSD ≤ 15.0% | 2.4% | |
| 5 mg | 2.0% | |||
| 10 mg | 3.8% | |||
| Precision (calibration curve) | Relative confidence interval | impurity A | ∆ ≤ 5.0% | 4.5% |
| impurity B | 3.7% | |||
| impurity C | 4.3% | |||
| impurity D | 4.2% | |||
| ramipril | 3.8% | |||
| Precision (spiked samples) | Standard deviation of the detected content between samples prepared by the same analyst | 2.5 mg | RSD ≤ 15.0% | ≤3.6% |
| 5 mg | ≤5.1% | |||
| 10 mg | ≤2.9% | |||
| Standard deviation of detected content between samples prepared by two analysts | 2.5 mg | RSDimp ≤ 15.0% | 3.1% | |
| 5 mg | 3.7% | |||
| 10 mg | 2.4% | |||
| The difference between the content obtained in the results by two analysts | 2.5 mg | ∆ ≤ 20.0% | 3.1% | |
| 5 mg | 1.9% | |||
| 10 mg | 2.0% | |||
| LOD | LOD | impurity A | LOD ≤ 0.15% | 0.03% |
| impurity B | 0.04% | |||
| impurity C | 0.07% | |||
| impurity D | 0.02% | |||
| ramipril | 0.03% | |||
| Signal to noise ratio for model solution L2 (0.1%) | impurity A | S/N ≥ 3 | ≥10.9 | |
| impurity B | ≥8.5 | |||
| impurity C | ≥5.6 | |||
| impurity D | ≥12.7 | |||
| ramipril | ≥10.0 | |||
| LOQ | LOQ | impurity A | LOQ ≤ 0.25% | 0.09% |
| impurity B | 0.11% | |||
| impurity C | 0.21% | |||
| impurity D | 0.09% | |||
| ramipril | 0.09% | |||
| Signal to noise ratio for model solution L3 (0.3%) | impurity A | S/N ≥ 10 | ≥36.9 | |
| impurity B | S/N ≥ 10 | ≥28.0 | ||
| impurity C | S/N ≥ 10 | ≥16.1 | ||
| impurity D | S/N ≥ 10 | ≥36.3 | ||
| ramipril | S/N ≥ 10 | ≥31.7 | ||
| Calculated conversion factor of identified impurities | Calculated conversion factor | impurity A | 0.8 ≤ k ≤ 1.2 | 1.0 |
| impurity B | 1.1 | |||
| impurity C | 2.5 | |||
| impurity D | 1.2 | |||
Appendix C
Table A2.
Summary of validation results of the quality control method of the drug “ramipril, with 2.5 mg, 5 mg and 10 mg tablets” according to the indicator “Quantitative determination”.
Table A2.
Summary of validation results of the quality control method of the drug “ramipril, with 2.5 mg, 5 mg and 10 mg tablets” according to the indicator “Quantitative determination”.
| Investigation | Parameter | Eligibility Criteria | Evaluation | |
|---|---|---|---|---|
| Specificity (ramipril, tablets 2.5 mg) | Peak purity of ramipril calculated for the peaks of the tested solution | 999.993 | ||
| The difference in the retention times of ramipril on the chromatogram of the reference solution and the tested solution | 1.52% | |||
| Linearity | Range of application | 70–130% (0.070–0.130 mg/mL) | 66–137% (0.066–0.137 mg/mL) | |
| Intercept | |a| ≤ 3 | 0.45 | ||
| Correlation coefficient | r ≥ 0.999 | 0.9999 | ||
| The response factor of the individual concentration level, in % to the response factor of the target concentration | 98.0% ≤ Z ≤ 102.0% | 99.2–100.5% | ||
| Residual standard deviation | RSD ≤ 3.0% | ≤0.42% | ||
| Accuracy | Deviation of the mean “found/put” value from the linearity study | δ ≤ 0.51% | 0.01% | |
| Content in samples with an additive | 2.5 mg | 97.0% ≤ Z ≤ 103.0% | 98.4–101.1% | |
| 5 mg | 98.1–101.5% | |||
| 10 mg | 97.0–101.2% | |||
| Standard deviation of the found content for each concentration level | 2.5 mg | RSD ≤ 3.0% | ≤1.1% | |
| 5 mg | ≤1.3% | |||
| 10 mg | ≤2.1% | |||
| Precision | Confidence interval of “found/put” values from the linearity study | ∆ ≤ 1.60% | 0.82% | |
| Standard deviation of the found content between samples | 2.5 mg | RSDr ≤ 2.0% | ≤0.45% | |
| 5 mg | ≤1.70% | |||
| 10 mg | ≤0.54% | |||
Appendix D
Table A3.
Summary of validation results of the quality control method of the drug “ramipril, with 2.5 mg, 5 mg and 10 mg tablets” according to the indicator “Dissolution test”.
Table A3.
Summary of validation results of the quality control method of the drug “ramipril, with 2.5 mg, 5 mg and 10 mg tablets” according to the indicator “Dissolution test”.
| Investigation | Parameter | Eligibility Criteria | Evaluation | |
|---|---|---|---|---|
| Specificity | Peak purity of ramipril calculated for the peaks of the tested solution | 999.989 | ||
| The difference in the retention times of ramipril on the chromatogram of the reference solution and the tested solution | 0.10% | |||
| Linearity | Range of application | 2.5 mg | 60–120% (0.003–0.006 mg/mL) | 50.8–508.0% (0.0025–0.0254 mg/mL) |
| 5 mg | 60–120% (0.006–0.0120 mg/mL) | 25.4–254% (0.0025–0.0254 mg/mL) | ||
| 10 mg | 60–120% (0.012–0.024 mg/mL) | 12.7–127.0% (0.0025–0.0254 mg/mL) | ||
| Intercept | 2.5 mg | |a| ≤ 5 | 0.09 | |
| 5 mg | 0.04 | |||
| 10 mg | 0.02 | |||
| Correlation coefficient | r ≥ 0.995 | 1.0000 | ||
| The response factor of the individual concentration level, in % to the response factor of the target concentration | 2.5 mg | 95.0% ≤ Z ≤ 105.0% | 98.5–101.0% | |
| 5 mg | 99.9–102.4% | |||
| 10 mg | 98.9–101.3% | |||
| Residual standard deviation | RSD ≤ 5.0% | ≤0.91% | ||
| Accuracy (ramipril, tablets 2.5 mg) | Deviation of the mean “found/put” value | 2.5 mg | δ ≤ 0.96% | 0.65% |
| 5 mg | 0.75% | |||
| 10 mg | 0.28% | |||
| Content in samples with an additive | 2.5 mg | 90.0% ≤ Z ≤ 110.0% | 96.4–100.7% | |
| 5 mg | 98.8–101.0% | |||
| 10 mg | 99.4–101.3% | |||
| Standard deviation of the found content for each concentration level | 2.5 mg | RSD ≤ 5.0% | ≤2.5% | |
| 5 mg | ≤0.6% | |||
| 10 mg | ≤0.6% | |||
| Precision (ramipril, tablets 2.5 mg) | Confidence interval of “found/put” values | 2.5 mg | ∆ ≤ 3.00% | 1.76% |
| 5 mg | 1.79% | |||
| 10 mg | 1.77% | |||
| Standard deviation of the found content between samples | 2.5 mg | RSDr ≤ 4.0% | ≤1.2% | |
| 5 mg | ≤2.1% | |||
| 10 mg | ≤0.8% | |||
References
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_3 (accessed on 22 March 2023).
- Ramipril. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Ramipril (accessed on 14 March 2023).
- European Pharmacopoeia, 11th ed.; 2022; Available online: https://www.edqm.eu/en/european-pharmacopoeia-ph.-eur.-11th-edition (accessed on 22 March 2023).
- Babu, K.A.; Kumar, G.V.; Sivasubramanian, L. Simultaneous estimation of ramipril and amlodipine in bulk and tablet dosage form by RP-HPLC method. Int. J. Pharm. Pharm. Sci. 2011, 3, 196–198. [Google Scholar]
- Dai, S.Y.; Qiu, S.T.; Wu, W.; Fu, C.M. Development and validation of an rp-hplc method for simultaneous determination of Ramipril and Amlodipine in tablets. J. Pharm. Anal. 2013, 3, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Elshanawane, A.A.; Mostafa, S.M.; Elgawish, M.S. Application of a validated, stability-indicating LC method to stress degradation studies of Ramipril and Moexipril. HCl Chromatogr. 2008, 67, 567–573. [Google Scholar] [CrossRef]
- Gupta, K.R.; Wankhede, S.B.; Tajne, M.R.; Wadodkar, S.G. Simultaneous determination of Amlodipine and Ramipril by high performance thin layer chromatography. Asian J. Chem. 2007, 19, 4177–4182. [Google Scholar]
- Logoyda, L. Analysis of approaches to the development and validation of the methods of analysis of some active pharmaceutical ingredients from the group of angiotensin converting enzyme inhibitors in drugs and biological liquids. Int. J. Appl. Pharm. 2019, 11, 1–7. [Google Scholar] [CrossRef]
- Kumar, A.M.; Kumar, P.V.; Nasare, M.; Rao, V.; Parasad, V.V.L.; Diwan, V.P. Isocratic RP-HPLC estimation of Ramipril and Amlodipine in pharmaceutical dosage form. J. Adv. Pharm. Educ. Res. 2012, 2, 137–145. [Google Scholar]
- Maste, M.M.; Kalekar, M.C.; Kadian, N.; Bhat, A.R. Development and validation of RP-HPLC method for simultaneous estimation of Amlodipine and Ramipril in bulk and tablet dosage form. Asian J. Res. Chem. 2011, 4, 1210–1213. [Google Scholar]
- Panchal, H.J.; Suhagia, B.N.; Patel, N.J.; Rathod, I.S.; Patel, B.H. Simultaneous estimation of Atorvastatin Calcium, Ramipril and Aspirin in capsule dosage form by RP-LC. Chromatographia 2009, 69, 91–95. [Google Scholar] [CrossRef]
- Patel, J.; Patel, M. RP-HPLC method development and validation for the simultaneous estimation of ramipril and amlodipine besylate in capsule dosage form. J. Chem. Pharm. Res. 2014, 6, 725–733. [Google Scholar]
- Patole, S.M.; Khodke, A.S.; Potale, L.V.; Damle, M.C. A validated HPLC method for analysis of atorvastatin calcium, ramipril and asprin as the bulk drug and in combined capsule dosage forms. Int. J. Pharm. Sci. Rev. Res. 2010, 4, 40–45. [Google Scholar]
- Rajput, P.S.; Kaur, A.; Gill, N.K.; Mittal, K.; Sarma, G.S. Simultaneous estimation of ramipril and amlodipine in bulk and tablet dosage form by RP-HPLC method. J. Appl. Pharm. Sci. 2012, 2, 160–165. [Google Scholar] [CrossRef]
- Ramadevi, K.; Saraswathi, Z.; Maniklal, D. RP-HPLC method development for simultaneous determination of the drugs Ramipril and Amlodipine. IJSR 2013, 2, 4–7. [Google Scholar]
- Sharma, R.; Khanna, S.; Mishra, G.P. Development and validation of RP-HPLC method for simultaneous estimation of Ramipril, aspirin and Atorvastatin in pharmaceutical preparations. J. Chem. 2012, 9, 2177–2184. [Google Scholar] [CrossRef]
- Srinivasa Rao, K.; Srinivas, K. RP-HPLC method for the determination of Losartan Potassium and Ramipril in combined dosage form. Indian J. Pharm. Sci. 2010, 72, 108–111. [Google Scholar]
- Lincy, J.; Mathew, G.; Venkata, R. Simultaneous estimation of Atorvastatin and Ramipril by RP-HPLC and spectroscopy. Pak. J. Pharm. Sci. 2018, 21, 282–284. [Google Scholar]
- Sharma, A.; Shah, B.; Patel, B. Scholars Research Library Simultaneous Estimation of Atorvastatin Calcium, Ramipril and Aspirin in Capsule Dosage Form Using HPTLC. Der. Pharma. Chem. 2010, 2, 10–16. [Google Scholar]
- Zuromska-Witek, B.; Stolarczyk, M.; Szlósarczyk, M.; Kielar, S.; Hubicka, U. Simple, Accurate and Multianalyte Determination of Thirteen Active Pharmaceutical Ingredients in Polypills by HPLC-DAD. Chemosensors 2023, 11, 25. [Google Scholar] [CrossRef]
- De Diego, M.; Godoy, G.; Mennickent, S.; Olivares, M.; Godoy, R. Stress degradation studies of ramipril by a validated stability-indicating liquid chromatographic method. J. Chil. Chem. Soc. 2010, 55, 450–453. [Google Scholar] [CrossRef]
- The United States Pharmacopeia. The National Formulary; United States Pharmacopeial Convention, Inc.: Rockville, MD, USA, 2021; Available online: https://www.uspnf.com (accessed on 22 March 2023).
- ICH Validation of Analytical Procedures: Text and Methodology, Q2 (R1), Geneva. 2005. Available online: https://www.ich.org/page/quality-guidelines (accessed on 22 March 2022).
- Snyder, R.L.; Kirckland, J.; Dolan, W.J. Introduction to Modern Liquid Chromatography, 3rd ed.; John Willey & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Gałuszka, A.; Migaszewski, Z.; Konieczka, P.; Namieśnik, J. Analytical Eco-Scale for assessing the greenness of analytical procedures. TrAC Trends Anal. Chem. 2012, 37, 61–72. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J.; Wojnowski, W. Complementary green analytical procedure index (ComplexGAPI) and software. R. Soc. Chem. 2021, 23, 8657–8665. [Google Scholar] [CrossRef]
- Rezk, M.; Monir, H.; Marzouk, H. Novel determination of a new antiviral combination; sofosbuvir and velpatasvir by high performance thin layer chromatographic method; application to real human samples. Microchem. J. 2019, 146, 828–834. [Google Scholar] [CrossRef]
- Tantawy, M.; Weshahy, S.; Wadie, M.; Rezk, M. A novel HPLC-DAD method for simultaneous determination of alfuzosin and solifenacin along with their official impurities induced: Via a stress stability study; Investigation of their degradation kinetics. Anal. Methods 2020, 12, 3368–3375. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).